

Abstract
Epoxyeicosatrienoic acids (EETs) contribute to haemodynamics, electrolyte homoeostasis and blood pressure regulation, leading to the concept that EETs can be therapeutically targeted for hypertension. In the present study, multiple structural EET analogues were synthesized based on the EET pharmacophore and vasodilator structure-activity studies. Four EET analogues with 91–119 % vasodilatory activity in the isolated bovine coronary artery (EC50: 0.18–1.6 μM) were identified and studied for blood-pressure-lowering in hypertension. Two EET analogues in which the COOH group at carbon 1 of the EET pharmacophore was replaced with either an aspartic acid (EET-A) or a heterocyclic surrogate (EET-X) were administered for 14 days [10 mg/kg per day intraperitoneally (i.p.)]. Both EET-A and EET-X lowered blood pressure in spontaneously hypertensive rats (SHRs) and in angiotensin II (AngII) hypertension. On day 14, the mean arterial pressures in EET analogue-treated AngII-hypertensive and SHRs were 30–50 mmHg (EET-A) and 15–20 mmHg (EET-X) lower than those in vehicle-treated controls. These EET analogues (10 mg/kg per day) were further tested in AngII hypertension by administering orally in drinking water for 14 days and EET-A lowered blood pressure. Additional experiments demonstrated that EET-A inhibits epithelial sodium channel (ENaC) activity in cultured cortical collecting duct cells and reduced renal expression of ENaC subunits in AngII hypertension. In conclusion, we have characterized EET-A as an orally active antihypertensive EET analogue that protects vascular endothelial function and has ENaC inhibitory activity in AngII hypertension.
Keywords: angiotensin II, Cyp2c44-knockout mouse, epithelial sodium channel (ENaC channel), epoxyeicosatrienoic acid analogue, hypertension
INTRODUCTION
Epoxyeicosatrienoic acids (EETs) are cytochrome P450 metabolites that have numerous biological actions suggesting their therapeutic potential in cardiovascular diseases [1–4]. The prominent cardiovascular actions known for EETs include their role as endothelium-derived hyperpolarizing factors and regulation of tubular sodium reabsorption by inhibiting epithelial sodium channel (ENaC) [3,5,6]. In line with these biological actions of EETs, previous studies have demonstrated that inhibition of soluble epoxide hydrolase (sEH), which hydrolyses EETs to their less biologically active dihydroxyeicosatetraenoic acid metabolite, increases plasma EET level and exerts therapeutically important cardiovascular effects including blood pressure reduction [2,7]. Indeed, antihypertensive therapeutic actions for sEH inhibitors (sEHis) have been repeatedly demonstrated in angiotensin II (AngII)-dependent hypertension [8–11]. Moreover, such antihypertensive effects of increased plasma EET levels by sEH inhibition have also been demonstrated in non-AngII models of hypertension [12,13].
Another approach to target EETs for cardiovascular diseases is the development of analogues for the EETs. EET analogues were developed because of the limited solubility and storage issues with endogenous EETs [14–16]. These EET analogues were designed to resist metabolism and improve solubility, and significantly facilitated the identification of structure–activity relationships for EETs [14–18]. As demonstrated in a number of studies, these EET analogues vasodilate coronary, renal and mesenteric arteries [18–21]. A number of experimental studies have supported the use of EET analogues in cardiovascular disease. For instance, EET analogues reduced blood pressure in hypertensive rats [21], decreased experimental cardiac reperfusion injury [22] and reduced the metabolic syndrome in mice [23]. Indeed, considering numerous promising biological actions including blood-pressure-lowering, it is important to develop orally active antihypertensive EET analogues, which could eventually be developed as novel cardiovascular therapeutics.
With this background, in the present study we tested the in vitro vasodilatory activity of four novel EET analogues. We further tested the antihypertensive effect of these EET analogues in vivo using rat models of hypertension. Finally, we identified an orally active antihypertensive EET analogue and delineated its possible antihypertensive mechanism.
MATERIALS AND METHODS
Chemicals
Unless and otherwise mentioned, all chemicals used in the present study were purchased from Sigma–Aldrich. The EET analogues (EET-A, EET-X, EET-Y and EET-Z) were designed and synthesized in J.R.F.’s laboratory.
Animals
Experiments were conducted using male spontaneously hypertensive rats (SHRs), Sprague–Dawley (SD) rats (225–275 g) and mice lacking cytochrome P450 (Cyp) 2c44 (Cyp2c44−/− mice, 20–25 g). Animal protocols were in accordance with National Institutes of Health guidelines and approved by the Institutional Animal Care and Use Committee. Throughout the experiment, animals were housed under conditions of constant temperature and humidity, with a 12/12 h light–dark cycle. Animals were allowed to adapt to these conditions for several days before starting any experimental procedures.
In vivo experiments
Telemetry blood pressure measurement
In order to measure blood pressure, telemetry transmitters (Data Sciences) were implanted 14 days before the experiment in rats and mice using methods described previously [21,24]. Baseline arterial pressure and heart rate were recorded for 3–5 days before the experimental period. Mean arterial pressure (MAP) or systolic blood pressure and heart rate were recorded continuously throughout the experimental period.
Antihypertensive effects of intraperitoneally administered EET analogues in SHRs and AngII-hypertensive rats
In the first set of experiments, telemetry transmitters were implanted into male SHRs. After the surgical recovery period, baseline MAP was recorded for 14 days. In this set of experiments, EET analogues (EET-A, EET-X, EET-Y and EET-Z) were administered intraperitoneally (i.p.) continuously using ALZET® osmotic pumps (DURECT) at a dose of 10 mg/kg per day, and blood pressure was monitored continuously. The vehicle-treated rats were administered a solution containing DMSO, ethanol and PEG-400 (40 %, 15 % and 45 % respectively) for 14 days using ALZET® osmotic pumps. In the second set of experiments, telemetry transmitters were implanted into male SD rats. After 1 week of basal blood pressure recording, ALZET® osmotic pumps were implanted subcutaneously (s.c.) to deliver AngII at a dose of 180 ng/kg per min for 14 days. On the first day of AngII pump implantation, the EET analogues, EET-A and EET-X were administered (i.p.) using ALZET® osmotic pumps continuously at a dose of 10 mg/kg per day for 14 days and blood pressure was monitored. The vehicle-treated rats were administered either distilled water or a solution containing DMSO, ethanol and PEG-400 (40 %, 15 % and 45 % respectively) for 14 days using ALZET® osmotic pumps. In an additional set of experiments, we implanted osmotic pumps filled with EET-A (10 mg/kg per day) or vehicle-containing DMSO, ethanol and PEG-400 (40 %, 15 % and 45 % respectively) in SD rats to determine the effect of EET-A on their blood pressure.
