

It is predicted that over half of all eukaryotic proteins are glycosylated, and it is now well-established that co- and post-translational modification of proteins with glycans can have dramatic consequences on their folding, stability, and ultimately their function.[1] Considerable effort has thus been invested in delineating the impact of appended carbohydrates on the conformational preferences of proteins and peptides in solution and vice versa,[2] and also in understanding their interactions with their cognate receptors.[3] These endeavours are not straightforward, and success in rationalizing such processes has been possible only in a handful of well-studied cases.[4] Important insights into such questions have been gleaned from the study of glycoconjugate mimetics, whose interactions with cellular targets can impact a wide range of physiological phenomena, including fertilization, immune response, host–pathogen interactions, cell growth, and tumor metastasis.[1] However, attempts to successfully correlate biological functions of structurally well-defined glycopeptides with their secondary structures have been relatively sparse,[2–4] despite the importance of such targets in the quest for carbohydrate-based therapeutics.[5]
Herein we examine the effects of appended sugar moieties on the conformational behavior of peptide foldamers derived from δ-sugar amino acids (δ-SAAs).[6] The study of foldamers has in the past helped enlighten our understanding of the origins of the preferred secondary structures and biological activities of biopolymers.[7] Considering the endogenous and therapeutic importance of glycoproteins, we were struck by the dearth of reports describing the impact of glycosylation on the secondary structures of peptide foldamers.[8] Appended sugars in the two families of newly synthesized δ-SAA-derived glycofoldamers indeed play a defining role on the preferred conformations of the peptide foldamer backbones and, far less commonly, are seen to do so even in water.[9] Furthermore, the differences in conformation manifested by each glycofoldamers are shown to be mirrored in their distinct and contrasting interaction with selected targets including the lectin Concanavalin A (ConA)[10a] and the bacterium Escher-ichia coli (E. coli).[10b]
The families of δ-SAA-derived foldamers targeted for investigation herein, annotated cis- and trans- in the text, differ from one another in the configuration of the stereo-center at C2 of the furanoid rings of their constituent δ-SAA moieties: those with the “2S” configuration designated cis-foldamers, and those with “2R”, trans-foldamers (Scheme 1). Previous work has shown that in organic solvents, cis-foldamers adopt conformations reminiscent of a conventional β-turn, whereas the secondary structures of trans-foldamers are dependent on the substituent pattern of their constituent furanoid rings.[11] The targeted families of δ-SAA-based hybrid peptides, 1a and 1b, were assembled following a convergent strategy from the corresponding monomeric δ-SAA precursors 6a and 7a (for 1a) or 6b and 7b (for 1b), respectively. The required monomeric δ-SAA building blocks were conveniently obtained from the same starting sugar, 2-deoxy-δ-ribose.[12]
Scheme 1.

Representation of δ-SAA based hybrids.
N-Boc deprotection of 6a and saponification of the methyl ester function of 7a gave precursors 8a and 9a, respectively, which when condensed under standard solution-phase peptide coupling conditions gave smoothly the corresponding cis-δ-SAA dimer 10a. The dimer 10a could be deprotected at either ends to give the precursors 11a and 12a which, when reacted together (standard peptide coupling conditions), gave the target cis-δ-SAA based tetramer 1a (Scheme 2). An identical reaction sequence starting from the trans-δ-SAA monomers 6b and 7b allowed the straightforward assembly of the corresponding trans-δ-SAA derived tetramer 1b (Scheme 2). It was envisaged that the azido functions, thus integrated, would subsequently allow ready conjugation of selected partners at precise positions along each δ-SAA-derived backbone post-assembly. As expected, reaction of either 1a or 1b with either non-sugar or sugar partners armed with propargyl functions proceeded smoothly by a CuI-catalyzed Huisgen click cycloaddition[13] to give the corresponding conjugates 2a,b or 3a,b, respectively (Scheme 2). This click conjugation method has found appeal previously in neoglycopeptide assembly, where the triazole moieties themselves can impact physiochemical properties or biological activities of the resulting glycoconjugates.[14]
Scheme 2.

