

Abstract
Hippocampal α4βδ GABAA receptors (GABAA-R) are increased following progesterone withdrawal (PWD) in a rodent model of premenstrual anxiety. This α4βδ receptor isoform uniquely responds to the GABA agonist gaboxadol (THIP) with a maximum current greater than that gated by GABA, and is potentiated more by pentobarbital than are other GABAA-R. We therefore investigated the anxiolytic effects of these drugs using the elevated plus maze. Gaboxadol (1.25 mg/kg) was markedly more anxiolytic in animals undergoing PWD than in controls. Pentobarbital (10 mg/kg) also produced a greater anxiolytic effect during PWD. These results suggest that the pharmacological properties of α4βδ GABAA-R following PWD are evident behaviorally. Alterations in the α4βδ GABAA-R population may have implications for the etiology and treatment of premenstrual syndrome.
Keywords: α4 subunit, Allopregnanolone, Anxiety, Extra-synaptic, δ subunit, GABAA receptor, Neurosteroid, Premenstrual syndrome, Progesterone, Tonic current
INTRODUCTION
The regulation of atypical GABAA receptor (GABAA-R) subunit expression has many implications for the etiology and treatment of anxiety disorders [1,2]. GABAA-R are composed of five subunits, and the pharmacological profile of the GABAA-R is strikingly dependent on the subunit composition, such that the efficacy and potency of several GABAA-R modulators is a function of the type of α, β, γ or δ subunits comprising the receptor.
Progesterone derivatives, such as the neurosteroid 3α,5α-ALLO (allopregnanolone or 3α-OH-5α-pregnan-20-one or 3α-5α-THP), are also potent positive modulators of the GABAA-R when acutely applied [3]. However, chronic exposure to and withdrawal from progesterone (PWD) in a rodent model of premenstrual syndrome increases anxiety and reduces total GABA-gated current in association with up-regulation of atypical GABAA-R subunits [4–8].
We have previously demonstrated increased expression of the α4-containing GABAA-R following PWD [4–6]. Furthermore, we have also shown profound changes in GABAA-R pharmacology following PWD that are consistent with up-regulation of α4βγ2 isoforms [9,10]. These include insensitivity to the GABA-modulatory effects of benzodiazepines and agonist-like effects of the benzodiazepine antagonist, flumazenil, in acutely isolated hippocampal neurons and in the elevated plus maze [4–6].
We have also recently demonstrated increased expression of the α4βδ GABAA-R isoform following PWD [7]. Although α4βδ receptors are also insensitive to benzodiazepines, this isoform has an increased response to pentobarbital [12,13] and uniquely responds to the GABA agonist, gaboxadol (THIP, 4,5,6,7- tetrahydroisoxazolo[5,4-c]pyridin-3-ol) with a maximum current greater than that gated by GABA itself [7,12,13]. This is a feature that distinguishes the α4βδ isoform from the α4βγ2 isoform, where gaboxadol generates maximum currents less than those gated by saturating concentrations of GABA [10,12,13]. Gaboxadol is also a partial agonist at most other GABAA-R isoforms [10,14]. These studies, however have been conducted in in vitro systems, where relevant behavioral outcomes cannot be assessed.
We therefore predicted that the anxiolytic effects of gaboxadol and pentobarbital might also be increased during PWD. To this end, we tested these the effects of these compounds in the elevated plus maze, an established animal model of anxiety, to determine whether increased expression of α4βδ receptors following PWD would be accompanied by an increased behavioral sensitivity to gaboxadol and pentobarbital.
MATERIALS AND METHODS
Animals
Female Long–Evans rats weighing 250 ± 25 g (~70 days old, Charles River) were housed in pairs under a 14:10 h light:dark cycle with food and water ad lib. All animals were tested during the light portion of the circadian cycle between 09:00 and 14:00 h. Estrous cycle stage was determined by microscopic examination of the vaginal lavage and by measures of vaginal impedance [6]. All animal care was conducted in accordance with guidelines provided by the Institutional Animal Care and Use Committee.
Drugs and hormone administration
Progesterone was administered rather than 3α5α ALLO because elevated circulating levels of of progesterone are converted to 3α5α ALLO in the brain and result in levels sufficient to potentiate GABAergic inhibition and modulate GABAR subunit expression [4,6]. These doses result in CNS levels of 3α5α ALLO in the high physiological range (6–12 ng/g) [15].
