

Abstract
The following findings concerning the structure of the cytochrome b6f complex and its component polypeptides, cyt b6, subunit IV and cytochrome f subunit are discussed:
Comparison of the amino acid sequences of 13 and 16 cytochrome b6 and subunit IV polypeptides, respectively, led to (a) reconsideration of the helix lengths and probable interface regions, (b) identification of two likely surface-seeking helices in cyt b6 and one in SU IV, and (c) documentation of a high degree of sequence invariance compared to the mitochondrial cytochrome. The extent of identity is particularly high (88% for conserved and pseudo- conserved residues) in the segments of cyt b6 predicted to be extrinsic on the n-side of the membrane.
The intramembrane attractive forces between trans-membrane helices that normally stabilize the packing of integral membrane proteins are relatively weak.
The complex isolated in dimeric form has been visualized, along with isolated monomer, by electron microscopy. The isolated dimer is much more active than the monomer, is the major form of the complex isolated and purified from chloroplasts, and is inferred to be a functional form in the membrane.
The isolated cyt b6f complex contains one molecule of chlorophyll a.
The structure of the 252 residue lumen-side domain of cytochrome f isolated from turnip chloroplasts has been solved by X-ray diffraction analysis to a resolution of 2.3 Å.
Keywords: Cytochrome bc1, electron transfer, energy transduction, membrane protein, structure
INTRODUCTION ANDGENERAL CONSIDERATIONS
The cytochrome2 b6f complex is one of the three integral oligomeric membrane protein complexes involved in linear or noncyclic electron transport in the chloroplast thylakoid and cyanobacterial membrane systems that participate in oxygenic photosynthesis. It is located electrochemically and in the pathways of electron transport between the two reaction center complexes (Cramer et al., 1991), and its photosynthetic electron transfer reactions can occur in the dark. It is phylogenetically related to the cytochrome bc1 complex of mitochondria and photosynthetic bacteria (Widger et al., 1984), with which there are many structure-function similarities.
Reviews have recently appeared on sequence-structure-function of the mitochondrial bc1 complex (Degli Esposti et al., 1993), mutational and mutagenesis studies of the bacterial bc1 complex (Gennis et al., 1993), and aspects of the function of the cytochrome b6f complex of oxygenic photosynthesis (Hope, 1993). The present article concerns the cytochrome b6f complex, new structural information on the complex, reconsideration of the larger number of compiled sequences of cytochrome b6 and subunit IV, and a discussion of the crystal structure determination of the cytochrome f subunit. It does not consider questions of structure-function related to the Rieske iron-sulfur protein subunit of the complex, aspects of which have been recently considered for the mitochondrial protein (Graham et al., 1993; Link et al., 1993). The present article has the underlying viewpoint that the set of data obtained on cytochrome bc1 complexes is not sufficient to describe all important properties related to the function of the b6f complex in oxygenic photosynthesis.
The four major (Mr > 15,000) subunit polypeptides of the cytochrome b6f complex that are readily detected using SDS-PAGE are cytochrome f (285 residues in spinach chloroplasts; MW = 31,372), cytochrome b6 (214 residues; MW = 24,038), the Rieske iron-sulfur protein (180 residues; MW = 18,922), and subunit IV (160 residues; MW = 17,444) (MW data for subunits from different sources compiled in Widger and Cramer, 1991). There are also 4–5 small (Mr < 5,000) subunits in the complex, one of which has been identified as a 37-residue pet G subunit V (Haley and Bogorad, 1989), whose function is not known.
SEQUENCE COMPARISONS AND STRUCTURAL INFERENCES FOR CYT b6 AND SUBUNIT IV
The assumption of major subunit and prosthetic and redox group similarity of the cytochrome b6f complex of oxygenic photosynthetic membranes to the cytochrome bc1 complex of mitochondria and purple photosynthetic bacteria (Hauska et al., 1983) was extended by the demonstration of significant amino acid sequence identity and hydrophobic segment alignment of the cyt b polypeptides (Widger et al., 1984). Hydropathy graphs derived from the amino sequence of the long hydrophobic putative membrane-spanning segments in the N-terminal half of the approximately 400-residue cyt b(bc1) could be aligned by cross-correlation analysis (Shiver et al., 1989) with a similar graph derived from the 214-residue spinach chloroplast cyt b6 sequence to (i) identify the four histidine residues apparently conserved and involved in heme coordination, and (ii) infer that cytochrome b6 is analogous to the hemebinding domain of cytochrome b of the bc1 complex, and subunit IV to its C-terminal half (Widger et al., 1984). This also implied that the information in the single cyt b gene product of the bc1 complex is contained in the split gene products, cyt b6 and subunit IV, of oxygenic photosynthesis. These genes are contiguous or close in higher plant chloroplasts and cyanobacteria, but separated by a large distance in the green alga Chlamydomonas (Büschlen et al., 1991).
The involvement of the same four histidine residues in ligation of the two hemes was also inferred from their conservation in six mitochondrial sequences (Saraste, 1984). The original models for folding of the cytochrome b heme binding domain across the membrane bilayer contained five trans-membrane helices, with heme coordination involving the second and fifth helices from the N-terminus (Widger et al., 1984; Saraste, 1984). It was subsequently proposed that the fourth helix in this model was not trans-membrane, but rather a surface-bound amphiphilic helix (Crofts et al., 1987). Support for the revised four-helix model of the cytochrome b heme-binding domain was provided for cyt b(bc1) by: (i) the consistent mapping pattern of cytochrome b mutants resistant to inhibitors known to act primarily at the quinone binding site on the n- (e.g., antimycin, funiculosin) and p- (e.g., stigmatellin, mythathiazol) sides (cf. discussion of notation in legend to Fig. 1) of the membrane (diRago and Colson, 1988; Howell and Gilbert, 1988; Daldal et al., 1989; Gennis et al., 1993); (ii) the location of both polypeptide termini of cyt b6 on the n-side of the membrane using protease accessibility of epitopes for peptide-directed antibodies (Szczepaniak and Cramer, 1990).
Fig. 1.

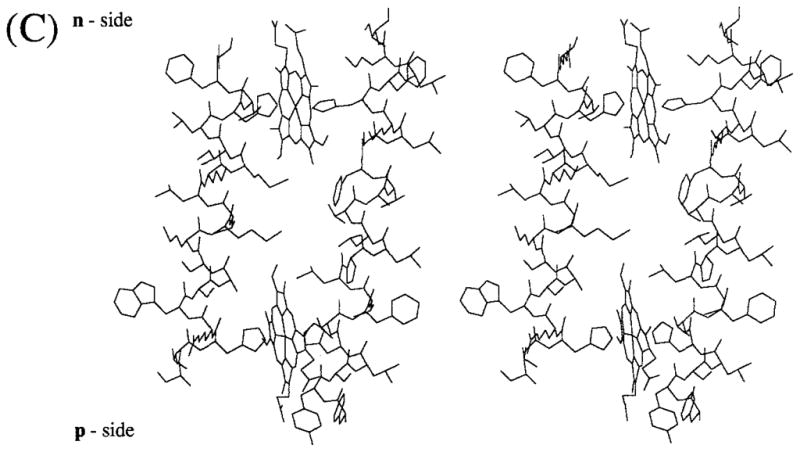
Folding pattern across the thylakoid membrane of (A) cytochrome b6 and (B) subunit IV predicted by the distribution and conservation in the 13 and 16 sequences shown in Table IA, B of long (ca. 20 residue) hydrophobic segments. The hydrophobic membrane bilayer is delineated by the rectangular boxes. Hatched circles represent residues that are not invariant or pseudo-invariant. (C) Molecular model (stereo view) of heme-bridged helices B and D of spinach cyt b6. The amino acid sequences of these helices are: B helix, NH2 (p-side)- SVHRWSASMMVLMMILHVF-COOH; D helix, NH2 (p-side)-FYSLHTFVLPLLTAVFMLMHFLMI-COOH. Nomenclature: (1) The two sides of the membrane are designated n- and p-, the electrochemically negative and positive sides, which correspond to stroma and lumen, respectively, in the case of chloroplast thylakoid membranes. This notation is chosen instead of i and o proposed in the recent review of Degli Esposti et al. (1993) because it is conceptually simple, which is important for teaching purposes, allows comparative discussion of bacteria, chloroplasts, and mitochondria without reference to the definition of all of the respective extramembrane compartments (lumen, matrix, cytoplasmic, stroma, intermembrane, periplasmic), and is unambiguous, unlike i and o which can readily be mistaken by nonspecialists for “inside” and “outside.” (2) Putative trans-membrane helices and connecting peripheral domains are labeled A – G and ab…fg, respectively, according to the proposal of Crofts et al. (1990). (3) Cytochrome b hemes on the n- and p-sides of membrane coordinated by His-99 and His-201 and His-85 and His-186, respectively, bridging helices B and D, are designated hemes bn and bp, respectively. The notation is preferred over that of bh and bl, for high- and low-potential hemes, proposed by Degli Esposti et al. (1993) because (a) there is experimental disagreement for cyt b6 in situ concerning the existence of experimentally resolvable high- and low-potential b hemes, and (b) the redox titration of cyt b of the bc1 complex from the PS3 thermophilic bacterium, which shows some similarity to the b6f complex, does not show a discernible difference in Em of the two hemes (Kutoh and Sone, 1988). Regarding (a), two reports (Furbacher et al., I989; Rich et al., 1991) show that the Em values of the two hemes are not separately resolved (ΔEm < 50mV) in thylakoid membranes in the presence of Mg2+; one (Kramer and Crofts, 1990) showed potentiometric resolution (bh = −15 mV, bl = −110 mV, ΔEm = 95 mV) of the two hemes. The latter result was obtained in the absence of Mg2+ but is in close agreement with the heme potentials measured in the isolated b6f complex (Hurt and Hauska, 1983; Nitschke et al., 1988). It was proposed (Kramer and Crofts, 1990) that the use of a continuous high-measuring light intensity in the redox titrations of Girvin and Cramer (1984) and Furbacher et al. (1989) might account for the discrepancy, but the intensity (0.6 μE · m−2 sec−1) found by Kramer and Crofts (1990) to cause cytochrome oxidation was at least 30 times that used (10–20 nE · m−2 · sec−1) by the latter authors.