Synthesis of SAA-based hybrids. Reagents and conditions: i) 30% TFA in CH2Cl2, 0°C–RT, 3 h; ii) LiOH, THF/MeOH/H2O (3:1:1), 0 °C–RT, 1 h; iii) 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI), 1-hydroxybenzotriazole (HOBt), diisopropylethylamine (DIPEA), CH2Cl2, 0 °C–RT, 12 h; iv) sodium ascorbate, CuSO4·5H2O, methyl propiolate, RT, CH2Cl2, 12 h, 79%; v) Sodium ascorbate, CuSO4·5H2O, 13, EtOH, H2O, microwave, 80 °C, 5 min, 84–86%; vi) NaOMe, MeOH, RT, 2 h, Amberlite 120H+, 91–94%; vii) H2, Pd-C, MeOH, RT, 12 h, 70–76%.
The data derived from NMR spectroscopy were used to establish the conformational preferences of the peptide foldamers, as this technique can provide discrete structural information at the atomic level.[2–4] The 1H NMR spectra of foldamers 1a,b and their clicked analogues 2a,b in CDCl3 feature sharp resonances and well-resolved amide proton signals, suggesting the presence of predominantly well-defined folded structures in solution. The downfield chemical shifts observed for the amide protons in the spectra of 1a,b and 2a,b further suggest their participation in internal hydrogen bonding (H-bonding), a feature that is also supported by the minimal perturbation of these signals upon titration in 33% of [D6]DMSO (v/v).[12] For peptide foldamers 1a and 2a, the small changes in chemical shifts (Δδ), observed for the NH2, NH3, and NH4 in solvent titration studies, and the invariance of their chemical shifts in variable concentration studies, are also consistent with the participation of these protons in intramolecular H-bonding.[15] The presence of sequential nOe cross-peaks between 2NH↔1C6H(pro-R), 1C3H(pro-S), 3NH↔2C6H(pro-R), 2C3H(pro-S), 4NH↔3C6H(pro-R), and 3C3H(pro-S) in the ROESY spectrum of foldamer 1a (Scheme 3) corroborates the participation of NH2, NH3, and NH4 in H-bonding, consistent with a 10-membered turn network implicating NH(i)-CO(i-2).[12] Furthermore, the medium-intensity nOes observed between 3NH↔1C3H(pro-R), 1C3H(pro-S) and 4NH↔2C3H(pro-S), 2C3H(pro-R) suggest that foldamer 1a adopts a compact secondary structure. The clicked foldamer analogue 2a afforded identical data to its precursor 1a, suggesting that these two compounds share related secondary structures.
Scheme 3.
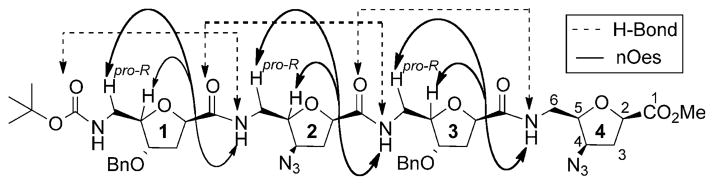
Characteristic nOes and H-bonding pattern of the 10-membered repeat structure of 1 a in CDCl3.
The NMR spectra of cis-glycofoldamer 3a, featuring two acetylated mannose units, display similar nOes and H-bonding networks as seen for 1a when recorded in CDCl3. However a very weak medium-range nOe between 4NH↔2C6H(pro-R) observable in the ROESY spectrum of cis-analogue 3a (Figure 1) indicates the presence of an additional bend around the second H-bonding, which is also suggested by the absence of the nOe between 3NH↔1C3H(pro-S) present in 1a. Titration studies carried out by sequential addition of 33% [D6]DMSO(v/v) to 3a gives a Δδ value for 3NH that is 0.26 ppm greater than those observed for the 2nd and 4th δ-SAA amide protons and further supports that in 3a local distortions at 3NH occur. Taken together, these observations suggest that 3a tends towards a higher-order structure than adopted by foldamer 1a.