Antihypertensive effects of orally administered EET analogues in AngII-hypertensive rats
This experiment was carried out in a set of SD rats implanted with radiotransmitters for continuous monitoring of blood pressure and AngII-filled ALZET® osmotic pumps (s.c.) to deliver AngII at a dosage of 180 ng/kg per minute for 14 days. The rats were treated with EET analogues (10 mg/kg per day), EET-A and EET-X given in drinking water ad libitum, and blood pressure was monitored continuously. Vehicle-treated groups received drinking water or a solution containing 1 % PEG-400 and 0.05 % ethanol ad libitum, while blood pressure was monitored continuously. At the end of the 14-day treatment period, 24-h urine samples were collected for the measurement of sodium and biochemical analysis.
Systolic blood pressure measurement in Cyp2c44−/− mice
Cyp2c44 is an important enzyme that generates renal EETs and Cyp2c44−/− mice develop salt-sensitive hypertension [6]. In mice, radiotransmitters were implanted for continuous monitoring of blood pressure. We investigated the antihypertensive effect of EET-A in Cyp2c44−/− mice receiving an 8 % NaCl diet for 28 days. Mice received drinking water or EET-A (10 mg/kg per day) in drinking water ad libitum during the 28-day treatment period. Blood pressure was monitored continuously and 24-h urine samples were collected at the end of the treatment period for the measurement of electrolytes and biochemical assays.
Biochemical measurements
Urinary electrolytes were measured using ion-selective electrode (ISE)-based method (EasyLyte Analyzer, Medica Corporation). sEHi activity was determined using a kit from Cayman Chemical.
In vitro experiments
Vascular reactivity studies
Three sets of vascular experiments were carried out. In the first set, measurements of isometric tone in bovine coronary artery rings were conducted as described previously [18,25]. The arterial rings were slowly stretched to a basal tension of 3.5 g and equilibrated for 1.5 h. KCl (40–60 mM) was repeatedly added and rinsed until reproducible stable contractions were observed. The thromboxane mimetic 9,11-dideoxy-11α,9α-epoxymethano-prostaglandin F2α (U46619; 20 nM) was added to increase basal tension to approximately 50–75 % of maximal KCl contraction. Relaxation responses to cumulative additions of the EET analogues (10−9–10−5 M) were recorded. Basal tension represents tension before the addition of U46619. Results are expressed as the percentage relaxation of the U46619-treated rings; 100 % relaxation represents basal tension.
In the second set of vascular experiments, second-order mesenteric arteries were excised from vehicle, EET-A and EET-X-treated AngII-hypertensive rats on day 14 of the experimental protocol to determine the effects of EET analogues on the acetylcholine vasodilator response. The third set of vascular experiments were also carried out with second-order mesenteric arteries of AngII-hypertensive rats treated with EET-A for 14 days, and acetylcholine vascular responses were studied. However, this set of experiments was carried out in the presence and absence of L-NG-nitroarginine methyl ester (L-NAME, 100 μM), an inhibitor of nitric oxide synthase. All these experiments were conducted as described previously [20]. In brief, the vessel diameter responses to acetylcholine (0.001–10 μM) were assessed after U46619 constriction. Vasodilatory responses were plotted as a percentage of relaxation from the maximum contraction. Sodium nitroprusside (100 μM) was added to the bath at the end of the experimental period to ensure vascular integrity.
Electrophysiological experiments
Effect of EET-A on sodium transport and on single-channel ENaC activity was measured in immortalized mouse cortical collecting duct principal (mpkCCDc14) cells as described previously [26–28]. In the first set of experiments, equivalent transepithelial sodium currents were calculated as the quotient of transepithelial voltage to transepithelial resistance under short-circuit conditions. The second set of experiments was carried out to study the effect of EET-A on ENaC activity using cell-attached patches made on the plasma membrane of mpkCCDc14 cells. NPo, the product of the number of channels (N) and the open probability (Po), or Po itself, was used to measure channel activity within a patch. Single-channel unitary current (i) was determined from the best-fit Gaussian distribution of amplitude histograms. Channel activity was assessed as NPo = I/i, where I is mean total current in a patch.
Immunohistochemical analysis
The kidney sections were embedded and cut into 4-μm-thick slices for use in immunohistochemistry protocols. Formalin-fixed paraffin-embedded kidney slices were deparaffinized, rehydrated and subjected to immunohistochemistry protocols. Kidney sections were immunostained with antibodies against α-, β- and γ-subunits of ENaC (α-ENaC, 1:1000 dilution; β-ENaC, 1:100 dilution; γ-ENaC, 1:500 dilution; StressMarq Biosciences) in order to determine the relative expression and localization of these ENaC subunits in the kidney. Biotinylated horse anti-goat or anti-rabbit secondary antibodies (1:200 or 1:300 dilution) were used for development with avidin–biotinylated horseradish peroxidase (HRP) complex (Vectastain ABC Elite kit, Vector Laboratories). Finally, the slides were counterstained with haematoxylin and mounted for image capturing (×200) and analysis using Nikon NIS Elements Software. Immunoreactivity of ENaC subunits was assessed by analysing ten images (×200) for each sample using Nikon NIS Elements Software, and the extent of immunoreactivity was calculated as the percentage area fraction of immunopositive area relative to the total area of an image.
Statistics
Data are expressed as means ± S.E.M. Statistical significance between two measurements was determined by the two-tailed unpaired Student’s t test (and among groups it was determined by repeated-measure one-way ANOVA followed by Tukey’s post-hoc test) using GraphPad Prism® Version 4.0 software. Probability values of P < 0.05 were considered significant where the critical value of P was two-sided.
RESULTS
EET analogues relaxed isolated bovine coronary artery
As shown in Table 1, all four EET analogues demonstrated marked vasorelaxation activity in isolated bovine coronary arteries. Maximum relaxations caused by these EET analogues were between 91 % and 119 % (EC50: 0.18–1.6 μM), and EET-Z was the most potent followed by EET-Y, EET-X and EET-A. In addition, it was observed that EET analogues have weak sEH inhibitory activity except EET-Z, which had an IC50 of 11 nM (Table 1).
Table 1. Structures and vasodilatory actions of four EET analogues on isolated bovine coronary arteries.