The original calculations of the approximate location and identity of the hydrophobic trans-membrane helices (Widger et al., 1984; Saraste, 1984; Crofts et al., 1987) could not predict the orientation of the cyt b polypeptide in the membrane. For cyt b(bc1), the orientation was subsequently established through (i) the distribution of sites of resistance for the n- and p-side inhibitors mentioned above, and (ii) for cyt b of the complex in the bacterium Rb. sphaeroides, the sites of fusion to alkaline phosphatase (Yun et al., 1991). (iii) For cyt b6, the orientation shown in Fig. 2A was determined by protease accessibility to specific epitopes (Szczepaniak and Cramer, 1990), also mentioned above, and is consistent with the statistical cis- positive rule of Von Heijne (1992) [cf. Gavel et al. (1991) for a specific application to thylakoid membrane proteins]. This rule states that for intrinsic membrane proteins the number of positively charged residues located on the side of the membrane (cis) from which the protein is imported is larger than the number on the opposite (trans) side (i.e., the translocation of positively charged residues across biological membranes is energetically costly).
Fig. 2.

Schematic diagram of the major subunits of the cytochrome b6f complex (A) as an organized cluster at neutral ambient pH, and (B) after lateral separation of the subunits in the membrane at alkaline pH (Cramer et al., 1992; figure reproduced with permission of The American Society for Biological Chemistry and Molecular Biology).
The number of (Arg + Lys) residues on the n-(cis) and p-(trans) sides of cyt b6 is 8 and 5 (not including the intramembrane Arg-86), and for SU IV it is 8 and 3. This identity of the basic residues participating in this asymmetric distribution is essentially conserved in both cyt b6 and SU IV. All of the basic residues are conserved in the 13 cyt b6 sequences shown in Table I with the exception of the substitution of Asn (N) for Lys (K) at position 111 in Proto- chorothrix hollandica. In the case of SU IV, the identity of 8 of the 11 residues contributing to the (+) charge bias is conserved (Table IB), P. hollandica has the two changes R-15 → L and K-20 → Q, the cyanobacterium A. quadruplicatum is also changed at the latter position, and 4 of 14 sequences are changed at position 367. [Note that the cis-positive rule does not apply to cytochrome f and the Rieske iron-sulfur protein of the cyt b6f complex because in each of these subunits the peripheral segment of the protein exceeds 60 residues (Von Heijne, 1992); in the case of the Rieske protein, there is also a question as to whether it has a membrane span or is totally extrinsic (Gonzalez-Halphen et al., 1988; Breyton et al., 1994).]
Table I.
Compilation of Aligned Amino Acid Sequences of (A) Cytochrome b6 and (B) Subunit IVa

|

|

|
AGMEN (Agmenellum quadruplicatum. Brand et al., 1992); NOSTOC (Cyanobacterium Nostoc strain PCC 7906. Kallas et al., 1988); HOLLANDICA (Prochlorothrix hollandica. Greer and Golden, 1992); BARLEY (Hordeum vulgare. Reverdatto et al., 1989); SPINACH (Spinacia oleracea, Heinemeyer et al., 1984); WHEAT (Triticum aestivum. Hird et al., 1991); MAIZE (Zea mays. Rock et al., 1987, PROTOTHECOI (Chlorella protothecoides. Reimann and Kueck, 1989); RICE (Oryza sativa. Cote et al., 1988; Hiratsuka et al., 1989); TOBACCO (Nicotiana tabacum. Shinozaki et al., 1986); GRACILIS (Euglena gracilis. Schlunegger and Stutz, 1984); S. 6803 (Synechocystis sp. PCC 6803. Osiewacz, 1992); EUGAMETOS (Chlamydomonas eugametos. Tunnel et al., 1989); REINHARDTII (Chlamydomonas reinhardtii. Büschlen et al., 1991); LVWORT (Marchantia polymorpha. Fukuzawa et al., 1987); PEA (Pisum sativum. Phillips and Gray, 1984); S. obliquus (Scenedesmus obliquus. Kück, 1989).
The four-helix model applied to cytochrome b6 is shown in Fig. 1A along with the folding model for SU IV. The helices of cyt b6 (Fig. 1A; in the rectangular boxes) and subunit IV (Fig. 1B) are designated A–D and E–F, respectively, and the n- and p-side extrinsic linking peptide segments “ab,” “bc,” and “cd” (segments of cyt b6), and “ef,” “fg” (segments of SU IV) according to the notation of Crofts et al. (1990). The models of Fig. 1A, B differ from earlier models of cyt b6 and SU IV (Widger and Cramer, 1991) by (i) inclusion of the tendency of the p-side surface segments “ab,” “cd” (cyt b6), and “ef” (SU IV) to form amphiphilic helices. The hydrophobic moments of helices “ab,” “cd,” and “ef” are 0.39, 0.53, and 0.45, respectively, using a segment length of 15 residues and the amino acid hydrophobicity data base of Eisenberg (1984), as in Shiver et al. (1989). This amphiphilic character has been noted previously for the “cd” helix of cyt b(bc1) (Crofts et al., 1987), and the latter amphiphilic helix has been explicitly included in models of the p-side quinone binding site (Robertson et al., 1990). Asp-155 or Glu-166 in the “cd” segment have been proposed to bind DCCD and to facilitate H+ translocation to the membrane surface associated with p-side quinol oxidation (Beattie, 1993). The “ef” segment in SU IV contains seven proline residues, three of which have been included in an “ef” helix because of its pronounced amphiphilicity. Precedents for such a helix are provided by surface helices in the photosynthetic bacterial reaction center: the “ab” helix with three or two (Rb. capsulatus), and the “cd” helix with three or two (C. aurantiacus) proline residues. (ii) The length of the F, G helices of SU IV has been extended one turn in order to take into account the tendency of basic residues Lys-333 and R-339, R-340 to provide punctuation for TM helices. (iii) The tendency of aromatic residues, and tryptophans in particular, to be found near the aqueous interface (Deisenhofer and Michel, 1989; Jacobs and White, 1989) was taken into account by placing Trp-79 on the p-side of helix B, and Trp-246 on the n-side of helix E in contact with the bilayer phase.
The orientation of the “fg” helix of subunit IV inferred by Li et al. (1991) from trypsin accessibility studies was opposite to that shown in Fig. 1B. The distribution of (Arg+Lys) in a SU IV with an orientation reversed from that shown in Fig. 1B would be decidedly opposite to that predicted by the cis-positive rule.
Two striking features of the comparison of sequences of the 13 cyt b6 and 16 SU IV polypeptides are (a) the high degree of overall sequence identity, as noted in a comparison of an initial set of three complete, and two partial b6-SU IV sequences (Hauska et al., 1988), and (b) the higher extent of identity on the n-side compared to the p-side of the membrane. The degree of conservation of the 13 sequences of the 214 cyt b6 polypeptide (Table IA) is 69% and 79% for identical and pseudo-identical (K↔R, D ↔ E, and S ↔ T) residues, and is 53% and 63%, respectively, for the 160 residue subunit IV (Table IB). These levels of identity appear to far exceed those seen in cyt b of the mitochondrial bc1 complex (Hauska et al., 1988; Degli Esposti et al., 1993). In the 18 mitochondrial sequences compiled by Hauska et al. (1988), 10% of the residues are invariant (Degli Esposti et al., 1993). Degli Esposti et al. (1993) have also noted that the result of sequence analysis of the cytochrome b protein from more than 800 species, the most extensive analysis for any integral membrane protein, is that there are only 9 invariant amino acids (Table II).