Figure 1.

Expanded ROESY spectrum of 3 a in CDCl3 (ca. 9 mM, 300 K). The nOes 2NH↔1C6H(pro-S), 2NH↔1C6H(pro-R), 4NH↔2C6H(pro-R), 4NH↔3C6H(pro-R), 2NH↔1C5H, 4NH↔3C5H, and 3NH↔2C5H are marked as 1–7.
It was critical that the glycofoldamers would be soluble in water once deprotected, so that their biological evaluation might be undertaken. That these glycofoldamers would maintain distinct secondary structures in water was also imperative. Although short linear δ-SAA-based backbones (non-glycosylated) are predisposed towards defined secondary structures in organic solvents,[9] these are invariably disrupted upon deprotection.[16] We were nevertheless hopeful that the glycofoldamers synthesized herein, once deprotected, would behave similarly to short natural glycopeptides in water, in which attached carbohydrates have been demonstrated to stabilize secondary structures or compact conformations.[17] Indeed, deacetylation or deacetylation/debenzylation of cis-glycofoldamer 3a afforded the corresponding analogues 4a or 5a, respectively, which proved to be soluble in either methanol (4a) or water (5a).
The NMR data for 4a in CD3OH features a sequence of i↔i +2 nOes along with those nOes observed in the spectra of 1a and 3a. The medium-range nOes between (i)NH↔(i-2)C6H(pro-R) and (i)NH↔(i–2)C3H(pro-S) (Scheme 4 and Figure 2) identified in the spectrum of 4a for the 3rd and 4th residues, with the data of solvent exchange studies and minimal temperature dependencies, confirms participation of these amide protons in H-bonding. The data taken together is consistent with the cis-glycofoldamer 4a existing predominantly in a 16-membered H-bonded turn structure involving 3NH↔Boc-CO and 4NH↔1CO.
Scheme 4.
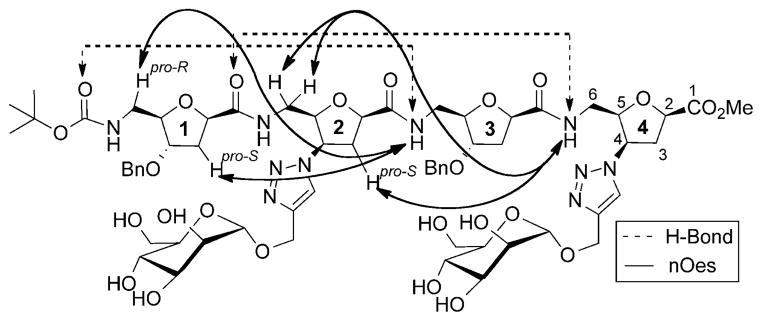
Characteristic nOes and H-bonding pattern of the 16-membered helical turn structure of 4 a in CD3OH.
Figure 2.

Expanded ROESY spectrum of 4 a in CD3OH (ca. 8 mM, 300 K). The nOes 2NH↔1C6H(pro-S), 2NH↔1C6H(pro-R), 4NH↔2C6H, 3NH↔1C6H(pro-R), 4NH↔2C3H(pro-S), 3NH↔1C3H(pro-S), 4NH↔3C6H(pro-R), 4NH↔3C5H, and 3NH↔2C5H are marked as 1–9.
The deacetylated/debenzylated cis-glycofoldamer 5a, studied in 94% H2O + 6% D2O, showed an almost identical nOe pattern and H-bonding network as that observed for 4a, suggesting similar secondary structures for both. As far as we are aware this is the first high-order 16-membered turn structure observed in water for any foldamer. The conformational switch evidenced for the cis-glycofoldamers 4a/5a is driven uniquely by their appended man-nosyl moieties and are not observed for the foldamer 2a, which features an alternate aliphatic ester-appended backbone.