The percentage relaxation and EC50 for EET-A, EET-X, EET-Y and EET-Z were calculated from five independent measurements with each EET analogue. For each EET analogue, sEHi activity was determined and IC50 was calculated from five independent measurements.
| EET analogue | Structure | Vascular relaxation
|
EC50 (μM) | sEHi activity IC50 (nM) |
|---|---|---|---|---|
| Percentage relaxation | ||||
| EET-A |
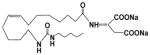
|
91 | 1.6 | 392 |
| EET-X |
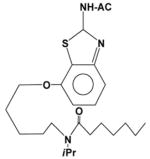
|
96 | 1.3 | >500 |
| EET-Y |
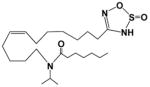
|
109 | 0.32 | >500 |
| EET-Z |
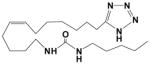
|
119 | 0.18 | 11 |
Intraperitoneal administration of EET-A and EET-X reduced MAP in SHRs and in AngII-hypertensive rats
The effects of EET analogues on blood pressure in SHRs are shown in Figure 1. EET-A and EET-X significantly blunted the development of hypertension in SHRs, and MAP was 15–20 mmHg lower on day 14. It is also observed that on day 14 these EET analogues lowered both systolic and diastolic blood pressures in SHRs (Supplementary Table S1 at http://www.clinsci.org/cs/127/cs1270463add.htm). On the other hand, EET-Y and EET-Z did not lower blood pressure in SHRs. EET analogues did not alter heart rate in SHRs. Heart rate remained unchanged on days 0, 7 and 14 [EET-A: 346 ± 11, 353 ± 3 and 353 ± 7 beats per min (BPM); EET-X: 347 ± 10, 332 ± 3 and 347 ± 7 BPM; EET-Y: 340 ± 6, 332 ± 7 and 330 ± 3 BPM; EET-Z: 327 ± 8, 321 ± 2 and 315 ± 6 BPM]. EET-A and EET-X were further studied in AngII hypertension. These EET analogues significantly blunted the development of AngII hypertension, and MAP was 30–50 mmHg lower on day 14 (Figure 2). Moreover, similar to SHRs, it is observed that on day 14 both of these EET analogues lowered systolic and diastolic blood pressures in AngII-hypertensive rats (Supplementary Table S1). Heart rate averaged 419 ± 23 BPM on day 0 and decreased to the same extent in AngII-vehicle (−55 ± 7 BPM), EET-A-treated (−45 ± 3 BPM) and EET-X-treated (−43 ± 9 BPM) rats on day 7. On day 14, heart rate returned near day 0 values and averaged 431 ± 19 BPM in AngII-vehicle, 435 ± 29 BPM in EET-A-treated and 429 ± 17 BPM in EET-X-treated rats.
Figure 1. Effects of EET analogues on the MAP of SHRs.

EET-A (a), EET-X (b), EET-Y (c) and EET-Z (d) were administered at a dose of 10 mg/kg per day (i.p.) for 14 days. Blood pressure data are presented as the average of 12-h readings that were recorded continuously throughout the experimental protocol. *P < 0.05 compared with vehicle; n = 8–10. All data are presented as means ± S.E.M.
Figure 2. Effects of EET analogues on the MAP in AngII hypertension.
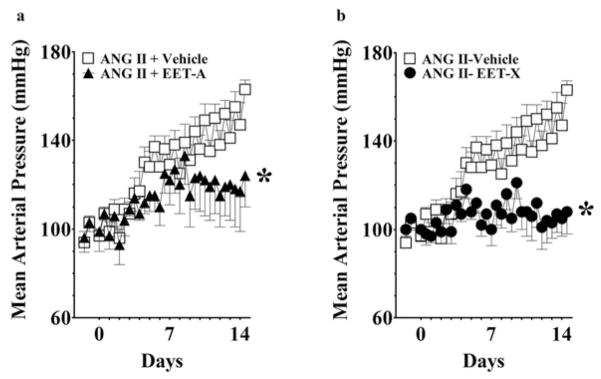
EET-A (a) and EET-X (b) were administered at a dose of 10 mg/kg per day (i.p.) for 14 days. Blood pressure data are presented as the average of 12-h readings that were recorded continuously throughout the experimental protocol. *P < 0.05 compared with vehicle; n = 8–10. All data are presented as means ± S.E.M.
Oral administration of EET-A, but not EET-X, reduced blood pressure and improved vascular function in AngII hypertension
Antihypertensive effects were studied further in AngII hypertension by administering EET-A and EET-X in drinking water. EET-A treatment markedly blunted the development of AngII hypertension without affecting heart rate (Figures 3a and 3b). Orally administered EET-X delayed the onset of hypertension in AngII-infused rats; however, blood pressure was not lower on day 14. Heart rate was unaltered by EET-X treatment (Figures 3c and 3d). Although EET-A effectively attenuated the development of hypertension in SHRs and AngII hypertension, it did not affect MAP (117 ± 4 mmHg compared with 115 ± 8 mmHg) or heart rate (395 ± 20 BPM compared with 378 ± 28 BPM) in normal SD rats.
Figure 3. Effects of orally administered EET analogues on the MAP and heart rate in AngII hypertension.

EET-A (a and d) and EET-X (b and c) were administered at a dose of 10 mg/kg per day in drinking water for 14 days. Blood pressure and heart rate data are presented as the average of 12-h readings that were recorded continuously. *P < 0.05 compared with vehicle; n = 8–10. All data are presented as means ± S.E.M.
In the present study, we determined the vasodilator response of mesenteric resistance arteries to acetylcholine in vehicle- and EET analogue-treated AngII-hypertensive rats. Vasodilation of mesenteric resistance arteries to acetylcholine was blunted in AngII-hypertensive rats compared with SD rats. The vasodilator response to 10 μM acetylcholine averaged 90 ± 9 % in SD rats and 45 ± 12 % in AngII-hypertensive rats. Interestingly, chronic EET-A treatment improved the vasodilator response in AngII-hypertensive rats and the mesenteric resistance artery response to acetylcholine increased to a maximum response of 75 ± 5 %. Unlike EET-A, the mesenteric resistance arteries, vasodilator response to acetylcholine was not improved in EET-X-treated AngII-hypertensive rats (Figure 4a). In the present study, we demonstrate that nitric oxide synthase blockade by L-NAME attenuated EET-A-mediated improvement in acetylcholine vasodilator response in AngII hypertension (61 ± 8 %). Although the EET-A-mediated improvement in the vascular response to 10 μM acetylcholine was attenuated in the presence of L-NAME, it was still greater than the acetylcholine response in vehicle-treated AngII-hypertensive rats in the presence of L-NAME (41 ± 9 %) (Figure 4b). These findings support the notion that mesenteric resistance artery endothelial function in AngII hypertension is improved by EET-A treatment. These results also indicate that the nitric oxide system, along with other vasodilatory components, contributes to improved acetylcholine-induced vasodilation in EET-treated AngII-hypertensive rats.
Figure 4. Effect of orally administered EET analogues on the vascular function in AngII hypertension.