Progesterone implants were made from silicone tubing as described previously (Silicone tubing and adhesive were obtained from Nalgene Co. and Dow Corning, respectively) and implanted s.c. under halothane anesthesia [4,6]. Control animals were implanted in a similar manner with empty (sham) silicone capsules. Following 21 days of implantation, animals were tested 24 h after removal of the implant (progesterone withdrawal or PWD).
On the day of testing, animals were injected with gaboxadol (THIP, 1.25 mg/kg), pentobarbital (10 mg/kg) or vehicle (1.8% polyethylene glycol 400 in propylene glycol) and were tested 30–45 min after injection. Chemicals were obtained from Sigma, Inc., unless otherwise indicated. Pentobarbital was obtained from Abbot Labs (Chicago, IL).
Electrophysiology
Pyramidal neurons were acutely isolated from CA1 hippocampus of sham implanted and progesterone withdrawn animals using a procedure described previously [4]. GABA-gated current was recorded at room temperature (20–25°C) in a 120 mM NaCl buffer and a pipette solution containing 120 mM N-methyl-D-glucamine. Maximum currents gated by GABA (10 mM) or gaboxadol (10 mM) were recorded with whole cell patch clamp techniques at a holding potential of −50 mV. Modulation of GABA (GABA EC20 = 10 μM)-gated current by 100 μM pentobarbital was also determined. Current was filtered at 1–2 kHz and digitally sampled at 500 Hz. Drug delivery was accomplished via a solenoid-activated gravity-feed super-fusion system positioned within 50 μm of the cell releasing drugs for 20 ms at 1–3 min intervals (exposure times in the 40–100 ms range) [5]. A background perfusion system (4 ml/ min) provided a wash-out flow in the opposite direction. Electrophysiological data were analyzed using an ANOVA followed by unpaired Student’s t-test.
Behavioural testing
The elevated plus maze consists of two enclosed arms (50 × 10 × 40 cm) and two open arms (50 × 10 cm) elevated 50 cm above the floor as described previously [6]. The floor of all four arms was marked with grid lines every 25 cm. On the day of testing each rat was placed in the testing room for 30–45 min prior to testing in order to acclimatize the animal. At the time of testing, each animal was tested for 10 min after exiting a start box in the center platform of the plus maze. To be considered as an entry into any arm, the rat must pass the line of the open platform with all four paws. The duration (s) of time spent in the open arm was recorded from the time of entry into the open arm. In order to measure general locomotor activity, the number of total grid crosses was counted. Data from the plus maze were analyzed in an ANOVA followed by a post-hoc t-test (Fisher’s PLSD).
RESULTS
Progesterone withdrawal increases the agonist effect of gaboxadol and the GABA-modulatory effects of pentobarbital
The maximum current gated by gaboxadol (10 mM) following PWD was increased by almost 50% relative to control values, assessed using whole cell patch clamp procedures in acutely isolated hippocampal pyramidal cells (Fig. 1a, p < 0.05). Furthermore, the maximum gaboxadol-gated current was 40% greater than the maximum GABA-gated current only during PWD (p < 0.05). As reported previously, the relative maximum currents generated by saturating concentrations of GABA:gaboxadol was 0.7, when assessed under control conditions [7]. In addition, pentobarbital (PTB, 100 μM) increased GABA-gated current following PWD by 250%, a > 2.5-fold increase over the 100% potentiation of GABA-gated current observed for this concentration of pentobarbital in control neurons (Fig. 1b, p < 0.01).
Fig. 1.
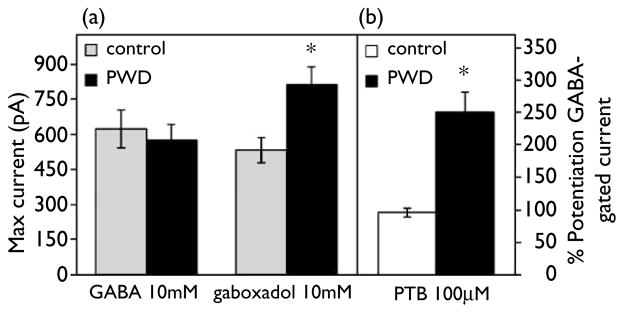
Progesterone withdrawal effects on peak GABA-gated current: increased agonist effects of gaboxadol and potentiation by pentobarbital. (a) Maximum currents gated by gaboxadol and GABA. In acutely isolated CA1pyramidal cells, the maximum GABAA-R current gated by gaboxadol (10 mM) was significantly increased (*p < 0.05, n control =15, n PWD =15) following progesterone withdrawal (PWD, dark bars) relative to control (shaded bars). In contrast, the maximum current gated by GABA (10 mM, n control =15, n PWD =15) was unaffected by PWD. (B) Potentiation of peak GABA-gated current by the GABAA-R modulator pentobarbital (PTB). PTB (100 μM, GABA EC20=10 μM) modulation of GABA-gated current was significantly (*p < 0.01) greater in hippocampal cells isolated from PWD rats (dark bar, n =20) than controls (open bar, n =20).
