

Abstract
Bacteriophage φ29 of Bacillus subtilis packages its double-stranded DNA into a preformed prohead during morphogenesis. The prohead is composed of the scaffold protein gp7, the capsid protein gp8, the portal protein gp10, and the dispensable head fiber protein gp8.5. Our objective was to elucidate the φ29 prohead assembly pathway and to define the factors that determine prohead shape and size. The structural genes of the φ29 prohead were cloned and expressed in Escherichia coli individually or in combination to study form determination. The scaffold protein was purified from E. coli as a soluble monomer. In vivo and in vitro studies showed that the scaffolding protein interacted with both the portal vertex and capsid proteins. When the scaffold protein interacted only with the capsid protein in vivo, particles were formed with variable size and shape. However, in the presence of the portal vertex protein, particles with uniform size and shape were produced in vivo. SDS–PAGE analysis showed that the latter particles contained the proteins of the scaffold, capsid, head fiber, and portal vertex. These results suggest that the scaffolding protein serves as the linkage between the portal vertex and the capsid proteins, and that the portal vertex plays a crucial role in regulating the Size and shape Of the prohead.
Introduction
In the tailed, double-stranded DNA bacteriophages, the DNA is packaged into a preformed prohead during morphogenesis. There are common features of the prohead structure and the DNA packaging machine. Minimally, the proheads contain three structural proteins: the scaffold, the shell (capsid), and the portal vertex (reviewed in Bazinet and King, 1985; Earnshaw and Casjens, 1980; Casjens and Hendrix, 1988; Black, 1988, 1989). The portal vertex, located at the junction of the head and tail, is a dodecamer with a 3 to 4-nm central hole through which viral genomic DNA passes during packaging and exits during infection (Bazinet and King, 1985; Casjens, 1985; Black, 1988, 1989). The mechanism of prohead assembly and the sequential interaction of the three structural components is an intriguing problem in viral morphogenesis. It was presumed that prohead assembly is initiated by a primer such as a vertex (Murialdo and Becker, 1978). Prohead elongation has historically been considered to be a copolymerization of both the coat and the scaffolding subunits onto the growing double shell (Fuller and King, 1982). Phage T4 can, however, form a naked core without the capsid protein from just gp22, gp67, and gp21; the three internal proteins Ipl, Ipll, and Iplll; and a 17k protein (Traub and Maeder, 1984; Traub et al., 1984). The portal vertex may function as the initiator of the phage P22 procapsid assembly (Bazinet and King, 1985), but recently it has been reported that the scaffolding protein and capsid protein of phage P22 were polymerized efficiently into particles in vivo and in vitro in the absence of the portal protein (Bazinet and King, 1988; Prevelige et al., 1988). Removal of the gene one product (gp1, portal protein) of P22 by mutation did not slow the rate of polymerization of coat and scaffolding subunits into shells. Thus, the portal ring is dispensable for the initiation of assembly of prohead-like particles (Bazinet and King, 1988).
Extensive studies of phage assembly have led to the dogma that macromolecular assembly almost invariably follows well-defined pathways (Epstein et al., 1963; Edgar and Wood, 1966; Wood et al., 1968; Casjens and Hendrix, 1988). For example, if one particle is composed of proteins ABC and A + B → AB is the initial reaction, then the pathway of the reaction should be A + B → AB, and AB + C → ABC. However, neither the complex of A and C, nor of B and C can be formed (Casjens and Hendrix, 1988). Accordingly, if the portal vertex protein is not the initiator of prohead assembly, then the prohead assembly should be initiated with the formation of the scaffold/capsid complex. The interaction of the scaffold and capsid proteins in prohead assembly could occur in one of two ways: (1) by formation of the scaffold–capsid particle with defined size to which the portal vertex is inserted subsequently; or (2) by initiation of the capsid and scaffold subunits to form heterodimers which then would assemble onto the portal vertex and subsequently onto the growing nascent structure. However, there is no evidence for such heterodimers for any of the DNA phages, nor has a prohead assembly initiation complex been observed directly. It seems as though the portal vertexes are subsequently inserted into a scaffold–capsid particle of a defined size. However, the prohead has yet to be assembled in vitro utilizing purified components. The sequential interactions of structural proteins in prohead assembly remains an unsolved problem. The mechanism for the prevention of uncontrolled polymerization is also unknown.
The prohead of bacteriophage φ29 of Bacillus subtilis is composed of four proteins: the scaffold, the shell, the portal vertex (connector or DNA translocating vertex), and the dispensable head fiber. We have constructed a defined in vitro DNA packaging system in which more than half of the added DNA molecules can be packaged into purified proheads with the aid of the cloned and purified DNA packaging protein gp16. The DNA filled heads prepared using this system can be converted into viruses after the addition of the neck and tail proteins and other factors required for neck and tail assembly (Guo et al., 1986, 1987c,d).