It is noteworthy that NMR data recorded for the members of the trans-foldamer family, 1b–3b, are very similar to those of their cis-counterparts, suggesting that they share related conformational properties. For example, the trans-foldamer 1b displayed sequential nOe peaks between (i)NH↔(i–1)C5H, (i)NH↔(i–1)C6H(pro-S) in its NMR spectrum together with those characteristic of a 10-membered H-bonded conformation for (i)NH↔(i–2)CO similar to that observed for the cis-foldamer 1a. Solvent titration studies further supported the adoption of a 10-membered H-bonding pattern by 1b.[12] Likewise, the trans-glycofoldamers 2b and 3b also exist in a stable 10-membered H-bonded conformation similar to that observed for the corresponding cis-glycofoldamers 2a and 3a. However, careful observation of the deacetylated, or deacetylated/debenzylated trans-glycofoldamers, 4b and 5b respectively, showed that they do not form the higher-order structures observed in their cis-counterparts 4a and 5a, but rather maintain the conformational preferences adopted prior to their being appended with sugar moieties.
Restrained molecular dynamics (MD) studies were carried out using experimental distance and torsional constraints derived from nOe volume integrals and vicinal coupling constants (3J), respectively. The average structures of the 5 ns MD run of each cis-glycofoldamers are presented in Figure 3. The average distance between the anomeric carbons of mannosyl residues, as calculated from the structure ensembles, is 6.73 Å for the cis-analogue, 5a (20 least-energy conformations range from 5.45 Å to 9.40 Å), and 6.58 Å for the trans-compound, 5b (20 least-energy conformations vary from 5.25 Å to 8.19 Å) respectively.
Figure 3.

10- and 16-membered cis-δ-foldamers and cis-δ-glycofoldamers. H-bonded average MD structure of A) 1 a, B) 3 a, C) 4 a, and D) 5 a.
We were curious to establish whether or not the mannosyl residues in glycofoldamers 5a and 5b interacted with selected biological targets differently, despite the fact that the average distance between these residues differs by less than 0.2 Å (from MD calculations). Any difference in binding between 5a and 5b was anticipated to provide additional insights into the impact of foldamer conformational properties. The protein ConA, which is known to bind mannopyranosyl moieties preferentially, and the bacterium, E. coli in which the lectin FimH contributes specifically to bladder colonization through binding to terminal α-δ-mannosyl units that are present on glycoproteins such as uroplakins, were selected for examination.[18,10b]
The predisposition of glycofoldamers 5a and 5b to bind our targets was initially probed in a superimposition study in which the structures of the glycofoldamers (determined experimentally) and those of the proteins were kept rigid.[19] Each of the two mannosyl residues in 5a and 5b were then superimposed onto the mannosyl residues of ligands[12] complexed in the binding pocket of ConA or that of FimH (PDB codes: 1CVN, 2VCO), respectively. This resulted in two possible poses for the pair of glycofoldamers with each protein, all of which were evaluated for steric collisions. Significant steric clashes were observed between ConA and both 5a and 5b for all but one pose.[12] The superposition of glycofoldamers 5a and 5b onto the Man3GlcNAc2 in FimH also resulted in unacceptable interactions.[12] The preliminary study indicates that some level of induced fit would be expected for binding of the glycofoldamers 5a or 5b with either of the selected targets.