Progesterone withdrawal increases the anxiolytic effects of gaboxadol and pentobarbital
PWD increased in anxiety-like behavior, as reflected by a decrease in time spent in the open arm of the elevated plus maze, compared to sham implanted controls (p < 0.01). When tested during PWD, a non-sedative dose of gaboxadol (1.25 mg/kg) resulted in a significant anxiolytic effect (Fig. 2), evident in the 2- to 3-fold increase in time spent in the open arm, the number of entries into the open arm and the prcentage open arm entries (Fig. 2). In contrast, this low concentration of gaboxadol did not significantly alter any parameters in control rats.
Fig. 2.

Progesterone withdrawal increases the anxiolytic effects of gaboxadol and pentobarbital in the elevated plus maze. Anxiety Measures in the EPM – The anxiolytic effect of gaboxadol (1.25 mg/kg) was increased during PWD (dark bars) relative to controls (hatched bars). For this and the following graphs, sample size is indicated at the base of the bars in panel D and significant effects relative to controls injected with the same substance at p < 0.005 is indicated by *. (a) Both gaboxadol and PTB significantly increased time spent in the open arm following PWD relative to control animals with similar injections. (b) Gaboxadol significantly increased the number of open arm entries only during PWD. (c) This figure depicts the number of open arm entries (OAE) corrected for locomotor activity by presenting these data as a percentage of grid crosses (% OAE). Gaboxadol injected animals have a higher % OAE only during PWD. (d) Locomotor activity is illustrated as total number of grid crosses in a 10 min test period. There were no significant effects of locomotor activity across treatment conditions.
Pentobarbital (10 mg/kg) also produced a greater anxiolytic effect in withdrawn animals than in controls (Fig. 2), increasing the time spent in the open arm roughly 2-fold above that observed for control animals (Fig. 2, p < 0.01). In contrast, there were no significant alterations in general locomotor activity (grid crosses) in withdrawn animals relative to controls across any of the treatment conditions.
DISCUSSION
These data demonstrate that PWD produces an increased sensitivity to the anxiolytic effects of the GABA partial agonist, gaboxadol, an effect consistent with the increased expression of the α4βδ isoform of the GABAA-R we have reported during PWD [7]. Furthermore, these changes were paralleled by increases in the maximum current generated by gaboxadol relative to GABA in hippocampal neurons isolated from PWD rats relative to controls. These results suggest that the pharmacological profile characteristic of elevated α4βδ expression [7] is manifested at the behavioral as well as the cellular level.
The primary effects of progesterone withdrawal include increased anxiety, possibly as a result of the decreased GABAergic inhibition and hippocampal hyperexcitability which we have demonstrated previously [4–6,8]. The majority of the effects of PWD are mediated via the GABA-modulatory neurosteroid, 3α,5α-ALLO and are prevented by suppression of α4 subunit expression using antisense administration [4,5]. In addition to increasing expression of the atypical α4 subunit, PWD also increases hippocampal expression of the GABAA-R δ subunit. α4 subunits preferentially co-express with δ subunits (in addition to γ2 subunits) [16], and we have previously demonstrated the co-expression of δ and α4 subunits in hippocampus during PWD by co-immunoprecipitation [7].
α4βδ GABAA-R exhibit distinctive biophysical and pharmacological properties [7,13,17] compared to the major native GABAA-R isoforms. For example, saturating concentrations of gaboxadol generate a maximum current (gaboxadolmax) greater than saturating concentrations of GABA (GABAmax) only at the α4βδ isoform [12,13]. This increase in the gaboxadolmax:GABAmax response ratio (1.25–1.4) is in striking contrast to the majority of native GABAA-R isoforms (α1–3β1–3γ2) for which the gaboxadolmax: GABAmax ratio is significantly less than 1 (~0.7) [10,12–14]. Although our earlier studies also suggest increased expression of α4βγ2 isoforms following PWD [4–6], the α4βγ2 isoform also exhibits a gaboxadolmax:GABAmax ratio < 1 [10,12,13], indicating that this is not the only isoform which is increased following PWD. In fact, in addition to exhibiting increased efficacy, gaboxadol is also more potent at α4β3δ receptors, as it is following PWD [7,12,13]. Therefore, the increase in gaboxadol sensitivity observed in the present study is consistent with increases in the α4βδ isoform. It is also noteworthy that this low dose of gaboxadol was without effect in control animals (although higher doses have been demonstrated to be anxiolytic by other groups [18]), suggesting that the concentration of gaboxadol is an important factor to distinguish between different GABAA-R populations.