Phage prohead assembly has generally been studied by the use of suppressor-sensitive mutations of the structural proteins (Murialdo and Becker, 1978; Thacker, Ph.D. Thesis, University of Minnesota), but with this technique structural protein fragments may still remain in the cell. The fragment may react atypically with other structural proteins and interfere with the assembly. To avoid this problem and to determine the possible role of other viral and B. subtilis proteins in prohead assembly, we cloned the structural proteins of the φ29 prohead individually, and in combination, to study the reactions. We report here that the φ29 scaffolding protein is expressed in E. coli as a soluble monomer. The scaffolding protein interacted with both the portal vertex and the capsid protein. We examined the cloned scaffold–capsid particles both with and without the portal protein using the technique of cryoelectron microscopy. With this technique the particles are frozen, unstained, in a thin layer of vitreous ice and are not distorted or flattened as is common when particles are negatively stained (Adrian et al., 1984; Milligan et al., 1984). Without the portal protein, scaffold–capsid particles were formed in vivo with variable size and shape. However, in the presence of the portal protein, particles with a uniform size and shape were produced. SDS–PAGE analysis has shown that the latter particles contain the proteins of the scaffold, capsid, head fiber, and portal vertex of typical φ29 proheads.
Materials and Methods
DNA preparation
Wild-type φ29 DNA was purified from the virion by heat extraction at 70° for 15 min in TE buffer (50 mM Tris–HCI, pH 7.8/10 mM EDTA). The DNA was then digested with the restriction enzymes SnaBI (cut site 11303), Scal (cut site 5192), and BstNI (cut site 2742), resulting in fragments of 2742, 2450, 6111, and 7977 bp. The 6111-bp fragment, which contained the structural genes 7, 8, 8.5, and 10, was purified by electroelution from a 0.8% agarose gel, concentrated by centrifugation in Centricon-30 tubes (Amicon) and purified by Geneclean (Bio 101). The DNA was cut with EcoRI (cut site 9862), resulting in fragments of 4670 and 1441 bp. The 1441-bp EcoRI/SnaBI fragment, which contained part of gene 10, was cloned into the EcoRI/Smal site of pBluescript (KS+), generating a plasmid pBlue10. The larger 4670-bp fragment containing genes 7, 8, 8.5, and 9 and a part of gene 10 had a Xhol linker ligated to the Scal blunt end. The fragment was then ligated into the vector pBluescript (KS+) at the EcoRI/Xhol site, generating a plasmid pBlueXhol. These two plasmids were used in subsequent constructions.
Construction of plasmid pARgp7
A Ndel/Nhel (5221–5660) fragment containing the gp7 coding sequence was removed from pBlueXhol and inserted into the Ndel/Nhel sites of the expression vector pET-3c (Rosenberg et al., 1987) that was modified prior to ligation by removal of one of the Nhel sites. The single T7 promotor of the plasmid pET-3c was used for transcription with the ATG sequence in the Ndel site (CATATG) of the vector pET-3c serving as the start codon of gene 7. We confirmed the DNA sequences at the junction by sequencing with the double-strand plasmid as templates (Guo et al., 1989; Guo, 1990). The resulting plasmid, pARgp7 (Fig. 1), was transferred into the expression host E. coli HMS-174(DE3) which contains an inducible bacteriophage T7 RNA polymerase gene (Rosenberg et al., 1987; Guo and Moss, 1990).
Fig. 1.
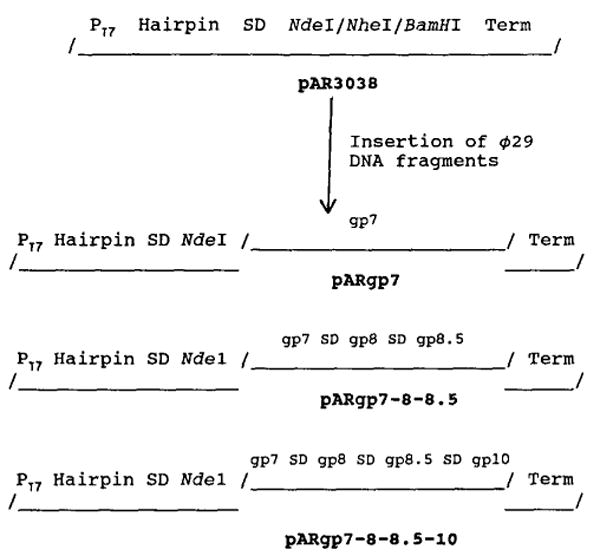
Outline of construction of plasmids pARgp7, pARgp7-8-8.5, and pARgp7-8-8.5-10 expressing or coexpressing the scaffolding protein gp7, the capsid protein gp8, the fiber protein gp8.5, and the portal protein gp10. Individual DNA fragments were cloned into the Ndel site of the plasmid pET-3c. PT7, bacteriophage T7 promotor; Hairpin, a dyad structure that produces a hairpin loop in RNA which protects the RNA transcripts from degradation by exonuclease; gp, gene product; Term, φ10 transcription termination sequence.