(a) Effects of orally administered EET analogues on the mesenteric resistance artery function in AngII hypertension. EET-A and EET-X were administered at a dose of 10 mg/kg per day in drinking water for 14 days. Mesenteric resistance artery diameter was not different between experimental groups, averaging 296 ± 19 μm (n = 30) under control conditions and 132 ± 11 μm after administration of the thromboxane mimetic U46619. Mesenteric resistance artery responses to acetylcholine were determined. *P < 0.05 compared with SD+vehicle and #P < 0.05 compared with AngII+vehicle; n = 8–10. All data are presented as means ± S.E.M. (b) Effects of orally administered EET-A (10 mg/kg per day in drinking water for 14 days) on the mesenteric resistance artery function in the presence and absence of nitric oxide synthase inhibition by L-NAME in AngII hypertension. Mesenteric resistance artery responses to acetylcholine were determined. Data are presented as the maximum percentage response to the highest concentration (10 μM) of acetylcholine. *P < 0.05 compared with SD+vehicle; #P < 0.05 compared with AngII+vehicle; †P < 0.05 compared with absence of L-NAME; n = 8–10. All data are presented as means ± S.E.M.
EET-A reduced kidney ENaC protein expression in AngII hypertension and inhibited the ENaC current in vitro
In immunohistochemical studies to determine the relative protein abundance of ENaC subunits (α, β and γ) in the kidney of AngII-hypertensive rats, renal expression of all ENaC subunits increased in AngII hypertension and EET-A reduced the expression of the ENaC subunits (Figure 5). In line with these findings, in electrophysiological study with immortalized mpkCCDc14 cells, we demonstrate further that EET-A markedly reduced equivalent I sc in mpkCCDc14 cells (Figure 6a). EET-A also reduced ENaC channel activity (NP0). NP0 averaged 0.6 ± 0.1 at baseline and decreased to 0.2 ± 0.1 in response to EET-A (Figures 6b and 6c).
Figure 5. Immunohistochemical staining of α-, β- and γ-subunits of ENaC in the kidney of rats from different experimental groups.

(a–c) Semi-quantitative assessment of α-, β- and γ-ENaC immunoreactivity in the kidney of different experimental groups. Immunoreactivity of ENaC subunits was assessed by analysing ten images (×200) for each sample using Nikon NIS Elements Software, and the immunoreactivity is presented as the percentage area fraction of immunopositive area relative to the total area of an image. *P < 0.05 compared with SD+vehicle and #P < 0.05 compared with AngII+vehicle; n = 6. All data are presented as means ± S.E.M. (d) Representative photomicrographs of kidney immunohistochemical staining of ENaC subunits in different experimental groups and immunopositive areas are indicated by arrows.
Figure 6. Effects of EET-A on sodium transport in mpkCCDc14 cells.

(a) Effect of EET-A on ENaC-mediated basal sodium transport in mp-kCCDc14 cells. The graph demonstrates equivalent Isc in mpkCCDc14 principal cells in response to EET-A. *P < 0.05 compared with Vehicle; n = 6. All data are presented as means ± S.E.M. (b) The graph demonstrates the results of experiments determining the acute effect of EET-A on ENaC NPo. *P < 0.05 compared with control; n = 6. All data are presented as means ± S.E.M. (c) Continuous current trace from a representative cell-attached patch that was made on the apical membrane of an mpkCCDc14 cell before and after treatment with 10 μM of EET-A. Patches were held at a 60-mV potential during the course of these experiments. The dashed lines demonstrate closed and open states of the current level.
EET-A increased natriuresis and attenuated the development of salt-sensitive hypertension in Cyp2c44−/− mice
Next, we determined natriuretic and antihypertensive effect of EET-A in Cyp2c44−/− mice that develop salt-sensitive hypertension. EET-A treatment exerted a natriuretic effect with a 23 % increase in sodium excretion in EET-A-treated (1.6 ± 0.1 mmol/day) compared with vehicle-treated (1.3 ± 0.1 mmol/day) Cyp2c44−/− mice fed on a high-salt diet. EET-A also markedly blunted the development of salt-sensitive hypertension in these mice without affecting the heart rate (Figure 7).
Figure 7. Effects of EET-A on blood pressure in Cyp2c44−/− mice fed on a high-salt diet.
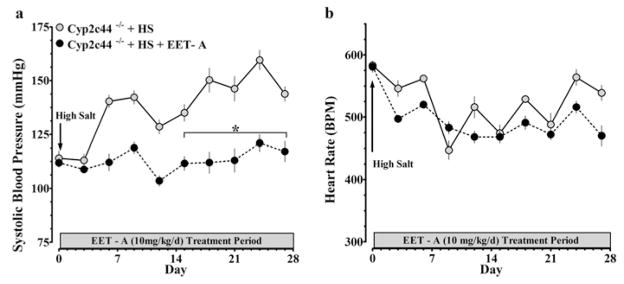
EET-A was administered in drinking water at a dose of 10 mg/kg per day for 28 days. Blood pressure and heart rate were recorded continuously throughout the 28-day protocol. EET-A lowered blood pressure (a) without altering heart rate (b) in Cyp2c44−/− mice fed on a high-salt diet. *P < 0.05 compared with EET-A; n = 4. All data are presented as means ± S.E.M.
DISCUSSION
It is well known that EETs have a number of cardiovascular actions that implicate them as important contributors to vascular function and blood pressure control [1–5]. One approach to target EET for combating diseases is the development of EET analogues that are designed to resist metabolism and also to have improved bioavailability [14–16]. Several of these EET analogues have been demonstrated cardiovascular actions in different experimental to have disease models [21–23]. Although they demonstrate great potential in experimental disease models, many of these previously developed EET analogues could not be administered orally. We have recently developed a series of EET analogues and have demonstrated that these can be administered orally, and protect the kidney from cisplatin-induced nephrotoxicity and in Dahl-salt sensitive hypertension [29,30]. Consequently, in the present study we have investigated the vascular reactivity and antihypertensive effects of a similar EET analogue series and investigated the mechanism by which orally administered EET-A lowered blood pressure.
A number of EET analogues have been used for many years to examine the cardiovascular actions attributed to EETs [21–23,30]. In particular, these EET analogues have been used to identify structure–activity relationships and cell signalling mechanisms that are responsible for vasodilation [14–18]. These studies have determined that an acidic carboxy group in carbon 1, a Δ8 double bond, 20-carbon chain length and a cis-epoxide are required for full vasodilator activity of EETs [14,18]. With this knowledge, attempts have been made in modifying the acidic carboxy group in carbon 1 to develop analogues with resistance to β-oxidation. Using this approach, we have designed a number of novel EET analogues in which the carboxy group in carbon 1 position of the EET pharmacophore is replaced with an aspartic acid (EET-A) or heterocyclic surrogates (EET-X, EET-Y and EET-Z). These EET analogues possess vasodilatory activity and were tested for their blood-pressure-lowering effect in experimental hypertension. We demonstrated that two of these four EET analogues, EET-A and EET-X, attenuated blood pressure elevation in SHRs and AngII hypertension when administered i.p. for 14 days. Blood-pressure-lowering effects of these two EET analogues were investigated further in AngII hypertension by administering them orally in drinking water for 14 days. We found that EET-A delivered in drinking water was available in the plasma (see Supplementary Online Data at http://www.clinsci.org/cs/127/cs1270463add.htm) and lowered blood pressure in AngII hypertension.