Table II.
Summary of Residues of Cyt b6 and SU IV in Sequence Compilations of Tables IA, B and Figs. 2A, B That Are Most Highly Conserved (Degli Esposti et al., 1993)
| Residue | Invariance | Location (Figs. 2A, B) | Function |
|---|---|---|---|
| Cytochrome b6 | |||
| Gly 36 | Completeb | n-side, A helix | Heme pocketa |
| Gly 50 | Essentially completeb | A helix | — |
| Gly 78 | Essentially completeb | “ab” region | Turn between “ab” and B helices |
| Arg 82 | (+) Residue essentially completeb | p-side interface, B helix | Stop transfer, B helix |
| His 85 | Completeb | p-side, B helix | Ligand of p-side hemec,d |
| Ser 90 | Incompleteb | B helix | — |
| His 99 | Completeb | n-side, B helix | Ligand of n-side hemec,d |
| Arg 102 | (+) Residue completeb | n-side interface, B helix | Stop transfer, B helix |
| Trp 117 | Completeb | n-side, C helix | — |
| Gly 120 | Essentially completeb | n-side, C helix | — |
| Gly 134 | Essentially completeb | p-side, C helix | — |
| Tyr 135 | Incompleteb | p-side, C helix | — |
| Trp 145 | Incompleteb | p-side, “cd” helix | — |
| Thr 178 | Essentially completeb | p-side interface—“cd” helix | — |
| His 186 | Completeb | p-side, D helix | Ligand of p-side hemec,d |
| His 201 | Completeb | n-side, D helix | Ligand of n-side hemec,d One extra residue between two His ligands on D helixc |
| SU IVe | |||
| Asp 249 | Essentially completeb | n-side interface, E helix | — |
| Ile 289 | Incompleteb | p-side, “ef” loop | — |
| Pro 291 | Completeb | p-side, “ef” loop | — |
| Glu 292 | Essentially completeb | p-side, “ef” loop | — |
| Trp 293 | Essentially completeb | p-side, “ef” loop | — |
| Leu 302 | Essentially completeb | p-side, “ef” loop | — |
| Lys 308 | Incompleteb | p-side interface, F helix | Stop transfer, F helix |
| Gly 311 | Essentially completeb | p-side interface, F helix | Turn between “ef” and F helix |
Tron et al., 1991; Yun et al., 1991;
note that the numbering of SU IV is different from that used by Degli Esposti et al. (1993) who omitted the first 30 residues of the SU IV sequence. Use of the numbering system of the latter authors would cause overlap of numbering with the cyt b6 sequence from residue 195–214. The basis of the numbering system used in the present work is based on the concept that SU IV is a functional extension of cyt b6.
Table II summarizes (a) the position in the sequence of cyt b6-SU IV, and (b) the likely or proposed function of the 24 most highly conserved residues in the mitochondrial cytochrome b (Degli Esposti et al., 1993). Sixteen of the 24 residues are in cyt b6, according to Degli Esposti et al. (1993). Eight are in SU IV, of which three are the consecutive Pro 291-Glu 292-Trp 293. Five of the residues listed as most highly conserved by Degli Esposti et al. (1993) are different although conserved in cyt b6, where they are Val-132 (p-side, C helix), Gly-143 (p-side, “cd” loop), Lys-148 (p-side, “cd” helix), Ile-149 (p-side, “cd” helix), and Arg-206 (n-side, C-terminal segment). No such residues occur in SU IV. Thus, four of the 16 most conserved residues in cyt b6, and four of the five that break the invariance of the superfamily, are in the “cd” segment of cyt b6. The fifth residue that breaks the invariance, Arg-206, is analogous to His-202 that is otherwise completely conserved in the mitochondrial and photosynthetic bacterial cytochrome. In the latter, it is His-217. The mutation His 217 → Arg in the bacteria results in an inhibition of the rate of heme bn oxidation by a factor of 3–4 (Gray and Daldal, 1993; Hacker et al., 1993). The fact that the Arg residue that results in inhibition of bacterial heme bn (cf. discussion of notation in legend of Fig. 1) oxidation is conserved in cyt b6 can be conservatively interpreted as indicating that the putative quinone binding site on the n-side of the membrane (Qn site) has a different structure compared to that in the mitochondria and bacteria. Arg 206-Lys 207 have also been shown to be accessible to trypsin in thylakoid membranes (Szczepaniak and Cramer, 1990). This implies that the heme coordinated by His-201 is close (ca. 7 – 9 Å ) to the n-side interface, in contrast to heme bn of the mitochondrial cytochrome that was inferred from EPR experiments with external paramagnetic complexes (Ohnishi et al., 1989) and electrochromic msec “slow” phase measurements (Glaser and Crofts, 1984; Robertson and Dutton, 1988) to be appreciably removed (approximately 25 Å) from the n-side interface. One other Arg residue that is uniquely conserved in the cyt b6 sequences is Arg-86 adjacent to His-85 in the B helix. It is the only conserved residue that carries a nominal charge in the designated trans-membrane helices of the cytochrome b polypeptides.
The difference in sequence and probable structure of the n-side of cyt b6 compared to cyt b(bc1) is further illustrated by the extremely high level of sequence identity of the n-side peripheral segments of cyt b6, 78% and 87% for invariance and pseudo-invariance (Table IA, Fig. 1). These levels are higher than the corresponding values of 61% and 73% for the p-side domains. Thirteen of the last fifteen residues in cyt b6, Met-200 to Leu-214 on the n-side of helix D, are invariant, with Arg-206 approximately in the middle of this segment. The greater invariance on the n-side of the membrane is a bias opposite to that in cytochrome b of the bc1 complex (Degli Esposti et al., 1993). The n-side of the b6fcomplex is known to be characterized by the lack of a high-affinity binding site for quinone-analogue inhibitors (Widger and Cramer, 1991; Hope, 1993). The lack of such a site for a potent n-side inhibitor such as antimycin A is one of the obvious differences between the b6f and bc1 complexes, and has been a major point in discussions about the possibility of different properties of the b6f complex compared to cyt b(bc1) (Cramer et al., 1991; Rich et al., 1992). [The inhibitors NQNO (Selak and Whitmarsh, 1982; Rich et al., 1991; Kramer and Crofts, 1992) and MOA-stilbene (Rich et al., 1992) may be weakly binding n-side inhibitors of cyt b6; see Hope (1993) for a discussion of some inconsistencies in this interpretation regarding NQNO.] How- ever, when contrasted with mitochondria, the higher extent of sequence identity of the n-side of cyt b6 and SU IV implies that (i) a much higher degree of sequence invariance is required to establish a binding site of plastoquinone to allow the same Q-cycle mechanism that has been documented for mitochondria and bacterial chromatophores (Crofts, 1985; Trumpower, 1990); (ii) alternatively, the n-side extrinsic regions of cyt b6 and SU IV are responsible for essential functions in addition to, or instead of, the Q cycle function. One such function might be the docking of redox proteins [e.g., the “G” protein component of Joliot and Joliot (1988)] that can supply reducing equivalents to heme bn from the n-side aqueous phase.
WEAKNESS OF HELIX–HELIX INTERACTIONS
The b6fcomplex is extracted from the membrane as an integral membrane protein complex in detergents such as cholate, MEGA-9, or β-D-dodecylmaltoside. As a membrane protein complex, the b6fcomplex has the unusual property that the subunits can be separated from the complex and from each other at alkaline pH (ca. 10.5–11.5) [Fig. 2; Szczepaniak et al., 1991; Cramer et al., 1992]. This implies that, unlike membrane proteins such as bacteriorhodopsin (Popot and Engelman, 1990) and the glycophorin dimer (Lemmon et al., 1992), the attractive forces arising from intramembrane hydrophobic, hydrogen bond, and van der Waals interactions between the subunits are relatively weak. As a consequence, the electrostatic repulsive force exerted between the subunits at alkaline pH, arising from net excess negative charge on the peripheral polypeptide segments, is sufficient to cause the subunits of the complex to separate in the membrane. The fact that the subunits of the spinach chloroplast b6f complex do undergo lateral separation in the membrane at high pH is inferred from the separate and sequential release of the subunits of the complex as a function of increasing pH. A satisfying explanation does not exist as to why the subunits are not just separated, but also extruded from the membrane. One may speculate that the release from the merebrane is a consequence of the partial deprotonation and neutralization of the basic residues that are proposed to form the defined “stops” for the predicted trans-membrane α-helices A, B, and D of cyt b6 (Fig. 1A) and E, F, and G of SU IV (Fig. 1B). Once the “stops” are lost, the positions of the helices in the membrane would have fewer thermodynamic constraints, and more freedom to move in the direction normal to the plane of the membrane.
The relative weakness of the attractive intramembrane forces between the subunits of the b6f complex may be partly a consequence of prosthetic groups such as the helix-bridging heroes (Fig. 1C) providing spacers between these helices that prevent close packing between some of the trans-membrane helices.