Construction of plasmid pARgp7-8-8.5
The genes coding for the scaffolding protein gp7, the major capsid protein gp8 and the dispensable head fiber gp8.5 were cloned together into plasmid pET-3c with a single phage T7 promotor. The Ndel fragment (5221–7757) containing the φ29 genes 7 and 8 was isolated from the plasmid pBlueXhol (see above) and inserted into the expression vector pET-3c at the Ndel site. The resulting plasmid, pARgp7-8-8.5 (Fig. 1), was transferred into E. coli HMS174(DE3). In this construction, the ribosomal binding site sequence of the vector pET-3c was used for the translation of gp7. Translation of gp8 and gp8.5 depended upon the use of their own ribosomal binding site.
Construction of plasmid pARgp7-8-8.5-10
The genes coding for the scaffolding protein gp7, the major capsid protein gp8, the dispensable head fiber gp8.5, and the portal vertex protein gp10 were cloned into plasmid pET-3c as a polycistronic structure. The complete sequence of the structural genes of the φ29 prohead was constructed by combining two fragments in plasmids pBlue10 and pBlueXhol (see above). A 1441-bp EcoRI/BamHI fragment was isolated and ligated into the plasmid pBlueXhol at the EcoRI/BamHI sites. The resulting plasmid pBlue6K contained all of the structural genes of the φ29 prohead. The Nhel/BamHI fragment (5643 bp) from plasmid pBlue6K was isolated and ligated to plasmid pARgp7 at the Nhel/BamHI sites, generating a plasmid pARgp7-8-8.5-10 (Fig. 1) which expressed all of the structural genes of the φ29 prohead. As with pAR7-8-8.5, the ribosome binding sequence from the vector pET-3c was used in the translation of gp7, but translation of genes 8, 8.5, and 10 was dependent on the use of their own ribosomal binding sites.
Expression and purification of the scaffolding protein gp7
The procedure for the purification of the vaccinia virus mRNA capping enzyme was modified for gp7 purification (Guo and Moss, 1990). Overnight cultures (100 ml) of transformed bacteria were inoculated into 2 liters of Super broth (Quality Biological, Gaithersburg, MD), and IPTG (isopropyl β-D-thiogalactoside) was added after 2 hr at 37°. The bacteria were incubated for an additional 2 hr, collected by centrifugation, resuspended in 50 ml of buffer A (50 mM Tris–HCI, pH 8.2/2 mM dithiothreitol/1 mM EDTA/0.01% Nonidet P-40/5% (v/v) glycerol/50 mM NaCI/1 mM phenylmethylsulfonyl fluoride), and broken by passage through a French pressure cell at 10,000 psi. After centrifugation at 10,000 rpm for 10 min, the supernatant was applied to a DEAE-Sephadex column equilibrated with buffer A and the proteins were eluted with a gradient of 0.05 to 1 M NaCI in buffer A. Fractions containing gp7 were pooled and precipitated with saturated ammonium sulfate added to a final concentration of 40%. The protein was dialyzed to remove ammonium sulfate before use.
Purification of the scaffold–capsid and the scaffold–capsid–portal vertex particles
E. coli HMS174(DE3) containing either plasmid pARgp7-8-8.5 or pARgp7-8-8.5-10 was grown in LB broth at 37° to a cell density of 109 cells/ml. The T7 promoter was induced with 0.5 mM IPTG, and the cells were incubated for another 2 hr. Cells were sedimented and lysed in the French press cell at 10,000 psi. The sample was clarified three times at 10,000 rpm and then sedimented at 35K rpm for 3 hr. Particles were resuspended and loaded onto a 5–20% sucrose gradient in MMS buffer (2 mM sodium azide/5 mM maleic acid, pH 5.6/0.1 M NaCI/15 mM MgCI2) and centri-fuged at 35K rpm for 3 hr at 20°. The opalescent band at the typical prohead position was collected, and the particles were sedimented at 35K for 3 hr. Particles were resuspended in MMS (pH 6.5) buffer.
Interaction of the scaffolding protein and the portal vertex in vitro
Purified gp7 and portal vertex (Ibanez et al., 1984; Carazo et al., 1986) were dialyzed on TBE buffer (89 mM Tris borate, pH 8.3/2.5 mM EDTA) for 30 min at ambient temperature and then on TMS buffer (0.05 M Tris–HCI, pH 7.8/0.01 M MgCI2/0.1 M NaCI) for 30 min at the same temperature. Samples were then run on a 1% agarose gel in TBE.