The pathophysiology of AngII hypertension involves elevated vascular resistance and impaired tubular sodium reabsorption [31]. Regarding these pathophysiological events, a number of studies have demonstrated increased vascular resistance and impaired endothelial function in AngII-dependent hypertension [32–34]. Indeed, studies have shown that, in hypertension, endothelial dysfunction is present in both resistance and conduit arteries and constitutes an early independent predictor of cardiovascular events [35,36]. Moreover, normalization of blood pressure, using conventional therapies, is associated with a larger reduction in the occurrence of cardiovascular events when endothelial function is restored [36]. Among other factors involved, reduced EET levels could contribute to the pathophysiology of endothelial dysfunction during arterial hypertension [37]. In line with these findings, in the present study we have demonstrated an impaired vascular response to acetylcholine in AngII hypertension that was markedly improved by EET-A treatment. Such EET-mediated endothelial vascular protection is also observed in an experimental model of hypertension. For instance, increased EET levels by sEH inhibition prevented coronary endothelial dysfunction in mice with two-kidney-one-clip (2K1C) renovascular hypertension [38]. The improved acetylcholine response in EET-A-treated AngII-hypertensive rats could be a result of blood pressure reduction by EET-A as impaired endothelial function has been shown to be associated with high blood pressure [33–36]. However, it is also possible that the improved vascular response by EET-A in the present study was a direct effect of EET-A. Indeed, we have demonstrated previously that an 11,12-EET analogue (11,12-EET-SI) reversed the enhanced renal vascular reactivity to AngII in AngII-hypertensive rats [39]. In the present study, our findings also indicate that improved endothelium-dependent vasodilation to acetylcholine in EET-treated AngII-hypertensive rats is partly dependent on the nitric oxide system along with other vasodilator components. Indeed, a previous report has demonstrated interactions between nitric oxide and EETs to vasodilation in mice mesenteric arteries [40]. Taken together, these findings support the notion that EET-A has antihypertensive actions and improves vascular function in AngII hypertension.
In addition to the vascular effects of AngII, among the other factors intricately involved in the pathophysiology of AngII hypertension are their antinatriuretic effects [41,42]. Previously, it was demonstrated that elevated renal microvascular reactivity occurs in AngII hypertension, and inappropriate elevation in renal microvascular reactivity contributes to the impaired natriuresis in AngII hypertension [32,43]. The impaired natriuresis in AngII hypertension has been associated with sodium retention caused by activated ENaC, and direct ENaC inhibition with amiloride attenuates AngII hypertension [44,45]. In relation to the role of sodium retention in the pathophysiology of AngII hypertension and attenuation of blood pressure in AngII hypertension by the EET analogue EET-A, it should be noted that EETs are known to possess strong natriuretic properties [5]. Indeed, it has been demonstrated that EETs lower blood pressure via inhibition of sodium reabsorption in the proximal tubule and cortical collecting duct (CCD) [6,46], and in the CCD sodium transport occurs via ENaC [47]. Therefore, we investigated the effects of the EET-A on ENaC activity and its expression in the kidney to determine whether this contributed to lower blood pressure in AngII hypertension.
A number of studies have reported that EETs inhibit ENaC channel activity, resulting in increased renal sodium excretion. For instance, electrophysiological patch–clamp studies have demonstrated that 11,12-EET but not 14,15-EET markedly reduced ENaC channel activity in the isolated rat CCD [48]. Using a similar electrophysiological approach, a relatively recent study, however, has comprehensively demonstrated that both 11,12-EET and 14,15-EET inhibit ENaC channel in immortalized mpkC-CDc14 cells [28]. In M1 cells derived from mouse collecting duct cells, it has also been shown that both EET and amiloride reduced the inward sodium current by acting on ENaC. The present study has further shown that, in the presence of 14,15-EET, amiloride becomes insensitive in regulating apical to basolateral sodium transport and suggested that EET is a functional analogue of amiloride [49]. In line with these findings, in the present study we used an electrophysiological approach and clearly demonstrated that EET-A reduced ENaC channel activity in mpkCCDc14 cells, and this led us to suggest a role of this novel analogue in renal tubular sodium transport. We also demonstrated that kidney protein abundance of all the three subunits of ENaC increased in AngII hypertension and EET-A prevented the increase in renal expression of ENaC subunits, typically expected in AngII hypertension. This is an important finding as a previous study also demonstrated that systemic AngII infusion increased kidney abundance of α-ENaC protein in rats [44], and α-ENaC is the rate-limiting subunit that regulates the assembly of the complete functional heterotrimeric channel [50]. These previous findings and our data in the present study led us to conclude that EET-A plays an important role in ENaC-mediated sodium transport in the kidney, and thereby blood pressure regulation in AngII hypertension.
To further support the notion that EET-A inhibits sodium absorption, we carried out experiments in Cyp2c44−/− mice. Cyp2c44 is the predominant EET-producing epoxygenase in the mouse kidney, up-regulated by high-salt intake and expressed in the CCD [6,46,51]. Because of the lack of a functional Cyp2c44 gene in Cyp2c44−/− mice, there is increased sodium absorption in the distal nephron, resulting in a diminished ability to excrete sodium that ultimately leads to hypertension development [6,46]. We demonstrate that, in Cyp2c44−/− mice with salt-sensitive hypertension, EET-A increased natriuresis and markedly lowered blood pressure. These findings suggest that EET-A possesses natriuretic activity that contributes to blood-pressure-lowering in Cyp2c44−/− mice fed on a high-salt diet. A recent study has shown that the salt-sensitive hypertension in Cyp2c44−/− mice is caused by an increase in ENaC gating and a decrease in the inhibitory phosphorylation of -ENaC subunit [6]. That same study also showed that amiloride lowered blood pressure in salt-sensitive Cyp2c44−/− mice, and exogenously added 11,12-EET-inhibited ENaC in the collecting duct cells of these mice [6]. These findings, along with our present data on EET-A-mediated inhibition of ENaC activity, lead us to conclude that the blood-pressure-lowering effect of EET-A in Cyp2c44−/− mice is associated with its ENaC inhibitory activity. Taken together, these studies demonstrate that the EET analogue EET-A has the ability to inhibit ENaC as revealed in electrophysiological studies with mpkCCDc14 cells and immunohistochemical studies in AngII hypertension. We further demonstrate that EET-A prevents salt-sensitive hypertension in Cyp2c44−/− mice that is known to be caused by hyperactive ENaC.