THE b6f COMPLEX AS A STRUCTURAL AND FUNCTIONAL DIMER
The relatively weak intramembrane forces between subunits could also facilitate interconversion of monomer and dimer forms of the complex. The existence of a dimer form of isolated cyt b6f reconstituted in vitro was indicated from its geometric cross-section in electron micrographs (Mörschel and Staehelin, 1983). The presence of both monomer and dimer forms in isolated cyt b6f complex was inferred from (a) the presence in a sucrose density gradient of two fractions of unknown, but different, relative molecular weight, (b) the conversion of the higher to the lower Mr form after longer centrifugation times or in higher Triton detergent concentrations, (c) inhibition of this conversion by cross-linking reagents, and (d) the generation of different cross-linked Mr forms from the high- and low-Mr fractions (Chain and Malkin, 1991). Mr values and distinct sizes and geometries corresponding to monomer and dimer forms of cyt b6f complex isolated from spinach thylakoids were determined by (a) FPLC molecular chromatography in β-D-dodecylmaltoside, (b) native gel electrophoresis according to the method of Schägger and von Jagow (1991), and (c) averaged images obtained by electron microscopy of negatively stained single particles (Huang et al., 1993). The dimer was shown to be much more abundant (80–90% of the total complex) than the monomer when assayed by native gel electrophoresis (Huang et al., 1993). When isolated by FPLC molecular sieve chromatography which, compared to the native gel technique, resulted in a higher fraction (≤ 30% of the total) of monomer, the dimer was also much more (~ 5-fold) active in electron transfer from plastoquinol-2 to plastocyanin. The level of activity of the monomer was low enough that it could be attributed to residual dimer (Huang et al., 1993). Single-particle images of the monomer and dimer are shown in Fig. 3. A discussion of structure determination by electron microscopy is presented elsewhere in this volume by Boekema et al.
Fig. 3.
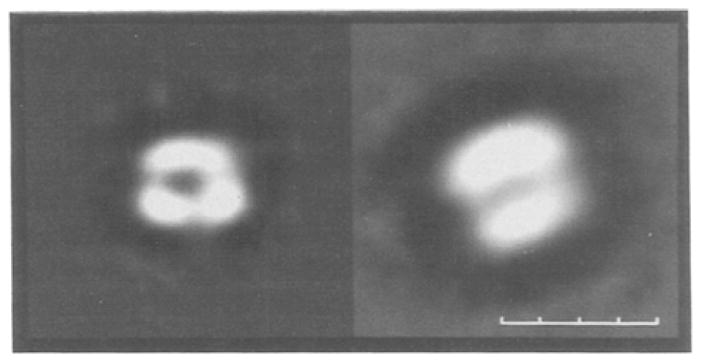
Electron micrographs of negatively stained (left) monomer and (right) dimer forms of the cytochrome b6f complex. Monomer and dimer images were computed as the average of 58 and 59 single-particle images, respectively (Huang et al., 1993). Size bar = 10 nm.
The existence of the dimeric form as the predominant form of the purified mitochondrial cytochrome bc1 complex is well established (Weiss and Kolb, 1979; Von Jagow and Sebald, 1980; Leonard et al., 1981; Nalecz and Azzi, 1985), although the functional significance of this dimer is still unclear (de Vries et al., 1983; Schmitt and Trumpower, 1990; Bechmann et al., 1992; Nieboer and Berden, 1992). It is known that oligomeric structures of membrane proteins are statistically likely in the two-dimensional space of the membrane (Grasberger et al., 1986), and that a dimeric structure and dimer–monomer transitions of the respective receptors are important in the mechanism of trans-membrane signaling in at least several systems (Pakula and Simon, 1992; Bormann and Engelman, 1992). Two of the more thoroughly studied examples include the bacterial aspartate chemoreceptor (Milburn et al., 1991), and the epidermal growth factor receptor-tyrosine kinase (Canals, 1992) signaling systems. We make the admittedly speculative suggestion that the cytochrome b6f and bc1 complexes are primitive trans-membrane signaling systems. In the case of the b6f complex, this is suggested by its apparent association with a protein kinase, the LHCP kinase, on the n-side of the membrane, and the ability of p-side quinone analogue inhibitors to block kinase activity (Gal et al., 1990a, b).
MOLECULE OF CHLOROPHYLL a IN b6f COMPLEX
The presence of chlorophyll in the spectrum of the cytochrome b6f complex isolated from spinach chloroplasts was noted in a benchmark paper on the purification and properties of the complex (Hurt and Hauska, 1983). The cyt b6f complex from the cyanobacterium Synechocystis PCC 6803, which was isolated in the monomeric form, was found to contain a single chlorophyll a molecule that was determined by linear dichroism analysis to be oriented in the complex (Bald et al., 1992). Electron microscopic analysis of this complex was carried out by Bald et al. (1992) and is reviewed in the present volume of this journal by Boekema et al. Analysis of the isolated b6f complex from spinach chloroplasts showed a chlorophyll a:cytochrome f stoichiometry > 1, but a smaller value close to 1 when the dimer was further purified from the complex by FPLC molecular sieve chromatography (Huang et al., 1993). The latter stoichiometry appeared to be the minimum value because it was not decreased further by an additional chromatographic step, implying that the one chlorophyll a molecule was not bound adventitiously.
CYTOCHROME f STRUCTURE: BIOCHEMICAL AND CELL BIOLOGICAL CONSEQUENCES
The understanding of structure/function of cytochrome f was advanced recently by the determination of its X-ray structure at a resolution of 2.3 Å (Martinez et al., 1991, 1992, 1993). The structure that was solved was that of the soluble 252-residue lumen-side domain of the 285-residue (MW = 31, 298 in turnip chloroplasts) mature polypeptide that spans the membrane once (Fig. 4C).
Fig. 4.
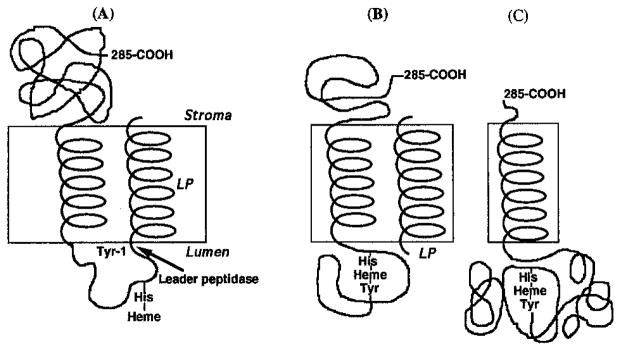
(A, B) Model for processing and assembly of intermediate forms of cyt f and (C) membrane topography of mature form. The schematic shows that complete heme ligation involving the α-amino group of Tyr-1 cannot occur until the 35-residue leader peptide (LP) has been removed by the processing peptidase that is believed to reside on the lumen side of the membrane (Johnson et al., 1991; Kirwin et al., 1991).
The use of the lumen-side polypeptide of the cytochrome arose from attempts to prepare intact cytochrome f from spinach chloroplast thylakoid membranes for crystallization. These attempts were abandoned because the cytochrome tended to aggregate. This had been noted previously by Ho and Krogmann (1980) who determined a molecular weight of 285,000 for the aggregate that approximately corresponded to an octamer. Preparations of cytochrome f from cruciferous plants, however, were reported to be soluble and monomeric with an Mr value of 27,000 instead of 32,000–33,000 (Gray, 1978). It was suggested that the decrease in apparent molecular size might arise from proteolysis at the C-terminus because the N-terminal sequence of cytochrome f from the cruciferous plant, rape, was found to be identical to that of spinach and pea (Willey et al., 1984). It was also inferred that the tendency of the spinach cytochrome f to aggregate in aqueous solution might arise from the 20-residue hydrophobic segment (residues Nos. 251–270, underlined in Table III) near the C-terminus, and that removal of the hydrophobic domain near the C-terminus might explain the water-soluble monomeric nature of the cruciferous cytochrome f fragment. Therefore, it was decided to purify cruciferous cytochrome f for structural analysis.
Table III.
Amino Acid sequence of Turnip Cytochrome f (Gray, 1992).