Frozen-hydrated electron microscopy
Individual samples (3–4 μl, 4 mg/ml in 12.5 mM Tris–HCI, pH 7.4) of purified scaffold–capsid and scaffold–capsid–portal vertex particles from E. coli were applied to 400-mesh electron microscope grids covered with perforated carbon films made hydrophilic by glow discharge in air. The grids were blotted with filter paper and then rapidly plunged into liquid ethane (Adrian et al., 1984; Milligan et al., 1984) before being transferred into liquid nitrogen and then into the specimen holder (Gatan Inc., Warrendale, PA) that was maintained at about −165°. The samples were viewed on a Philips EM420 transmission electron microscope (Philips Electronics Instruments, Mahwah, NJ) and photographed under minimal dose conditions.
Results
Cloning of gene 7 of φ29 and purification of the prohead scaffolding protein gp7
The scaffolding protein gp7 of φ29 was expressed from the plasmid pARgp7 in E. coli and purified to homogeneity (Fig. 2). The purification of the scaffolding protein from the recombinant plasmid pARgp7 was verified by (1) molecular weight and sedimentation rate determinations (Figs. 3 and 4); (2) analysis of the amino acid composition and comparison to the predicted sequence; (3) failure to stain with silver; and (4) low ultraviolet absorbance, due to the absence of tryptophan.
Fig. 2.
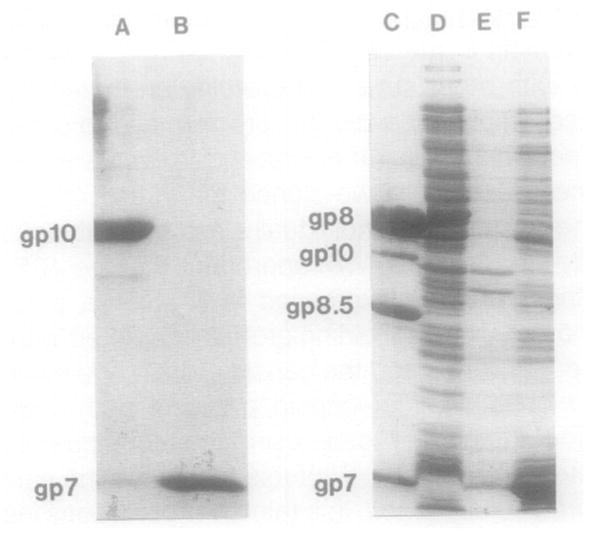
SDS–PAGE of the scaffolding protein demonstrating solubility and purity. (Lane A) Purified portal protein gp10; (lane B) purified scaffolding protein gp7; (lane C) the four structural proteins of purified proheads; (lane D) lysate of E. coli HMS174(DE3) as a control; (lanes E and F) pellets and supernatant, respectively, of induced HMS174(DE3)pARgp7 lysate after 16,000 g sedimentation. The gp7 expressed in E. coli was present in the supernatant, indicating it is in a soluble form.
Fig. 3.
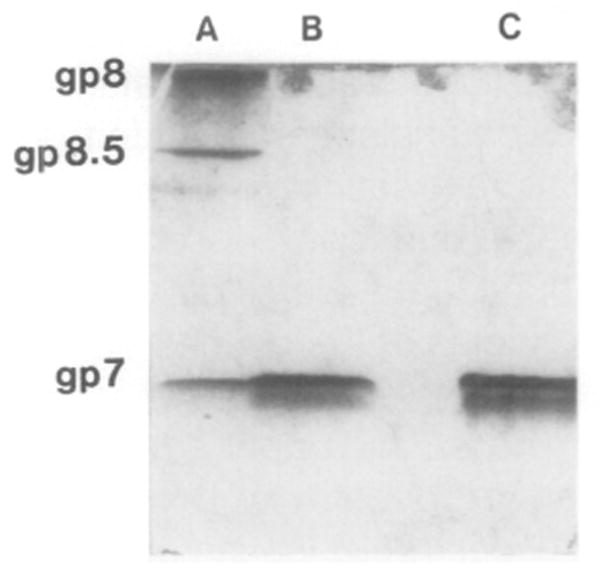
Native 15% polyacrylamide gel of purified gp7, showing the monomeric form of gp7 in solution. (Lane A) Structural proteins of purified proheads denatured in 1% SDS and 0.1% β-mercaptoethanol; (lane B) denatured gp7, prepared as above; (lane C) gp7 prepared in nondenaturing buffer. No difference in the migration rate between the denatured and nondenatured gp7 was detected, indicating that gp7 is present in a monomeric form in solution.
Fig. 4.