In summary, EET analogues were initially evaluated for their vasodilatory and blood-pressure-lowering action in SHRs and AngII-hypertensive rats. Further studies administering EET-A orally demonstrate that the vasodilatory and sodium channel inhibitory activities contribute to the antihypertensive action of EET-A.
Supplementary Material
CLINICAL PERSPECTIVES.
Hypertension affects approximately a quarter of all adults worldwide. This cardiovascular disease is a leading risk factor for heart disease or stroke and has significant health care costs.
A number of studies have demonstrated the therapeutic potential for EET analogues in cardiovascular diseases including hypertension. In line with these efforts, we characterized a novel orally active antihypertensive EET analogue, EET-A, that protects vascular endothelial function and has ENaC inhibitory activity in AngII hypertension.
EET-A can be developed further as a drug for the treatment of hypertension
Acknowledgments
Jessica Myers, Katherine A. Walsh and Rachel J. McClure provided excellent technical assistance.
FUNDING
These studies were supported by the National Institutes of Health [grant number DK38226] and Advancing a Healthier Wisconsin grant to J.D.I.; the National Institutes of Health [grant number HL-83297] to W.B.C.; Robert A. Welch Foundation [grant number GL625910], the National Institutes of Health [grants numbers GM31278 and DK38226] to J.R.F.; the National Institutes of Health [grant number HL108880] to A.S.; and a postdoctoral fellowship from the Midwest Affiliate of the American Heart Association to Md. A.H.K.
Abbreviations
- AngII
angiotensin II
- BPM
beats per min
- CCD
cortical collecting duct
- Cyp
cytochrome P450
- EET
epoxyeicosatrienoic acid
- ENaC
epithelial sodium channel
- i.p
intraperitoneally
- Isc
short-circuit current
- L-NAME
L-NG-nitroarginine methyl ester
- MAP
mean arterial pressure
- s.c
subcutaneously
- SD
Sprague–Dawley
- sEH
soluble epoxide hydrolase
- sEHi
soluble epoxide hydrolase inhibitor
- SHR
spontaneously hypertensive rat
Footnotes
AUTHOR CONTRIBUTION
Md. Abdul Hye Khan primarily conceived and designed the study, performed experiments, analysed the data and wrote the manuscript. John Imig conceived and designed the study, examined and interpreted the data and also wrote the manuscript. Jan Neckář designed, performed and interpreted immunohistochemical data. John Falck designed and synthesized EET analogues. William Campbell and Kathryn Gauthier designed and Sarah Christian performed the vascular studies with bovine coronary artery. Alexander Staruschenko designed and Tengis Pavlov performed the electrophysiological experiments. Jorge Capdevila generated the Cyp2c44−/− mice. All authors have thoroughly read the manuscript, agreed on the experimental findings, data interpretation and presentation and approved for publication.
References
- 1.Fleming I. Epoxyeicosatrienoic acids, cell signaling and angiogenesis. Prostaglandins Other Lipid Mediat. 2007;82:60–67. doi: 10.1016/j.prostaglandins.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 2.Fleming I. Vascular cytochrome p450 enzymes: physiology and pathophysiology. Trends Cardiovasc Med. 2008;18:20–25. doi: 10.1016/j.tcm.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 3.Imig JD. Epoxides and soluble epoxide hydrolase in cardiovascular physiology. Physiol Rev. 2012;92:101–130. doi: 10.1152/physrev.00021.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zeldin DC. Epoxygenase pathways of arachidonic acid metabolism. J Biol Chem. 2001;276:36059–36062. doi: 10.1074/jbc.R100030200. [DOI] [PubMed] [Google Scholar]
- 5.Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002;82:131–185. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- 6.Capdevila JH, Pidkovka N, Mei S, Gong Y, Falck JR, Imig JD, Harris RC, Wang W. The cyp2c44 epoxygenase regulates epithelial sodium channel activity and the blood pressure responses to increased dietary salt. J Biol Chem. 2014;289:4377–4386. doi: 10.1074/jbc.M113.508416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Imig JD. Targeting epoxides for organ damage in hypertension. J Cardiovasc Pharmacol. 2010;56:329–335. doi: 10.1097/FJC.0b013e3181e96e0c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Imig JD, Zhao X, Capdevila JH, Morisseau C, Hammock BD. Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension. 2002;39:690–694. doi: 10.1161/hy0202.103788. [DOI] [PubMed] [Google Scholar]
- 9.Imig JD, Zhao X, Zaharis CZ, Olearczyk JJ, Pollock DM, Newman JW, Kim IH, Watanabe T, Hammock BD. An orally active epoxide hydrolase inhibitor lowers blood pressure and provides renal protection in salt-sensitive hypertension. Hypertension. 2005;46:975–981. doi: 10.1161/01.HYP.0000176237.74820.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao X, Yamamoto T, Newman JW, Kim IH, Watanabe T, Hammock BD, Stewart J, Pollock JS, Pollock DM, Imig JD. Soluble epoxide hydrolase inhibition protects the kidney from hypertension-induced damage. J Am Soc Nephrol. 2004;15:1244–1253. [PubMed] [Google Scholar]
- 11.Jung O, Brandes RP, Kim IH, Schweda F, Schmidt R, Hammock BD, Busse R, Fleming I. Soluble epoxide hydrolase is a main effector of angiotensin II-induced hypertension. Hypertension. 2005;45:759–765. doi: 10.1161/01.HYP.0000153792.29478.1d. [DOI] [PubMed] [Google Scholar]
- 12.Loch D, Hoey A, Morisseau C, Hammock BD, Brown L. Prevention of hypertension in DOCA-salt rats by an inhibitor of soluble epoxide hydrolase. Cell Biochem Biophys. 2007;47:87–98. doi: 10.1385/cbb:47:1:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manhiani M, Quigley JE, Knight SF, Tasoobshirazi S, Moore T, Brands MW, Hammock BD, Imig JD. Soluble epoxide hydrolase gene deletion attenuates renal injury and inflammation with DOCA-salt hypertension. Am J Physiol Renal Physiol. 2009;297:F740–F748. [Google Scholar]