|
Cytochrome f was purified from turnip leaves and found to display an Mr value of 31,000 on SDS-PAGE, compared to 33,800 in the turnip thylakoid membrane (Fig. 5). After promising crystals had been obtained, truncation of the turnip cytochrome f near the C-terminus during purification was confirmed by C-terminal sequencing of the turnip cytochrome polypeptide with carboxypeptidases Y and A. Three of the last four residues were found to be Leu 249-Arg 250-Val 251 (Martinez et al., 1991). The fourth C-terminal residue was inferred to be a serine at position 252, instead of the glutamine that was conserved in the several chloroplast cytochrome f sequences known at that time. It was therefore thought that the explanation of the susceptibility of cytochrome f to lumen-side cleavage by an endogenous protease was a Gin → Set substitution at residue 252 (Martinez et al., 1991). Determination of the nucleotide sequence of the turnip cytochrome f gene by J. C. Gray showed that residue 252 is, in fact, a glutamine in the turnip (Brassica campestris) cytochrome f (Gray, 1992). Therefore, the explanation of the “natural” proteolysis in the Cruciferae cytochrome f is the existence of a protease in chloroplasts from these plants that is activated or made accessible to the lumen-side of the cytochrome f during its extraction from the thylakoid membranes. The same cleavage was subsequently reported for cytochrome f from charlock (Gray, 1992). It is perhaps interesting that the cleavage predominantly occurs two residues into the 20-residue, hydrophobic trans-membrane α-helix. This suggests that the protease might be related to a processing or leader peptidase. The thylakoid processing peptidase of cytochrome f is known to be membrane-bound (Johnson et al., 1991), and the active site of that for plastocyanin is on the lumen side of the membrane (Kirwin et al., 1991). On the other hand, the cleavage site of a processing protease should be unique, whereas analysis of the charlock (Gray, 1992) cytochrome f polypeptide by ESMS showed that the C-terminus is somewhat ragged, with small populations of fragments resulting from cleavage at residues adjacent to Glu-252. Preliminary ESMS of the turnip cyt f fragment indicated that it is ≥ 90% of the fragment population (D. Huang, D.L. Smith, and W.A. Cramer, unpublished data). The residue closest to the C-terminus for which electron density can be resolved in the structure is Leu-249 adjacent to Arg-250 (Fig. 6).
Fig. 5.
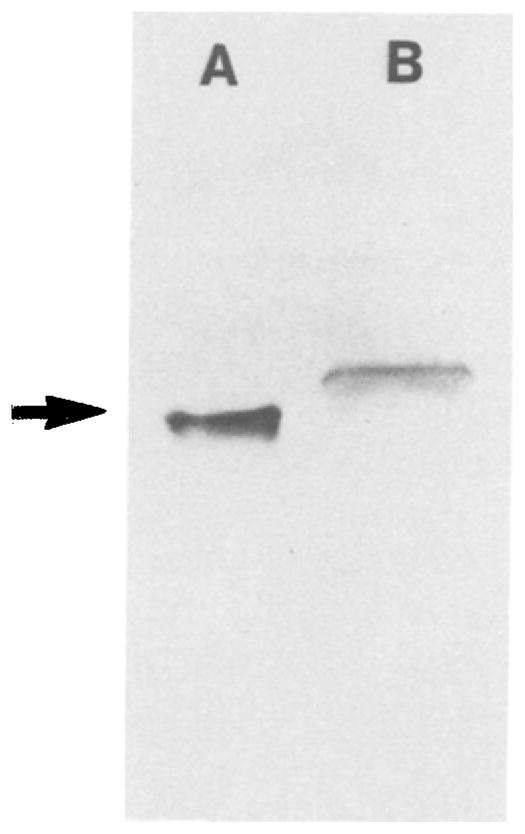
SDS-PAGE of cytochrome f purified from turnip thylakoid membranes (lane A), compared to that of the cytochrome in membranes that were dissolved in SDS and run on the get without prior extraction (lane B). ΔMr = 3,000 between the two bands that correspond to polypeptides with (A) the 252 residue turnip cyt f fragment (arrow) and (B) the complete turnip cyt f polypeptide with 285 residues. The molecular weight of the 252-residue polypeptide is 27,500 including the covalently bound heme. The purification procedure was essentially as described by Gray (1978), except for the use of PMSF (0.5 mM), benzamidine (2 mM), and ε-aminocaproic acid (2 mM) (Martinez et al., 1992, 1993). For electrophoresis, the cytochrome was solubilized in 50 mM Tris-HCl, pH 8.6, 4 M urea, 4% SDS, 10% glycerol, and 5% β-mercaptoethanol, and run with a gel system containing 15% acrylamide (Piccioni et al., 1982).
Fig. 6.
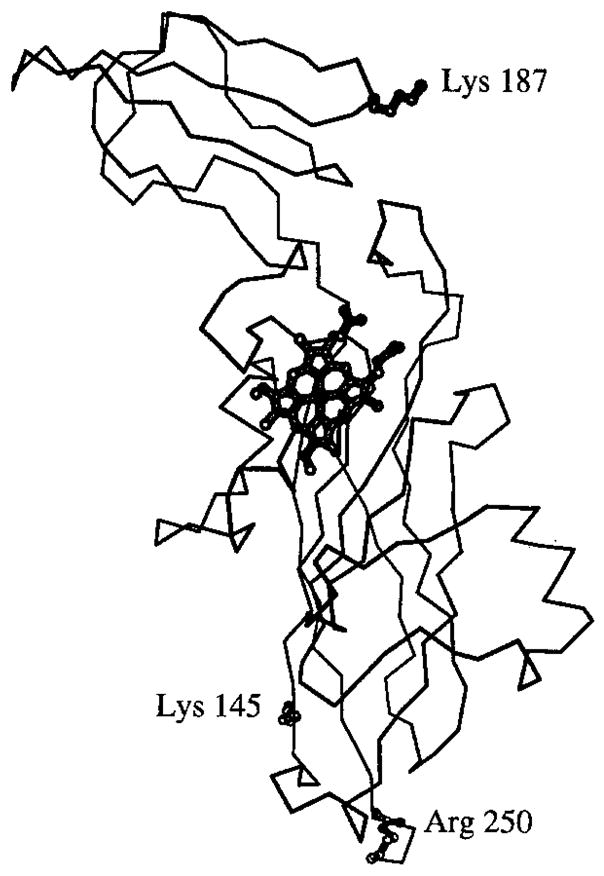
α-Carbon model of the lumen-side extrinsic polypeptide of turnip (French, “le navet”; German, “die Steckrübe” or “der Kohlrabi”) cytochrome f (SDS-PAGE in Fig. 5) showing the large and small domains of the cytochrome, the heme, and residues Lys-145 and Lys-187 in the large and small domains, and Arg-250 at the C-terminus that are separated by 33, 28, and 45 Å, respectively, from the heme iron.
The successful crystallization of the soluble turnip cyt f polypeptide that contains a small part (Val 251-Gln 252) of the putative hydrophobic membrane-spanning helical domain suggests a strategy for the systematic genetic engineering for purposes of crystallization of redox proteins such as cytochrome c1 or the Rieske iron-sulfur protein that have the same kind of general topography: generate a polypeptide fragment of the peripheral soluble fragment that is as long as possible to allow proper folding of the protein. This fragment could include a few residues of the hydrophobic helical domain, in order to increase the chances for correct folding of the fragment without affecting solubility.
The turnip cytochrome f polypeptide was crystallized at 5°C in the reduced form in the presence of dithioerythreitol using 40–42% acetone as the precipitant (Martinez et al., 1992). The structure has been solved by multiple isomorphous replacement and anomalous scattering to a resolution of 2.3 Å (Martinez et al., 1993). Some of the major features of the structure are:
Unlike the prototypical structure of soluble cytochrome c that has one mostly α-helical domain, the elongate (25 × 35 × 75 Å) cytochrome f structure is made of two domains whose major secondary structure motif is a β-sheet.
The heme is near the interdomain interface in the larger domain, the Fe 45 Å from the C-terminus of Arg-250 that is connected to the TM α-helix.
The smaller domain, consisting of residues 169–231, contains Lys-187, previously shown to cross-link to Asp-44 of plastocyanin (Morand et al., 1989; cf. Redinbo et al., this volume), that is solvent-exposed and 28 Å from the heme Fe (Fig. 6). Lys-187 is in a positively charged region that includes Lys-185 and Arg-209 on the small domain. Lys-66, Lys-65, and Lys-58 are neighboring basic residues on the large domain.
The axial sixth heme ligand is the α-amino group of the N-terminal tyrosine residue. A number of spectroscopic studies had noted similarities between spectra of cytochrome f and soluble mammalian cytochrome c at alkaline pH (> 10) in which the methionine ligand is exchanged for a lysine (Siedow et al., 1980; Davis et al., 1988; Rigby et al., 1988; Simpkin et al., 1989). The conserved Lys-145 was proposed as the axial sixth ligand (Davis et al., 1988). However, the ε-amino group of Lys-145 is 33 Å from the heme iron. The amino group function inferred from the spectroscopic studies can be fulfilled by the tyrosine α-amino group, and in one respect is fulfilled even more readily because the pK of the latter is close to neutrality.
Unlike mammalian cytochrome c, the heme is insulated from the aqueous phase by a shield including three aromatic residues of which one is Tyr-1. This may explain why the midpoint redox potential of cyt f is approximately 100 mV more positive than that of mammalian cyt c.