An enzyme-linked lectin assay (ELLA)[20] providing information on binding affinity between the sugar ligand and a single CRD in the lectin indeed confirms that both glycofoldamers are recognized by ConA, with the cis-compound, 5a giving an IC50 (inhibitory concentration 50) some 1.3 fold lower (IC50 = 347 ± 10 μM) than its trans-counterpart 5b, (IC50 = 440 ± 10 μM).[12] However, nearly four- to sixfold affinity enhancement was found for Man3 (IC50 = 75 ±6 μM), which supports the hypothesis that induced fit would be required for an increase in binding of either glycofoldamers with ConA, as suggested also by the findings of the superimposition studies. Peptide glycofoldamers 5a and 5b were also evaluated as inhibitors for FimH-expressing K-12 E. coli adhesion.[21] In this study, the cis-analogue 5a gave an inhibition titer (IT) fourfold lower (IT= 0.087 mM) than the trans-compound 5b (IT=0.35 mM) in the yeast agglutination assay.[12] The IT was considered as the lowest compound concentration able to inhibit agglutination. Likewise, both glycofoldamers were seen to reduce the binding of E. coli to T24 bladder cells, the cis-glycofoldamer 5a giving an IC50 of some 2.2-fold lower (IC50 = 0.02986 mM) than its trans-counterpart 5b (IC50 = 0.06621 mM). Binding to an E. coli strain not expressing FimH was not seen.[12] The differences observed between glycofoldamers 5a and 5b, in assays with both ConA as well as E. coli, are consistent with their contrasting conformational preferences. The origin of these differences may at present only be conjectured upon.
The data presented is the first demonstration that appended carbohydrates can have a defining influence on the structural preferences of δ-SAA-derived foldamer backbones in water. Of particular significance is the observation that the grafted mannoside moieties provoke a conformational switch in the cis-foldamer backbone (and not in its trans-foldamer counterpart) from a 10-membered H-bonded turn structure, when not appended with sugars, to an unprecedented 16-membered helical one when mannosyl units are grafted. The variations are seen to be exquisitely dependent on the particular backbone to which sugars are appended and is consistent with a scenario in which the mannosyl units modulate the backbone torsional preferences of a given foldamer as defined by its backbone stereochemistry (cis- or trans-) and amplified by variations in its constituent δ-SAA furanoid (2R- or 2S-) conformations. Our findings mirror those of Imperiali et al.,[22] who observed a chitobiose-driven conformational switch of a short peptide backbone and further showed by NMR that this was not due to any observable interaction between the peptide backbones and their constituent sugar moieties.[2,22] Likewise, in neither glycofoldamer 5a or 5b are any interactions observable between the peptide backbone and the constituent mannosyl residues by NMR spectroscopy.
Of particular significance is the demonstration that the interactions of the cis- and trans-glycofoldamers 5a and 5b with both the lectin ConA and the bacterium E. coli are distinct, suggesting that these differences may have their seeds in the underlying conformational preferences of this pair of neoglycopeptides. These data further confirm the notion that subtle differences in solution conformation, and it is likely that dynamics preferences should indeed have consequences on the underlying biology of natural glycopeptides even when these might be structurally closely related.[2–4]
Footnotes
T.K.C., R.S.A., A.S., S.R., and O.B. acknowledge financial support from the CEFIPRA-ICPAR. K.K.P. and P.S.K. are thankful to CSIR, New Delhi, for financial support. A.S., S.R., and O.B. acknowledge support from the CNRS. R.J.W. and K.I. thank the NIH (GM094919) (EUREKA) and the Science Foundation of Ireland (08/IN.1/B2070) for financial support. The SAIF-CDRI and the LG, Amiens are thanked for analytical facilities. The authors wish to thank Profs I. Huc (Bordeaux) and J. Jiménez-Barbero (Madrid) for their critical reading of the manuscript. This is CSIR-CDRI communication No. 8490.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201304239.
Contributor Information
Dr. Aloysius Siriwardena, Email: aloysius.siriwardena@u-picardie.fr, Laboratoiredes Glucides, FRE-3517, Université de Picardie Jules Verne, Amiens 80039 (France)
Kiran Kumar Pulukuri, Medicinal & Process Chemistry Division, CSIR-Central Drug Research Institute, Lucknow 226031 (India).