- 14.Falck JR, Krishna UM, Reddy YK, Kumar PS, Reddy KM, Hittner SB, Deeter C, Sharma KK, Gauthier KM, Campbell WB. Comparison of vasodilatory properties of 14,15-EET analogs: structural requirements for dilation. Am J Physiol Heart Circ Physiol. 2003;284:H337–H349. doi: 10.1152/ajpheart.00831.2001. [DOI] [PubMed] [Google Scholar]
- 15.Falck JR, Kodela R, Manne R, Atcha KR, Puli N, Dubasi N, Manthati VL, Capdevila JH, Yi XY, Goldman DH, et al. 14,15-epoxyeicosa-5,8,11-trienoic acid (14,15-EET) surrogates containing epoxide bioisosteres: influence upon vascular relaxation and soluble epoxide hydrolase inhibition. J Med Chem. 2009;52:5069–5075. doi: 10.1021/jm900634w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang W, Holmes BB, Gopal VR, Kishore RV, Sangras B, Yi XY, Falck JR, Campbell WB. Characterization of 14,15-epoxyeicosatrienoyl-sulfonamides as 14,15-epoxyeicosatrienoic acid agonists: use for studies of metabolism and ligand binding. J Pharmacol Exp Ther. 2007;321:1023–1031. doi: 10.1124/jpet.107.119651. [DOI] [PubMed] [Google Scholar]
- 17.Falck JR, Reddy LM, Reddy YK, Bondlela M, Krishna UM, Ji Y, Sun J, Liao JK. 11,12-epoxyeicosatrienoic acid (11,12-EET): structural determinants for inhibition of TNF-alpha-induced VCAM-1 expression. Bioorg Med Chem Lett. 2003b;13:4011–4014. doi: 10.1016/j.bmcl.2003.08.060. [DOI] [PubMed] [Google Scholar]
- 18.Gauthier KM, Falck JR, Reddy LM, Campbell WB. 14,15-EET analogs: characterization of structural requirements for agonist and antagonist activity in bovine coronary arteries. Pharmacol Res. 2004;49:515–524. doi: 10.1016/j.phrs.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 19.Imig JD, Inscho EW, Deichmann PC, Reddy KM, Falck JR. Afferent arteriolar vasodilation to the sulfonimide analog of 11,12-epoxyeicosatrienoic acid involves protein kinase A. Hypertension. 1999;33:408–413. doi: 10.1161/01.hyp.33.1.408. [DOI] [PubMed] [Google Scholar]
- 20.Dimitropoulou C, West L, Field MB, White RE, Reddy LM, Falck JR, Imig JD. Protein phosphatase 2A and Ca2+-activated K+ channels contribute to 11,12-epoxyeicosatrienoic acid analog mediated mesenteric arterial relaxation. Prostaglandins Other Lipid Mediat. 2007;83:50–61. doi: 10.1016/j.prostaglandins.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 21.Imig JD, Elmarakby A, Nithipatikom K, Wei S, Capdevila JH, Tuniki VR, Sangras B, Anjaiah S, Manthati VL, Sudarshan RD, Falck JR. Development of epoxyeicosatrienoic acid analogs with in vivo anti-hypertensive actions. Front Physiol. 2010;1:157. doi: 10.3389/fphys.2010.00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seubert JM, Zeldin DC, Nithipatikom K, Gross G. Role of epoxyeicosatrienoic acids protecting the myocardium following ischemia/reperfusion injury. Prostaglandins Other Lipid Mediat. 2007;82:50–59. doi: 10.1016/j.prostaglandins.2006.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sodhi K, Inoue K, Gotlinger KH, Canestraro M, Vanella L, Kim DH, Manthati VL, Koduru SR, Falck JR, Schwartzman ML, Abraham NG. Epoxyeicosatrienoic acid agonist rescues the metabolic syndrome phenotype of HO-2-null mice. J Pharmacol Exp Ther. 2009;331:906–916. doi: 10.1124/jpet.109.157545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mills PA, Huetteman DA, Brockway BP, Zwiers LM, Gelsema AJ, Schwartz RS, Kramer K. A new method for measurement of blood pressure, heart rate, and activity in the mouse by radiotelemetry. J Appl Physiol. 2000;88:1537–1544. doi: 10.1152/jappl.2000.88.5.1537. [DOI] [PubMed] [Google Scholar]
- 25.Bukhari IA, Gauthier KM, Jagadeesh SG, Sangras B, Falck JR, Campbell WB. 14,15-Dihydroxyeicosa-5(Z)-enoic acid selectively inhibits 14,15-epoxyeicosatrienoic acid-induced relaxations in bovine coronary arteries. J Pharmacol Exp Ther. 2011;336:47–55. doi: 10.1124/jpet.110.169797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pavlov TS, Levchenko V, Karpushev AV, Vandewalle A, Staruschenko A. Peroxisome proliferator-activated receptor gamma antagonists decrease Na+ transport via the epithelial Na+ channel. Mol Pharmacol. 2009;76:1333–1340. doi: 10.1124/mol.109.056911. [DOI] [PubMed] [Google Scholar]
- 27.Levchenko V, Zheleznova NN, Pavlov TS, Vandewalle A, Wilson PD, Staruschenko A. EGF and its related growth factors mediate sodium transport in mpkCCDc14 cells via ErbB2 (neu/HER-2) receptor. J Cell Physiol. 2010;223:252–259. doi: 10.1002/jcp.22033. [DOI] [PubMed] [Google Scholar]
- 28.Pavlov TS, Ilatovskaya DV, Levchenko V, Mattson DL, Roman RJ, Staruschenko A. Effects of cytochrome P-450 metabolites of arachidonic acid on the epithelial sodium channel (ENaC) Am J Physiol Renal Physiol. 2011;301:F672–F681. doi: 10.1152/ajprenal.00597.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khan MA, Liu J, Kumar G, Skapek SX, Falck JR, Imig JD. Novel orally active epoxyeicosatrienoic acid (EET) analogs attenuate cisplatin nephrotoxicity. FASEB J. 2013;27:2946–2956. doi: 10.1096/fj.12-218040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hye Khan MA, Neckár J, Manthati V, Errabelli R, Pavlov TS, Staruschenko A, Falck JR, Imig JD. Orally active epoxyeicosatrienoic acid analog attenuates kidney injury in hypertensive Dahl salt-sensitive rat. Hypertension. 2013;62:905–913. doi: 10.1161/HYPERTENSIONAHA.113.01949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fyhrquist FI, Metsärinne K, Tikkanen I. Role of angiotensin II in blood pressure regulation and in the pathophysiology of cardiovascular disorders. J Hum Hypertens. 1995;5:S19–S24. [PubMed] [Google Scholar]