Because the structure of plastocyanin is known (Gross, 1993; Redinbo et al., this volume), the question immediately arises as to its competent binding configuration with cytochrome f. Information on the sites of interaction was provided by covalent cross-linking of plastocyanin to cytochrome f using the water-soluble 1-ethyl-3-[3-(dimethyl-amino)propyl]carbodiimide (EDC). One site of linkage involved Asp-44 of plastocyanin and Lys-187 of cyt f. A second linkage involved Glu-59 and/or 60 to an unknown, presumably basis, residue on cyt f. As noted above, Lys-187 is in a basic “patch” of residues (Fig. 6). The transfer of electrons from cytochrome f to plastocyanin is thought to be mediated as well by the Tyr-83 residue of plastocyanin which is exposed on the surface of PC and whose ring is approximately 9–10 Å and 10 Å from Asp-44 and the Cu atom, respectively. The distance of Asp-44 from the copper is 20 Å. With cyt f in the orientation shown in Fig. 6, PC would bind through its acidic regions (residues 42–45 and 59–61 of spinach plastocyanin) on the side of the barrel to the basic region of cyt f around Lys-187, the copper would be at the bottom of the PC barrel in this binding geometry, and Tyr-83 would then be located closer to the heme than Asp-44. It should be noted that covalent cross-linking of the PC–cyt f pair renders it incompetent for intermolecular electron transfer (Qin and Kostic, 1993), and the PC inactive in the reduction of photosystem I (Morand et al., 1989).
Orientation of Cytochrome f Relative to the Membrane Surface
It would seem that interaction of cytochrome f with the Rieske protein would be facilitated if the long axis of the large domain of cyt f was oriented with not too large an angle relative to the plane of the membrane. Interaction between the proposed surface “cd” helix of cytochrome b and the analogous cytochrome c1 is implied by the existence of a second site suppressor mutation in cyt c1 to a primary inhibitor-resistant mutation in the “cd” loop of cyt b(bc1) of Rb. capsulatus (F. Daldal, personal communication). It has been observed in EPR studies on magnetically oriented chloroplast membranes (Bergstrom and Vanngard, 1982; Crowder et al., 1982) and dehydrated two-dimensional lattices of cytochrome b6f complex oriented after centrifugation on Mylar sheets (Riedel et al., 1991) that the cyt f heme has a broad (Bergstrom and Vanngard, 1982; Riedel et al., 1991) or narrow (Crowder et al., 1982) distribution of orientations relative to the membrane surface. The most probable orientation of the heme plane in both kinds of distributions would have it sharply tilted at an angle of approximately 25–30° relative to the plane of the membrane. Although there may be some flexibility in the orientation of cyt f, the most probable orientation of the protein appears to be favorable for interaction with surface-bound or extrinsic segments of the Rieske iron-sulfur protein and cyt b6.
Sixth Heme Ligand of Cytochrome f: Consequences for Protein Translocation and Assembly
The consequence of the sixth heme ligand being the α-amino group of the N-terminal Tyr residue of the mature protein is that the complete ligation of the heme that results in formation of the low-spin coordination state, and assembly of the final folded state of the protein, cannot occur until the signal peptide of the intermediate translocated preprotein has been cleaved by the processing peptidase. This peptidase is believed to be located on the lumen side of the membrane (Johnson et al., 1991; Kirwin et al., 1991). This implies that the protein folding could not be completed until translocation has proceeded at least to the point where a significant part of the cytochrome including Tyr-1 and His-25 has crossed the membrane. In addition, the ligation of the heme and associated folding, as in the translocation pathway of apocytochrome c (Planner and Neupert, 1990), may provide additional free energy needed to complete the translocation process.
The use of the Tyr-1 α-amino group as the sixth heme ligand can be viewed as a control mechanism to ensure a proper delay in the timing of protein refolding, and is an example of the general principle of membrane protein translocation that the protein to be translocated be unfolded (Verner and Schatz, 1988; Pfanner and Neupert, 1990). Although the use of amino terminal ligation thus seems to be a rational mechanism, it appears that it is not necessarily ubiquitous for metalloproteins even for the closely related cytochrome c1 protein: (i) in the nitrogen-fixing microaerobic endosymbiont, Bradyrhizobium japonicum, cytochromes b and c1 can be made by mutagenesis as a single two-domain polyprotein connected by a 31-residue internal signal peptide (Thony-Meyer et al., 1991). Although it was shown in the latter work that covalent binding of the c1 heme was required for formation of the bc1 polyprotein, cleavage of the internal signal peptide was not. The use of a sixth ligand different from the N-terminal α-amino group, for at least some cytochromes c1, is also suggested by the appearance of a normal reduced α-band spectrum in a mutant of yeast cytochrome c1 that is blocked in cleavage of the signal peptide (Yang and Trumpower, 1993).
Acknowledgments
This research was supported by grants NIH GM-18457 (WAC), USDA 9101624 (JLS and WAC), and NSF DMB-8905062 (TSB). We thank P.N. Furbacher for helpful discussions, B. L. Trumpower for communication of a manuscript before publication, and J. Hollister and V. Livingston for careful work in preparing the manuscript.
Footnotes
Abbreviations: Cyt, cytochrome; cyt b(bc1), cytochrome b of the bc1 complex; ESMS, electrospray mass spectrometry; MOA- stilbene, E-β-methoxyacrylate-stilbene; MW, molecular weight; NQNO, 2-n-nonyl-4-hydroxyquinoline-N-oxide; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; SU IV, subunit IV; TM, transmembrane.
References
- Bald D, Kruip J, Boekema EJ, Rögner M. In: Research in Photosynthesis. Murata N, editor. I. Kluwer; Dordrecht: 1992. pp. 629–632. [Google Scholar]
- Beattie DS. J Bioenerg Biomembr. 1993;25:233–244. doi: 10.1007/BF00762585. [DOI] [PubMed] [Google Scholar]
- Bechmann G, Weiss H, Rich PR. Eur J Biochem. 1992;208:315–325. doi: 10.1111/j.1432-1033.1992.tb17189.x. [DOI] [PubMed] [Google Scholar]
- Bergstrom J, Vanngard T. Biochim Biophys Acta. 1982;682:452–456. [Google Scholar]
- Bormann BJ, Engelman DE. Annu Rev Biophys Biophys Chem. 1992;19:369–403. [Google Scholar]
- Brand SN, Tan X, Widger WR. Plant Mol Biol. 1992;20:481–491. doi: 10.1007/BF00040607. [DOI] [PubMed] [Google Scholar]
- Breyton C, de Vitry C, Popot J-L. J Biol Chem. 1994 in press. [PubMed] [Google Scholar]
- Büschlen S, Choquet Y, Kuras R, Wollman FA. FEBS Lett. 1991;284:257–262. doi: 10.1016/0014-5793(91)80698-3. [DOI] [PubMed] [Google Scholar]
- Canals F. Biochemistry. 1992;31:4493–4501. doi: 10.1021/bi00133a016. [DOI] [PubMed] [Google Scholar]
- Chain R, Malkin R. Photosynth Res. 1991;28:59–68. doi: 10.1007/BF00033715. [DOI] [PubMed] [Google Scholar]