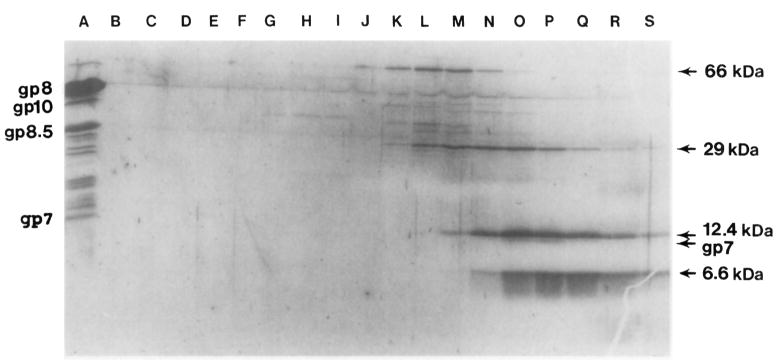
Size determination of purified gp7 by sucrose gradient sedimentation. Purified gp7 was sedimented in a 5–20% linear sucrose gradient with molecular weight markers. Samples from every other fraction were run on SDS–PAGE, which was first stained with Coomassie blue and then with silver stain after decoloration. Sedimentation was from right to left. (Lane A) Purified proheads; (lane B–S) fractions of the sucrose gradient; (lanes O and P) gp7 migrated with a rate close to the 12-kDa marker. gp7 was clearly identified by its typical color reaction, staining with Coomassie blue but not with silver stain. The 12-kDa molecular marker was only stained by silver stain.
We have found that the amino acid sequences of the scaffolding proteins of the phages φ29, λ, and T7 and of the herpes virus to contain the characteristic leucine zipper motif. The leucine zipper is reportedly involved in the formation of dimers in DNA binding proteins (Landschulz et al., 1988; Vinson et al., 1989; Gentz et al., 1989; Turner and Tjian, 1989). Interestingly, we found by sucrose density gradient sedimentation and native gel electrophoresis that the scaffolding protein of φ29, even though it constitutes up to 20–30% of the total cell protein, is a soluble monomer (Fig. 3 and 4). Large amounts of homogeneous gp7 were obtained with only two steps of purification (see Materials and Methods).
Interaction of the scaffolding protein with the portal vertex in vitro
The scaffolding protein plays an important role in φ29 prohead assembly. To elucidate the assembly pathway, we studied the interaction of the scaffolding protein gp7 with the portal vertex, the DNA packaging protein gp 16 (Guo et al., 1986, 1987b), the major capsid protein gp8 and the prohead small RNA (Guo et al., 1987a,c). Interaction of the scaffold protein and capsid protein in vivo was observed (see below), but no binding of scaffold to gp16 or the small RNA was detected. However, the scaffolding protein interacted strongly with the portal vertex dodecamers (Carazo et al., 1986). By mixing a fixed concentration of portal vertex with increasing amounts of scaffold protein, we observed an increase in the migration rate of product in a nondenaturing agarose gel (Fig. 5). In the electrophoresis conditions used, the scaffold protein is more negatively charged than the portal vertex as determined from the binding capacities to DEAE-cellulose and phosphocellulose. Interaction of the scaffolding protein with the portal vertex resulted in the formation of product with a migration rate faster than the portal vertex due to the change of charge.
Fig. 5.

Interaction of the scaffolding protein gp7 with the portal vertex gp10. Varying concentrations of gp7 and a fixed concentration of portal vertex were mixed and dialyzed together against TBE. After 30 min at ambient temperature, the mixture was transferred to TMS and allowed to dialyze another 30 min. The mixture was then run on a 1.0% native agarose gel. Lane A contained only the portal vertex and lane H only gp7. The final concentration of portal vertex in lanes A–G was 1 mg/ml and gp7 in Lanes B to H was 0.08, 0.16, 0.32, 0.48, 0.64, 0.80, and 1.6 mg/ml, respectively. The migration rate of the products increased with the increasing concentration of gp7, indicating the interaction of gp7 with the portal vertex.
Particles assembled in vivo from the scaffold and capsid proteins vary in size and shape
The genes coding for the scaffold protein gp7 and the capsid protein gp8 were both strongly expressed in the E. coli strain HMS174(DE3) after IPTG induction (Figs. 6 and 7). The gene 8.5 that encodes the head fibers was expressed at a lower level. We used cryoelectron microscopy to determine if the gp7 and gp8 proteins were assembled in vivo. Subtle structural differences are often not detectable in samples that are negatively stained, but frozen-hydrated virus particles retain their native morphology. Complexes of gp7 and gp8 were observed in the frozen-hydrated preparations but the size and shape of the particles varied greatly (Fig. 8A).
Fig. 6.

Coexpression of gp7 and gp8 after IPTQ induction as demonstrated by SDS–PAGE and Coomassie blue staining. (Lanes A and C) Crude lysate of E. coli HMS174(DE3)pARgp7-8-8.5 overexpressing both gp7 and gp8; (lane B) lysate of E. coli HMS174(DE3) as a control.
Fig. 7.