Pancham S. Kandiyal, Centre for Nuclear Magnetic Resonance, SAIF, CSIR-Central Drug Research Institute (India), Lucknow 226031 (India)
Dr. Saumya Roy, Laboratoiredes Glucides, FRE-3517, Université de Picardie Jules Verne, Amiens 80039 (France)
Dr. Omprakash Bande, Laboratoiredes Glucides, FRE-3517, Université de Picardie Jules Verne, Amiens 80039 (France)
Dr. Subhash Ghosh, Organic Chemistry Division III, CSIR-Indian Institute of Chemical Technology, Hyderabad 500 007 (India)
Dr. José Manuel Garcia Fernández, Instituto de Investigaciones, Quìmicas(IIQ), CSIC-Universidad de Sevilla, Américo Vespucio 49, 41092 Sevilla (Spain)
Dr. Fernando Ariel Martin, Institut Pasteur, Unité de Génétique des Biofilms, 25 rue du Dr. Roux, 75724 Paris cedex 15 (France)
Dr. Jean-Marc Ghigo, Institut Pasteur, Unité de Génétique des Biofilms, 25 rue du Dr. Roux, 75724 Paris cedex 15 (France)
Dr. Christophe Beloin, Institut Pasteur, Unité de Génétique des Biofilms, 25 rue du Dr. Roux, 75724 Paris cedex 15 (France)
Dr. Keigo Ito, The Complex Carbohydrate Research Center, The Department of Biochemistry and Molecular Biology, The University of Georgia, Athens, 30602 GA (USA)
Dr. Robert J. Woods, The Complex Carbohydrate Research Center, The Department of Biochemistry and Molecular Biology, The University of Georgia, Athens, 30602 GA (USA). The School of Chemistry, National University of Ireland, Galway University Road, Galway (Ireland)
Dr. Ravi Sankar Ampapathi, Email: ravi_sa@cdri.res.in, Centre for Nuclear Magnetic Resonance, SAIF, CSIR-Central Drug Research Institute (India), Lucknow 226031 (India)
Dr. Tushar Kanti Chakraborty, Email: chakraborty@cdri.res.in, Medicinal & Process Chemistry Division, CSIR-Central Drug Research Institute, Lucknow 226031 (India)
References
- 1.a) Hart GW, Copeland RJ. Cell. 2010;143:672–676. doi: 10.1016/j.cell.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Helenius A, Aebi M. Science. 2001;291:2364–2369. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]; c) Varki A. Glycobiology. 1993;3:97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Dwek RA. Chem Rev. 1996;96:683– 720. doi: 10.1021/cr940283b. [DOI] [PubMed] [Google Scholar]
- 2.a) Larkin A, Imperiali B. Biochemistry. 2011;50:4411–4426. doi: 10.1021/bi200346n. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shental-Bechor D, Levy Y. Curr Opin Struct Biol. 2009;19:524–533. doi: 10.1016/j.sbi.2009.07.002. [DOI] [PubMed] [Google Scholar]; c) Ellis CR, Maiti B, Noid WG. J Am Chem Soc. 2012;134:8184– 8193. doi: 10.1021/ja301005f. [DOI] [PubMed] [Google Scholar]
- 3.a) Corzana F, Busto JH, Marcelo F, García de Luis M, Asensio JL, Martín-Santamaría S, Jiménez-Barbero J, Avenoza A, Peregrina JM. Chem Eur J. 2011;17:3105–3110. doi: 10.1002/chem.201003124. [DOI] [PubMed] [Google Scholar]; b) Corzana F, Busto JH, Marcelo F, Luis MG, Asensio JL, Martín-Santamaría S, Sáenz Y, Torres C, Jiménez-Barbero J, Avenozaa A, Peregrina JM. Chem Commun. 2011;47:5319–5321. doi: 10.1039/c1cc10192g. [DOI] [PubMed] [Google Scholar]