- 32.Imig JD. Afferent arteriolar reactivity to angiotensin II is enhanced during the early phase of angiotensin II hypertension. Am J Hypertens. 2000;13:810–818. doi: 10.1016/s0895-7061(00)00264-8. [DOI] [PubMed] [Google Scholar]
- 33.Wang D, Luo Z, Wang X, Jose PA, Falck JR, Welch WJ, Aslam S, Teerlink T, Wilcox CS. Impaired endothelial function and microvascular asymmetrical dimethylarginine in angiotensin II-infused rats: effects of tempol. Hypertension. 2010;56:950–955. doi: 10.1161/HYPERTENSIONAHA.110.157115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schäfer SC, Pellegrin M, Wyss C, Aubert JF, Nussberger J, Hayoz D, Lehr HA, Mazzolai L. Intravital microscopy reveals endothelial dysfunction in resistance arterioles in angiotensin II-induced hypertension. Hypertens Res. 2012;35:855–861. doi: 10.1038/hr.2012.58. [DOI] [PubMed] [Google Scholar]
- 35.Modena MG, Bonetti L, Coppi F, Bursi F, Rossi R. Prognostic role of reversible endothelial dysfunction in hypertensive postmenopausal women. J Am Coll Cardiol. 2002;40:505–510. doi: 10.1016/s0735-1097(02)01976-9. [DOI] [PubMed] [Google Scholar]
- 36.Perticone F, Ceravolo R, Pujia A, Ventura G, Iacopino S, Scozzafava A, Ferraro A, Chello M, Mastroroberto P, Verdecchia P, Schillaci G. Prognostic significance of endothelial dysfunction in hypertensive patients. Circulation. 2001;104:191–196. doi: 10.1161/01.cir.104.2.191. [DOI] [PubMed] [Google Scholar]
- 37.Taddei S, Versari D, Cipriano A, Ghiadoni L, Galetta F, Franzoni F, Magagna A, Virdis A, Salvetti A. Identification of a cytochrome P450 2C9-derived endothelium-derived hyperpolarizing factor in essential hypertensive patients. J Am Coll Cardiol. 2006;48:508–515. doi: 10.1016/j.jacc.2006.04.074. [DOI] [PubMed] [Google Scholar]
- 38.Gao J, Bellien J, Gomez E, Henry JP, Dautreaux B, Bounoure F, Skiba M, Thuillez C, Richard V. Soluble epoxide hydrolase inhibition prevents coronary endothelial dysfunction in mice with renovascular hypertension. J Hypertens. 2011;29:1128–1135. doi: 10.1097/HJH.0b013e328345ef7b. [DOI] [PubMed] [Google Scholar]
- 39.Imig JD, Zhao X, Falck JR, Wei S, Capdevila JH. Enhanced renal microvascular reactivity to angiotensin II in hypertension is ameliorated by the sulfonamide analog of 11,12-epoxyeicosatrienoic acid. J Hypertens. 2001;19:983–992. doi: 10.1097/00004872-200105000-00020. [DOI] [PubMed] [Google Scholar]
- 40.Hercule HC, Schunck WH, Gross V, Seringer J, Leung FP, Weldon SM, da Costa Goncalves ACH, Huang Y, Luft FC, Gollasch M. Interaction between P450 eicosanoids and nitric oxide in the control of arterial tone in mice. Arterioscler Thromb Vasc Biol. 2009;29:54–60. doi: 10.1161/ATVBAHA.108.171298. [DOI] [PubMed] [Google Scholar]
- 41.Zhao D, Seth DM, Navar LG. Enhanced distal nephron sodium reabsorption in chronic angiotensin II-infused mice. Hypertension. 2009;54:120–126. doi: 10.1161/HYPERTENSIONAHA.109.133785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ortiz RM, Graciano ML, Seth D, Awayda MS, Navar LG. Aldosterone receptor antagonism exacerbates intrarenal angiotensin II augmentation in ANG II-dependent hypertension. Am J Physiol Renal Physiol. 2007;293:F139–F147. doi: 10.1152/ajprenal.00504.2006. [DOI] [PubMed] [Google Scholar]
- 43.Cowley AW, Jr, Roman RJ. The role of the kidney in hypertension. JAMA. 1996;275:1581–1589. [PubMed] [Google Scholar]
- 44.Beutler KT, Masilamani S, Turban S, Nielsen J, Brooks HL, Ageloff S, Fenton RA, Packer RK, Knepper MA. Long-term regulation of ENaC expression in kidney by angiotensin II. Hypertension. 2003;41:1143–1150. doi: 10.1161/01.HYP.0000066129.12106.E2. [DOI] [PubMed] [Google Scholar]
- 45.Gonzalez AA, Liu L, Lara LS, Seth DM, Navar LG, Prieto MC. Angiotensin II stimulates renin in inner medullary collecting duct cells via protein kinase C and independent of epithelial sodium channel and mineralocorticoid receptor activity. Hypertension. 2011;57:594–599. doi: 10.1161/HYPERTENSIONAHA.110.165902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakagawa K, Holla VR, Wei Y, Wang WH, Gatica A, Wei S, Mei S, Miller CM, Cha DR, Price E, Jr, et al. Salt-sensitive hypertension is associated with dysfunctional Cyp4a10 gene and kidney epithelial sodium channel. J Clin Invest. 2006;116:1696–1702. doi: 10.1172/JCI27546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hamm LL, Feng Z, Hering-Smith KS. Regulation of sodium transport by ENaC in the kidney. Curr Opin Nephrol Hypertens. 2010;19:98–105. doi: 10.1097/MNH.0b013e328332bda4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wei Y, Lin DH, Kemp R, Yaddanapudim GS, Nasjletti A, Falck JR, Wang WH. Arachidonic acid inhibits epithelial Na channel via cytochrome P450 (CYP) epoxygenase-dependent metabolic pathways. J Gen Physiol. 2004;124:719–727. doi: 10.1085/jgp.200409140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pidkovka N, Rao R, Mei S, Gong Y, Harris RC, Wang WH, Capdevila JH. Epoxyeicosatrienoic acids (EETs) regulate epithelial sodium channel activity by extracellular signal-regulated kinase 1/2 (ERK1/2)-mediated phosphorylation. J Biol Chem. 2013;288:5223–5231. doi: 10.1074/jbc.M112.407981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.May A, Puoti A, Gaeggeler HP, Horisberger JD, Rossier BC. Early effect of aldosterone on the rate of synthesis of the epithelial sodium channel alpha subunit in A6 renal cells. J Am Soc Nephrol. 1997;8:1813–1822. doi: 10.1681/ASN.V8121813. [DOI] [PubMed] [Google Scholar]
- 51.Capdevila J, Wang W. Role of cytochrome P450 epoxygenase in regulating renal membrane transport and hypertension. Curr Opin Nephrol Hypertens. 2013;22:163–169. doi: 10.1097/MNH.0b013e32835d911e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.