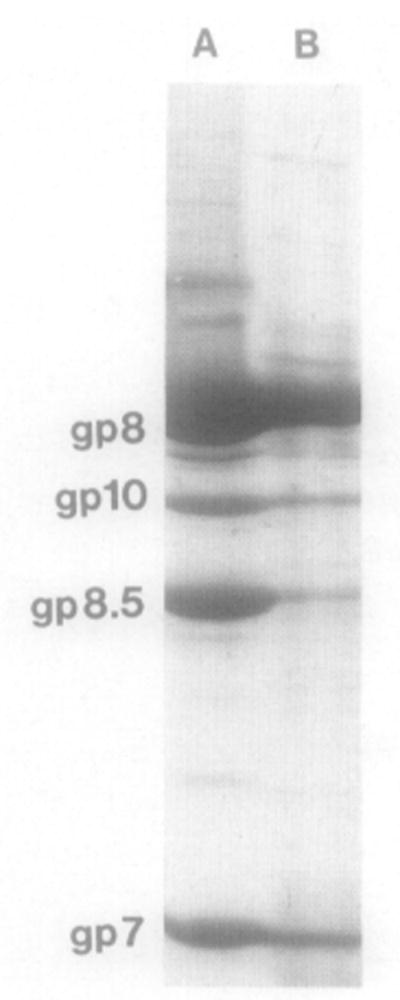
Identification of the components of purified particles by SDS–PAGE and Coomassie blue staining. (Lane A) Proheads from infected B. subtilis cells; (lane B) particles from pARgp7-8-8.5-10. Comigration of gp7, gp8, gp8.5 with gp10 at the prohead position in the gradient indicates incorporation of the portal vertex into particles.
Fig. 8.

Micrographs of frozen-hydrated φ29 particles. (A) Scaffold–capsid particles from E. coli that coexpressed gp7 and gp8. The particles vary greatly in size and shape. (B) Uniform scaffold–capsid–portal vertex particles from E. coli that expressed gp7, gp8, and gp10. (C) Purified proheads from φ29-infected B. subtilis cells. In (B) and (C), some particles are oriented with their long axes perpendicular to the plane of view and observed as circles (arrows). Bar = 200 nm.
Assembly of scaffold–capsid–portal vertex particles with uniform size and shape
To determine whether the structural proteins of φ29 can be assembled into typical φ29 proheads independent of other φ29 and B. subtilis factors, the genes coding for the scaffolding protein gp7, the major capsid protein gp8, the dispensable head fiber gp8.5, and the portal vertex protein gp10 were cloned into the plasmid pET-3c, resulting in the plasmid pARgp7-8-8.5-10. The proteins gp7, gp8, gp8.5, and gp10 were coexpressed in the E. coli strain HMS174(DE3) after IPTG induction. The particles, purified from the lysate, possessed a uniform size and shape (Fig. 8B) and closely resembled native proheads (Fig. 8C). This indicates that the portal protein regulates the size and shape of the prohead. The typical prohead composition, including the portal protein, was confirmed by SDS–PAGE (Fig. 7).
Although both plasmids pARgp7-8-8.5 and pARgp7-8-8.5-10 contain the gene coding for the head fiber of φ29 prohead, the fiber gene was expressed at a lower level compared to the expression of gp7 and gp8. This might result from the difference between the ribosome binding sites of individual genes. The head fibers are dispensable for prohead morphogenesis (Reilly et al., 1977) and will not be discussed further.
Discussion
Our results indicate that prohead-like particles could be formed, though with varied size and shape, in the absence of portal protein. This is in agreement with the results of Bazinet and King (1988) which showed that the portal proteins were dispensable for initiation of prohead assembly. The interaction of the scaffolding protein with the portal protein, and the production of uniform particles in the presence of the portal proteins, suggest that: (1) the scaffold protein serve as the linkage between the portal vertex and the capsid proteins, and (2) the portal vertex plays a crucial role in the regulation of prohead size and shape. The interaction of scaffold protein with both the capsid and portal vertex implies a process that deviates from the traditional single assembly pathway. The single assembly pathway allows control over each step such that the process will not continue unless the previous step is complete (see Introduction and Casjens and Hendrix, 1988). The production of scaffold–capsid particles with varying size and shape in the absence of the portal protein obviates the possibility that the scaffold–capsid particle is formed first and then the portal vertex is subsequently inserted into this structure. If this were the case, varying sizes of mature proheads would be produced. The scaffold protein must contain two or more active domains for protein binding or interaction. One domain interacts with the portal vertex and another with the capsid protein. This could provide a mechanism for the prevention of uncontrolled polymerization. A defined number of scaffolding molecules that interact with one dodecamic portal vertex might subsequently determine the number of molecules of capsid protein that interact with the scaffolding proteins; this would in turn determine the prohead size. The observation on prohead structural protein interactions cannot be explained by the branched pathway either (Wood et al., 1968; Berget, 1985). Continuation of these studies may provide new insight into macromolecular assembly.