- 4.a) Meyer B, Mçller H. Top Curr Chem. 2007;267:187–251. [Google Scholar]; b) Westerlind U. Beilstein J Org Chem. 2012;8:804– 818. doi: 10.3762/bjoc.8.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Muthana SM, Campbell CT, Gildersleeve JC. ACS Chem Biol. 2012;7:31–43. doi: 10.1021/cb2004466. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ernst B, Magnani JL. Nat Rev Drug Discovery. 2009;8:661– 677. doi: 10.1038/nrd2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Chakraborty TK, Srinivasu P, Tapadar S, Mohan BK. Glycoconjugate J. 2005;22:83–93. doi: 10.1007/s10719-005-0844-x. [DOI] [PubMed] [Google Scholar]; b) Jiménez Blanco JL, Ortega-Caballero F, Ortiz Mellet C, García Fernández JM. Beilstein J Org Chem. 2010;6(20) doi: 10.3762/bjoc.6.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Gellman SH. Acc Chem Res. 1998;31:173–180. [Google Scholar]; b) Goodman CA, Choi S, Shandler S, De Grado WF. Nat Chem Biol. 2007;3:252–262. doi: 10.1038/nchembio876. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Guichard G, Huc I. Chem Commun. 2011;47:5933– 5941. doi: 10.1039/c1cc11137j. [DOI] [PubMed] [Google Scholar]
- 8.a) Simpson GL, Gordon AH, Lindsay DM, Promsawan N, Crump MP, Mulholland K, Hayter BR, Gallagher T. J Am Chem Soc. 2006;128:10638–10639. doi: 10.1021/ja0614565. [DOI] [PubMed] [Google Scholar]; b) Norgren AS, Geitmann M, Danielson UH, Arvidsson PI. J Mol Recognit. 2007;20:132–138. doi: 10.1002/jmr.821. [DOI] [PubMed] [Google Scholar]; c) Norgren AS, Arvidsson PI. J Org Chem. 2008;73:5272–5278. doi: 10.1021/jo8003265. [DOI] [PubMed] [Google Scholar]; d) Norgren AS, Arvidsson PI. Org Biomol Chem. 2005;3:1359– 1361. doi: 10.1039/b503237g. [DOI] [PubMed] [Google Scholar]
- 9.a) Chakraborty TK, Jayaprakash S, Diwan PV, Nagaraj R, Jampani SRB, Kunwar AC. J Am Chem Soc. 1998;120:12962–12963. [Google Scholar]; b) Chakraborty TK, Ghosh S, Jayaprakash S, Sharma JARP, Ravikanth V, Diwan PV, Nagaraj R, Kunwar AC. J Org Chem. 2000;65:6441–6457. doi: 10.1021/jo000408e. [DOI] [PubMed] [Google Scholar]; c) Long DD, Smith MD, Marquess DG, Claridge TDW, Fleet GWJ. Tetrahedron Lett. 1998;39:9293–9296. [Google Scholar]; d) Claridge TDW, Long DD, Baker CM, Odell B, Grant GH, Edwards AA, Tranter GE, Fleet GWJ, Smith MD. J Org Chem. 2005;70:2082– 2090. doi: 10.1021/jo0480040. [DOI] [PubMed] [Google Scholar]
- 10.a) Morris TA, Peterson AW, Tarlov MJ. Anal Chem. 2009;81:5413–5420. doi: 10.1021/ac900715d. [DOI] [PubMed] [Google Scholar]; b) Knight SD, Bouckaert J. Top Curr Chem. 2009;288:67– 107. doi: 10.1007/128_2008_13. [DOI] [PubMed] [Google Scholar]
- 11.Baron R, Bakowies D, Van Gunsteren WF. Angew Chem. 2004;116:4147– 4151. doi: 10.1002/anie.200454114. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2004;43:4055–4059. doi: 10.1002/anie.200454114. [DOI] [PubMed] [Google Scholar]
- 12.See the Supporting Information.