- Cote JC, Wu NH, Wu R. Plant Mol Biol. 1988;11:873–874. doi: 10.1007/BF00019528. [DOI] [PubMed] [Google Scholar]
- Cramer WA, Black MT, Widger WR, Girvin ME. In: The Light Reactions. Barber J, editor. Elsevier; Amsterdam: 1987. pp. 446–493. [Google Scholar]
- Cramer WA, Furbacher PN, Szczepaniak A, Tae G-S. In: Current Topics in Bioenergetics. Lee CP, editor. Vol. 16. Academic Press; Orlando: 1991. pp. 179–222. [Google Scholar]
- Cramer WA, Engelman DM, von Heijne G, Rees DC. FASEB J. 1992;6:3397–3402. doi: 10.1096/fasebj.6.15.1464373. [DOI] [PubMed] [Google Scholar]
- Crofts AR. In: The Enzymes of Biological Membranes. 2. Martinosi A, editor. Vol. 4. Plenum Press; New York: 1985. pp. 347–382. [Google Scholar]
- Crofts AR, Robinson H, Andrews K, Van Doren S, Berry E. In: Cytochrome Systems: Molecular Biology and Bioenergetics. Papa S, Chance B, Ernster L, editors. Plenum Press; New York: 1987. pp. 617–624. [Google Scholar]
- Crofts AR, Wang Z, Chen Y, Mahalingam S, Yun CH, Gennis RB. In: Highlights in Ubiquinone Research. Lenaz G, editor. Taylor and Francis; London: 1990. pp. 98–103. [Google Scholar]
- Crowder MS, Prince RC, Bearden A. FEBS Lett. 1982;144:204–208. [Google Scholar]
- Daldal F, Tokito MK, Davidson E, Faham M. EMBO J. 1989;8:3951–3961. doi: 10.1002/j.1460-2075.1989.tb08578.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis DJ, Frame MK, Johnson DA. Biochim Biophys Acta. 1988;936:61–66. doi: 10.1016/0005-2728(88)90251-4. [DOI] [PubMed] [Google Scholar]
- Degli Esposti M, DeVries S, Crimi M, Ghelli A, Patarnello T, Meyer A. Biochim Biophys Acta. 1993;1143:243–271. doi: 10.1016/0005-2728(93)90197-n. [DOI] [PubMed] [Google Scholar]
- Deisenhofer J, Michel H. EMBO J. 1989;8:2149–2169. doi: 10.1002/j.1460-2075.1989.tb08338.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries S, Albracht SPJ, Berden JA, Marres CAM, Slater EC. Biochim Biophys Acta. 1983;723:91–103. doi: 10.1016/0005-2728(83)90013-0. [DOI] [PubMed] [Google Scholar]
- diRago JP, Colson AM. J Biol Chem. 1988;263:12564–12570. [PubMed] [Google Scholar]
- Eisenberg D. Annu Rev Biochem. 1984;53:595–623. doi: 10.1146/annurev.bi.53.070184.003115. [DOI] [PubMed] [Google Scholar]
- Fukuzawa H, Yoshida T, Kohchi T, Okumura T, Sawano Y, Ohyama K. FEBS Lett. 1987;220:61–66. [Google Scholar]
- Furbacher PN, Girvin ME, Cramer WA. Biochemistry. 1989;28:8990–8998. doi: 10.1021/bi00449a006. [DOI] [PubMed] [Google Scholar]
- Gal A, Mets LJ, Ohad I. In: Current Research in Photosynthesis. Baltscheffsky M, editor. II. Kluwer Academic; Dordrecht: 1990a. pp. 779–781. [Google Scholar]
- Gal A, Mor TS, Hauska G, Herrmann R, Ohad I. In: Current Research in Photosynthesis. Baltscheffsky M, editor. II. Kluwer Academic; Dordrecht: 1990b. pp. 783–785. [Google Scholar]
- Gavel Y, Steppuhn J, Herrmann R, von Heijne G. FEBS Lett. 1991;282:41–46. doi: 10.1016/0014-5793(91)80440-e. [DOI] [PubMed] [Google Scholar]
- Gennis RB, Barquera B, Hacker B, Van Doren SR, Arnaud S, Crofts AR, Davidson E, Gray KR, Daldal F. J Bioenerg Biomembr. 1993;25:195–209. doi: 10.1007/BF00762582. [DOI] [PubMed] [Google Scholar]
- Girvin ME, Cramer WA. Biochim Biophys Acta. 1984;767:29–38. doi: 10.1016/0005-2728(84)90076-8. [DOI] [PubMed] [Google Scholar]
- Glaser EA, Crofts AR. Biochim Biophys Acta. 1984;766:322–333. doi: 10.1016/0005-2728(84)90248-2. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Halphen D, Lindorfer MA, Capaldi RA. J Biol Chem. 1988;27:7021–7031. doi: 10.1021/bi00418a053. [DOI] [PubMed] [Google Scholar]
- Graham LA, Brandt U, Sargent JS, Trumpower BL. J Bioenerg Biomembr. 1993;25:245–257. doi: 10.1007/BF00762586. [DOI] [PubMed] [Google Scholar]
- Grasberger B, Minton AP, DeLisi C, Metzger H. Proc Natl Acad Sci USA. 1986;83:6258–6262. doi: 10.1073/pnas.83.17.6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JC. Eur J Biochem. 1978;82:133–141. doi: 10.1111/j.1432-1033.1978.tb12004.x. [DOI] [PubMed] [Google Scholar]
- Gray JC. Photosynth Res. 1992;34:359–374. doi: 10.1007/BF00029811. [DOI] [PubMed] [Google Scholar]
- Gray KA, Daldal F. Biophys J. 1993;64:105a. [Google Scholar]
- Greet KL, Golden SS. Plant Mol Biol. 1992;19:355–365. doi: 10.1007/BF00023383. [DOI] [PubMed] [Google Scholar]
- Gross EL. Photosyn Res. 1993;37:103–116. doi: 10.1007/BF02187469. [DOI] [PubMed] [Google Scholar]
- Hacker B, Barquera B, Crofts AR, Gennis RB. Biophys J. 1993;64:105a. [Google Scholar]
- Haley J, Bogorad L. Proc Natl Acad Sci USA. 1989;86:1534–1538. doi: 10.1073/pnas.86.5.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauska G, Hurt E, Gabellini N, Lockau W. Biochim Biophys Acta. 1983;726:97–133. doi: 10.1016/0304-4173(83)90002-2. [DOI] [PubMed] [Google Scholar]
- Hauska G, Nitschke W, Herrmann RG. J Bioenerg Biomem. 1988;20:211–228. doi: 10.1007/BF00768395. [DOI] [PubMed] [Google Scholar]
- Heinemeyer W, Alt J, Herrmann RG. Curr Genet. 1984;8:543–549. doi: 10.1007/BF00410442. [DOI] [PubMed] [Google Scholar]
- Hiratsuka J, Shimada H, Whittier R, Ishibashi T, Sakamoto M, Mori M, Kondo C, Honji Y, Sun CR, Meng BY, Li YQ, Kanno A, Nishizawa Y, Hirai A, Shinozaki K, Sugiura M. Mol Gen Genet. 1989;217:185–194. doi: 10.1007/BF02464880. [DOI] [PubMed] [Google Scholar]
- Hird SM, Webber AN, Wilson RJ, Dyer TA, Gray JC. Plant Mol Biol. 1991;16:745–747. doi: 10.1007/BF00023441. [DOI] [PubMed] [Google Scholar]
- Ho KK, Krogmann DW. J Biol Chem. 1980;255:3855–3861. [PubMed] [Google Scholar]
- Hope AB. Biochim Biophys Acta. 1993;1143:1–22. doi: 10.1016/0005-2728(93)90210-7. [DOI] [PubMed] [Google Scholar]
- Howell N, Gilbert K. J Mol Biol. 1988;203:607–618. doi: 10.1016/0022-2836(88)90195-7. [DOI] [PubMed] [Google Scholar]
- Huang DH, Everly M, Cheng RH, Heymann JB, Schägger H, Baker TS, Cramer WA. 1993 doi: 10.1021/bi00180a038. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurt EC, Hauska G. FEBS Lett. 1983;153:413–419. [Google Scholar]
- Jacobs RE, White SH. Biochemistry. 1989;8:3421–3437. doi: 10.1021/bi00434a042. [DOI] [PubMed] [Google Scholar]
- Johnson EM, Schabelrauch LS, Sears BB. Mol Gen Genet. 1991;225:106–112. doi: 10.1007/BF00282648. [DOI] [PubMed] [Google Scholar]
- Joliot P, Joliot A. Biochim Biophys Acta. 1988;933:319–333. [Google Scholar]
- Kallas T, Spiller S, Malkin R. J Biol Chem. 1988;263:14334–14342. [PubMed] [Google Scholar]
- Kirwin PM, Elderfield PD, Williams RS, Robinson C. J Biol Chem. 1991;263:18128–18132. [PubMed] [Google Scholar]
- Kramer DM, Crofts AR. In: Curr Res Photosyn. Baltscheffksy M, editor. III. Kluwer Academic; Dordrecht: 1990. pp. 283–286. [Google Scholar]
- Kramer DM, Crofts AR. In: Res Photosyn. Murata N, editor. II. Kluwer; Dordrecht: 1992. pp. 491–494. [Google Scholar]
- Kück U. Mol Gen Genet. 1989;218:257–265. doi: 10.1007/BF00331276. [DOI] [PubMed] [Google Scholar]
- Kutoh E, Sone N. J Biol Chem. 1988;263:9020–9026. [PubMed] [Google Scholar]
- Lemmon MA, Flanagan JM, Hunt JF, Adair BD, Bormann BJ, Engelman DE. J Biol Chem. 1992;267:7683–7689. [PubMed] [Google Scholar]