The scaffold–capsid–portal vertex particles produced in E. coli were fully competent to package φ29 DNA in both extracts and the defined in vitro DNA packaging system that includes RNA (Guo et al., manuscript in preparation). This provides an ideal system for studies of the pathway of prohead assembly and the mechanism of prohead size determination.
Acknowledgments
We thank Drs. Larry Glickman and Terry Bowersock for their comments and suggestions, Mr. Seth Glickman and Miss Deanna Drew for typing of the manuscript, and Mary Crouch for photographic assistance. This work was supported by a subcontract to P.G. from an NIH Grant to D.A. (DE03606), an NIH Grant to T.S.B. (GM33050), and a grant to the Center for Macromolecular Structure Research from the Lucille P. Markey Trust.
References
- Adrian M, Dubochet J, Lepault J, McDowall AW. Cryo-electron microscopy of viruses. Nature (London) 1984;308:32–36. doi: 10.1038/308032a0. [DOI] [PubMed] [Google Scholar]
- Bazinet C, King J. The DNA translocating vertex of dsDNA bacteriophages. Annu Rev Microbiol. 1985;39:109–130. doi: 10.1146/annurev.mi.39.100185.000545. [DOI] [PubMed] [Google Scholar]
- Bazinet C, King J. Initiation of p22 procapsid assembly in vivo. J Mol Biol. 1988;202:77–86. doi: 10.1016/0022-2836(88)90520-7. [DOI] [PubMed] [Google Scholar]
- Berget P. Pathways in viral morphogenesis. In: Casjens S, editor. Virus Structure and Assembly. Jones & Bartlett; Boston: 1985. pp. 149–168. [Google Scholar]
- Black LW. DNA packaging in dsDNA bacteriophagse. In: Calendar R, editor. The Bacteriophages. Vol. 2. Plenum; New York: 1988. pp. 321–373. [Google Scholar]
- Black LW. DNA packaging in dsDNA bacteriophages. Annu Rev Microbiol. 1989;43:267–292. doi: 10.1146/annurev.mi.43.100189.001411. [DOI] [PubMed] [Google Scholar]
- Carazo JM, Donate LE, Herranz L, Secilla JP, Carrascosa JL. Three-dimensional reconstruction of the connector of bacteriophage φ29 at 1.8 nm resolution. J Mol Biol. 1986;192:853–867. doi: 10.1016/0022-2836(86)90033-1. [DOI] [PubMed] [Google Scholar]
- Casjens S. Nucleic acid packaging by viruses. In: Casjens S, editor. Virus Structure and Assembly. Jones & Bartlett; Boston: 1985. pp. 75–147. [Google Scholar]
- Casjens S, Hendrix R. Control mechanisms in dsDNA bacteriophage assembly. In: Calendar R, editor. The Bacteriophages. Vol. 1. Plenum; New York: 1988. pp. 15–92. [Google Scholar]
- Earnshaw WC, Casjens SR. DNA packaging by the double-stranded DNA bacteriophages. Cell. 1980;21:319–331. doi: 10.1016/0092-8674(80)90468-7. [DOI] [PubMed] [Google Scholar]
- Edgar R, Wood W. Morphogenesis of bacteriophage T4 in extracts of mutant-infected cell. Proc Natl Acad Sci USA. 1966;55:498–505. doi: 10.1073/pnas.55.3.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein R, Bolle A, Steinberg C, Lielausis I. Physiological studies of conditional lethal mutants of bacteriophage T4D. Cold Spring Harbor Symp Quant Biol. 1963;28:375. [Google Scholar]
- Fuller MT, King J. Assembly in vitro of bacteriophage P22 procapsids from purified coat and scaffolding subunits. J Mol Biol. 1982;156:633–665. doi: 10.1016/0022-2836(82)90270-4. [DOI] [PubMed] [Google Scholar]
- Gentz R, Rauscher FJ, III, Abate C, Curran T. Parallel association of Fos and Jun leucine zippers juxtaposes DNA binding domains. Science. 1989;243:1695–1699. doi: 10.1126/science.2494702. [DOI] [PubMed] [Google Scholar]
- Guo P. Characterization of the gene and a antigenic determinant of equine herpesvirus 1 glycoprotein 14 with homology to gB-equivalent glycoprotein of other herpesvirus. Gene. 1990;87:249–255. doi: 10.1016/0378-1119(90)90309-f. [DOI] [PubMed] [Google Scholar]
- Guo P, Bailey S, Bodley JW, Anderson D. Characterization of the small RNA of the bacteriophage φ29 DNA packaging machine. Nucleic Acid Res. 1987a;15:7081–7090. doi: 10.1093/nar/15.17.7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo P, Erickson S, Anderson D. A small viral RNA is required for in vitro packaging of bacteriophage φ29 DNA. Science. 1987b;236:690–694. doi: 10.1126/science.3107124. [DOI] [PubMed] [Google Scholar]