- 13.a) Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem. 2002;114:2708–2711. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; b) Tornoe CW, Christensen C, Meldal M. J Org Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]; c) Wu P, Feldman AK, Nugent AK, Hawker CJ, Scheel A, Voit B, Pyun J, Frechet JMJ, Sharpless KB, Fokin VV. Angew Chem. 2004;116:4018– 4022. doi: 10.1002/anie.200454078. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2004;43:3928– 3932. doi: 10.1002/anie.200454078. [DOI] [PubMed] [Google Scholar]
- 14.a) Kümin M, Sonntag LS, Wennemers H. J Am Chem Soc. 2007;129:466–467. doi: 10.1021/ja067148o. [DOI] [PubMed] [Google Scholar]; b) Erdmann RS, Wennemers H. J Am Chem Soc. 2010;132:13957–13959. doi: 10.1021/ja103392t. [DOI] [PubMed] [Google Scholar]; c) Lin H, Walsh CT. J Am Chem Soc. 2004;126:13998–14003. doi: 10.1021/ja045147v. [DOI] [PubMed] [Google Scholar]; d) Schwardt O, Rabbani S, Hartmann M, Abgottspon D, Wittwer M, Kleeb S, Zalewski A, Smieško M, Cutting B, Ernst B. Bioorg Med Chem. 2011;19:6454– 6473. doi: 10.1016/j.bmc.2011.08.057. [DOI] [PubMed] [Google Scholar]
- 15.Ohnishi M, Urry DW. Biochem Biophys Res Commun. 1969;36:194– 202. doi: 10.1016/0006-291x(69)90314-3. [DOI] [PubMed] [Google Scholar]
- 16.Chakraborty TK, Jayaprakash S, Srinivasu P, Govardhana Chary M, Diwan PV, Nagaraj R, Ravi Sankar A, Kunwar AC. Tetrahedron Lett. 2000;41:8167– 8171. [Google Scholar]
- 17.a) Laczko I, Hollosi M, Urge L, Ugen KE, Weiner DB, Mantsch HH, Thurin JL, Jr, Otvos Biochemistry. 1992;31:4282–4288. doi: 10.1021/bi00132a019. [DOI] [PubMed] [Google Scholar]; b) Andreotti AH, Kahne D. J Am Chem Soc. 1993;115:3352–3353. [Google Scholar]; c) Live DH, Kumar RA, Beebe X, Danishefsky SJ. Proc Natl Acad Sci USA. 1996;93:12759–12761. doi: 10.1073/pnas.93.23.12759. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) O’Connor SE, Imperiali B. J Am Chem Soc. 1997;119:2295–2296. [Google Scholar]; e) Slynko V, Schubert M, Numao S, Kowarik M, Aebi M, Allain FHT. J Am Chem Soc. 2009;131:1274– 1281. doi: 10.1021/ja808682v. [DOI] [PubMed] [Google Scholar]
- 18.a) Sharon N, Lis H. Glycobiology. 2004;14:53R–62R. doi: 10.1093/glycob/cwh122. [DOI] [PubMed] [Google Scholar]; b) Pieters RJ. Org Biomol Chem. 2009;7:2013– 2025. doi: 10.1039/b901828j. [DOI] [PubMed] [Google Scholar]
- 19.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004;25:1605– 1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 20.Gómez-García M, Benito JM, Butera AP, Ortiz Mellet C, García Fernández JM, Jiménez Blanco JL. J Org Chem. 2012;77:1273– 1288. doi: 10.1021/jo201797b. [DOI] [PubMed] [Google Scholar]
- 21.a) Korea CG, Badouraly R, Prevost MC, Ghigo JM, Beloin C. Environ Microbiol. 2010;12:1957–1977. doi: 10.1111/j.1462-2920.2010.02202.x. [DOI] [PubMed] [Google Scholar]; b) Barras A, Martin FA, Bande O, Baumann JS, Ghigo JM, Boukherroub R, Beloin C, Siriwardena A, Szunerits S. Nanoscale. 2013;5:2307– 2316. doi: 10.1039/c3nr33826f. [DOI] [PubMed] [Google Scholar]
- 22.Bosques CJ, Tschampel SM, Woods RJ, Imperiali B. J Am Chem Soc. 2004;126:8421– 8425. doi: 10.1021/ja049266. [DOI] [PMC free article] [PubMed] [Google Scholar]