- Leonard K, Wingfield P, Arod T, Weiss H. J Mol Biol. 1981;149:259–274. doi: 10.1016/0022-2836(81)90301-6. [DOI] [PubMed] [Google Scholar]
- Li LB, Zou YP, Yu L, Yu CA. Biochim Biophys Acta. 1991;1057:215–222. doi: 10.1016/s0005-2728(05)80104-5. [DOI] [PubMed] [Google Scholar]
- Link TA, Haase U, Brandt U, von Jagow G. J Bioenerg Biomembr. 1993;25:221–232. doi: 10.1007/BF00762584. [DOI] [PubMed] [Google Scholar]
- Martinez SE, Szezepaniak A, Smith JL, Cramer WA. Biophys J. 1991;59:524a. [Google Scholar]
- Martinez SE, Smith JL, Huang D, Szczepaniak A, Cramer WA. In: Research in Photosynthesis. Murata N, editor. II. Kluwer Academic; Dordrecht: 1992. pp. 495–498. [Google Scholar]
- Martinez SE, Huang D, Szczepaniak A, Cramer WA, Smith JL. 1993 Manuscript in preparation. [Google Scholar]
- Milburn MV, Prive GG, Mittigan DL, Scott WG, Yeh J, Jancarik J, Koshland DE, Jr, Kim SH. Science. 1991;254:1342–1347. doi: 10.1126/science.1660187. [DOI] [PubMed] [Google Scholar]
- Morand L, Frame MK, Colvert KK, Johnson DA, Krogmann DW, Davis DJ. Biochemistry. 1989;28:8039–8047. doi: 10.1021/bi00446a011. [DOI] [PubMed] [Google Scholar]
- Mörschel E, Staehelin A. J Cell Biol. 1983;97:301–310. doi: 10.1083/jcb.97.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalecz MJ, Azzi A. Arch Biochem Biophys. 1985;240:921–931. doi: 10.1016/0003-9861(85)90101-8. [DOI] [PubMed] [Google Scholar]
- Nieboer P, Berden JA. Biochim Biophys Acta. 1992;1101:90–96. doi: 10.1016/0167-4838(92)90472-p. [DOI] [PubMed] [Google Scholar]
- Nitschke W, Hauska G, Crofts AR. FEBS Lett. 1988;232:204–208. [Google Scholar]
- Ohnishi T, Schägger H, Meinhardt SW, LoBrutto R, Link TA, von Jagow G. J Biol Chem. 1989;264:735–744. [PubMed] [Google Scholar]
- Osiewacz HD. Arch Microbiol. 1992;157:336–342. doi: 10.1007/BF00248678. [DOI] [PubMed] [Google Scholar]
- Pakula A, Simon M. Nature. 1992;355:496–497. doi: 10.1038/355496a0. [DOI] [PubMed] [Google Scholar]
- Pfanner N, Neupert W. Annu Rev Biochem. 1990;59:331–353. doi: 10.1146/annurev.bi.59.070190.001555. [DOI] [PubMed] [Google Scholar]
- Phillips AC, Gray JC. Mol Gen Genet. 1984;194:477–484. [Google Scholar]
- Piccioni R, Bellemare G, Chua N-H. In: Methods in Chloroplast Molecular Biology. Edelman M, et al., editors. Chap 80. 1982. pp. 985–1015. [Google Scholar]
- Popot JL, Engelman DE. Biochemistry. 1990;29:4031–4039. doi: 10.1021/bi00469a001. [DOI] [PubMed] [Google Scholar]
- Qin L, Kostic NM. Biochemistry. 1993;32:6073–6080. doi: 10.1021/bi00074a019. [DOI] [PubMed] [Google Scholar]
- Redinbo MR, Yeates TO, Merchant S. J Bioenerg Biomem. 1994 doi: 10.1007/BF00763219. in press. [DOI] [PubMed] [Google Scholar]
- Reimann A, Kueck U. Plant Mol Biol. 1989;13:255–256. doi: 10.1007/BF00016144. [DOI] [PubMed] [Google Scholar]
- Reverdatto SV, Andreeva AV, Buryakova AA, Chakhmakhcheva OG, Efimov VA. Nucleic Acids Res. 1989;17:2859–2860. doi: 10.1093/nar/17.7.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich PR, Madgwick SA, Moss DA. Biochim Biophys Acta. 1991;1058:312–328. [Google Scholar]
- Rich P, Madgwick SA, Brown S, von Jagow G, Brandt U. Photosynth Res. 1992;34:465–477. doi: 10.1007/BF00029819. [DOI] [PubMed] [Google Scholar]
- Riedel A, Rutherford W, Hauska G, Müller A, Nitschke W. J Biol Chem. 1991;266:17838–17844. [PubMed] [Google Scholar]
- Rigby SEJ, Moore GR, Gray JC, Godsby PMA, George SJ, Thomson AJ. Biochem J. 1988;256:571–577. doi: 10.1042/bj2560571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson DE, Dutton PL. Biochim Biophys Acta. 1988;935:273–291. doi: 10.1016/0005-2728(88)90223-x. [DOI] [PubMed] [Google Scholar]
- Robertson DE, Daldal F, Dutton PL. Biochemistry. 1990;29:11249–11260. doi: 10.1021/bi00503a014. [DOI] [PubMed] [Google Scholar]
- Rock CD, Barkan A, Taylor WC. Curt Genet. 1987;12:69–77. doi: 10.1007/BF00420729. [DOI] [PubMed] [Google Scholar]
- Saraste M. FEBS Lett. 1984;166:367–372. doi: 10.1016/0014-5793(84)80114-3. [DOI] [PubMed] [Google Scholar]
- Schägger H, von Jagow G. Anal Biochem. 1991;199:223–231. doi: 10.1016/0003-2697(91)90094-a. [DOI] [PubMed] [Google Scholar]
- Schlunegger B, Stutz E. Curr Genet. 1984;8:629–634. doi: 10.1007/BF00395709. [DOI] [PubMed] [Google Scholar]
- Schmitt ME, Trumpower BL. J Biol Chem. 1990;265:17005–17011. [PubMed] [Google Scholar]
- Selak MA, Whitmarsh J. FEBS Lett. 1982;150:286–292. [Google Scholar]
- Shinozaki K, Ohme M, Tanaka M, Wakasugi T, Hayashida N, Matsubayashi T, Zaita N, Chunwongse J, Obokata J, Yamaguchi-Shinozaki K, Ohto C, Torazawa K, Meng BY, Sugita M, Deno H, Kamogashira T, Yamada K, Kusuda J, Takaiwa F, Kato A, Tohdoh N, Shimda H, Sugiura M. EMBO J. 1986;5:2043–2049. doi: 10.1002/j.1460-2075.1986.tb04464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiver JW, Peterson AA, Widger WR, Cramer WA. Methods Enzymol. 1989;172:439–461. doi: 10.1016/s0076-6879(89)72028-0. [DOI] [PubMed] [Google Scholar]
- Siedow JN, Vickery LE, Palmer G. Arch Biochem Biophys. 1980;203:101–107. doi: 10.1016/0003-9861(80)90157-5. [DOI] [PubMed] [Google Scholar]
- Simpkin D, Palmer G, Devlin FJ, McKenna MC, Jensen GM, Stephens PJ. Biochemistry. 1989;28:8033–8039. doi: 10.1021/bi00446a010. [DOI] [PubMed] [Google Scholar]
- Szczepaniak A, Cramer WA. J Biol Chem. 1990;265:17720–17726. [PubMed] [Google Scholar]
- Szczepaniak AD, Huang D, Keenan TW, Cramer WA. EMBO J. 1991;10:2757–2764. doi: 10.1002/j.1460-2075.1991.tb07824.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thony-Meyer L, James P, Hennecke H. Proc Natl Acad Sci USA. 1991;88:5001–5005. doi: 10.1073/pnas.88.11.5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trumpower BL. J Biol Chem. 1990;265:11409–11412. [PubMed] [Google Scholar]
- Turmel M, Boulanger J, Bergeron A. Nucleic Acids Res. 1989;17:3593–3593. doi: 10.1093/nar/17.9.3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verner K, Schatz G. Science. 1988;241:1307–1313. doi: 10.1126/science.2842866. [DOI] [PubMed] [Google Scholar]
- Von Jagow G, Sebald W. Ann Rev Biochem. 1980;49:281–314. doi: 10.1146/annurev.bi.49.070180.001433. [DOI] [PubMed] [Google Scholar]
- Von Heijne G. J Mol Biol. 1992;225:487–494. doi: 10.1016/0022-2836(92)90934-c. [DOI] [PubMed] [Google Scholar]
- Weiss H, Kolb HJ. Eur J Biochem. 1979;99:139–149. doi: 10.1111/j.1432-1033.1979.tb13240.x. [DOI] [PubMed] [Google Scholar]
- Widger WR, Cramer WA, Herrmann R, Trebst A. Proc Natl Acad Sci, USA. 1984;81:674–678. doi: 10.1073/pnas.81.3.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widger WR, Cramer WA. In: Cell Culture and Somatic Cell Genetics of Plants: The Molecular Biology of Plastids and the Photosynthetic Apparatus. Vasil IK, Bogorad L, editors. Academic Press; Orlando: 1991. pp. 149–176. [Google Scholar]
- Willey DL, Auffret AD, Gray JC. Cell. 1984;36:555–562. doi: 10.1016/0092-8674(84)90248-4. [DOI] [PubMed] [Google Scholar]
- Yang M, Trumpower BL. J Biol Chem. 1993 in press. [PubMed] [Google Scholar]
- Yun CH, Van Doren SR, Crofts AR, Gennis RB. J Biol Chem. 1991;266:10967–10973. [PubMed] [Google Scholar]