- Guo P, Goebel PS, Davis S, Perkus M, Lanquet B, Desmettre P, Allen G, Paoletti E. Expression of the equine herpesvirus type 1 gene encoding glycoprotein gp13 in recombinant vaccinia virus and protection of immunized animals. J Virol. 1989;63:4189–4198. doi: 10.1128/jvi.63.10.4189-4198.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo P, Grimes S, Anderson D. A defined system for in vitro packaging of DNA-gp3 of the Bacillus subtilis bacteriophage φ29. Proc Natl Acad Sci USA. 1986;83:3505–3509. doi: 10.1073/pnas.83.10.3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo P, Moss B. Interaction and mutual stabilization of the two subunits of vaccinia virus mRNA capping enzyme co-expressed in Escherichia coli. Proc Natl Acad Sci USA. 1990;87:4023–4027. doi: 10.1073/pnas.87.11.4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo P, Peterson C, Anderson D. Prohead and DNA-gp3-dependent ATPase activity of the DNA packaging protein gp16 of bacteriophage φ29. J Mol Biol. 1987c;197:229–236. doi: 10.1016/0022-2836(87)90121-5. [DOI] [PubMed] [Google Scholar]
- Guo P, Peterson C, Anderson D. Initiation events in in vitro packaging of bacteriophage φ29 DNA-gp3. J Mol Biol. 1987d;197:219–228. doi: 10.1016/0022-2836(87)90120-3. [DOI] [PubMed] [Google Scholar]
- Ibanez C, Garcia JA, Carrascosa JL, Salas M. Overproduction and purification of the connector protein of Bacillus subtilis phage φ29. Nucleic Acids Res. 1984;12:2351–2365. doi: 10.1093/nar/12.5.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landschulz WH, Johnson PF, McKnight SL. The DNA binding domain of the rat liver nuclear protein C/EBP is bipartite. Science. 1989;240:1759–1764. doi: 10.1126/science.2494700. [DOI] [PubMed] [Google Scholar]
- Milligan RA, Brisson A, Unwin PNT. Molecular structure determination of crystalline specimens in frozen aqueous solutions. Ultramicroscopy. 1984;813:1–10. doi: 10.1016/0304-3991(84)90051-2. [DOI] [PubMed] [Google Scholar]
- Murialdo H, Becker A. Head morphogenesis of complex double-stranded deoxyribonucleic acid bacteriophages. Microbiol Rev. 1978;42:529–576. doi: 10.1128/mr.42.3.529-576.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevelige PE, Jr, Thomas D, King J. Scaffolding protein regulates the polymerization of P22 coat subunits into icosahedral shell in vitro. J Mol Biol. 1988;202:743–757. doi: 10.1016/0022-2836(88)90555-4. [DOI] [PubMed] [Google Scholar]
- Reilly BE, Nelson RA, Anderson DL. Morphogenesis of bacteriophage φ29 of Bacillus subtilis: Mapping and functional analysis of the head fiber gene. J Virol. 1977;24:363–377. doi: 10.1128/jvi.24.1.363-377.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg AH, Lade BN, Chui DO, Lin SW, Dunn JJ, Studier TW. Vectors for selective expression of cloned DNAs by T7 RNA polymerase. Gene. 1987;56:125–135. doi: 10.1016/0378-1119(87)90165-x. [DOI] [PubMed] [Google Scholar]
- Traub F, Keller B, Kuhn A, Maeder M. Isolation of the prohead core of bacteriophage T4 after cross-linking and determination of protein composition. J Virol. 1984;49:902–908. doi: 10.1128/jvi.49.3.902-908.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub F, Maeder M. Formation of the prohead core of bacteriophage T4 in vivo. J Virol. 1984;49:892–901. doi: 10.1128/jvi.49.3.892-901.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner R, Tjian R. Leucine repeats and an adjacent DNA binding domain mediate the formation of junctional cFoscJun heterodimers. Science. 1989;243:1689–1694. doi: 10.1126/science.2494701. [DOI] [PubMed] [Google Scholar]
- Vinson C, Sigler PB, McKnight SL. Scissors-grip model for DNA recognition by a family of leucine zipper proteins. Science. 1989;246:911–916. doi: 10.1126/science.2683088. [DOI] [PubMed] [Google Scholar]
- Wood WB, King J. Genetic control of complex bacteriophage assembly. Comprehensive Virol. 1979;13:581–633. [Google Scholar]
- Wood W, Edgar R, King J, Henninger M, Lielausis I. Bacteriophage assembly. Fed Proc. 1968;27:1160–1166. [PubMed] [Google Scholar]