In addition to demonstrating a distinctive anxiolytic role of gaboxadol following PWD, the present results also demonstrate an increase in the anxiolytic effects of a barbiturate during PWD. This is in striking contrast to the benzodiazepine insensitivity we have previously demonstrated in rats undergoing PWD [6] and that has also been demonstrated in patients with premenstrual dysphoric disorder [1]. The increased anxiolytic effects of pentobarbital during PWD are also consistent with increased expression of the α4βδ GABAA-R, since pentobarbital is more potent at this isoform than at α4βγ2, α1-3βγ2 or α1βδ isoforms [10,12–14].
It is likely that α4βδ receptors substitute for, rather than add to the ambient GABAA-R population because peak GABA-gated current is unchanged during PWD. The α4βδ receptor isoform is thought to be primarily peri- or extra-synaptic [19]. Increases in the gating of these receptors by gaboxadol would increase tonic inhibitory current [20], an effect which would decrease neuronal excitability [21] and might be expected to decrease behavioral manifestations of hyperexcitability, such as the anxiety state reported here. In fact, hippocampal tonic current has been shown to play a role in behavior [22], and extra-synaptic GABAA-R may be a predominant target for anxiolytic and anesthetic drug actions [20,23].
CONCLUSIONS
The results from the present study demonstrate enhanced anxiolytic potency of gaboxadol and pentobarbital following PWD in a rodent model of premenstrual anxiety. These effects are consistent with the pharmacological profile for α4βδ GABAA-R, which are also increased in hippocampus during PWD. In light of these data it is interesting that altered sensitivity to the anxiolytic and sedative effects of GABA-modulatory agents are correlated with negative mood symptoms and anxiety [1,11]. Taken together, these data suggest that both the manifestation of anxiety-like symptoms and the pharmacological response to GABAA-R subunit-selective drugs are regulated by similar underlying mechanisms, i.e. selectively regulated expression of specific GABAA-R isoforms. These results may have implications for alterations in anxiety state that have been reported across naturally occurring fluctuations in endogenous steroids, such as premenstrual syndrome.
Acknowledgments
This work was supported by a NIH grants DA09618 and AA12958 and a contract from Lundbeck to SSS. We would like to thank Bjarke Ebert for helpful discussions.
References
- 1.Sundström I, Nyberg S, Bäckström T. Neuropsychopharmacology. 1997;17:370–381. doi: 10.1016/S0893-133X(97)00086-9. [DOI] [PubMed] [Google Scholar]
- 2.Low K, Crestani F, Keist R, et al. Science. 2000;290:131–134. doi: 10.1126/science.290.5489.131. [DOI] [PubMed] [Google Scholar]
- 3.Majewska MD, Harrison NL, Schwartz RD, et al. Science. 1986;232:1004–1007. doi: 10.1126/science.2422758. [DOI] [PubMed] [Google Scholar]
- 4.Smith S, Gong QH, Hsu FC, et al. Nature. 1998;392:926–930. doi: 10.1038/31948. [DOI] [PubMed] [Google Scholar]
- 5.Smith S, Gong QH, Li X, et al. J Neurosci. 1998;18:5275–5284. doi: 10.1523/JNEUROSCI.18-14-05275.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gulinello M, Gong Q, Smith S. Neuropharmacol. 2002;43:702–715. doi: 10.1016/s0028-3908(02)00171-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sundström-Poromaa I, Smith DH, Gong QH, et al. Nature Neurosci. 2002;5:721–722. doi: 10.1038/nn888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsu F-C, Smith SS. J Neurophysiol. 2002 (in press) [Google Scholar]
- 9.Wisden W, Herb A, Wieland H, et al. FEBS Lett. 1991;289:227–230. doi: 10.1016/0014-5793(91)81076-k. [DOI] [PubMed] [Google Scholar]
- 10.Wafford K, Thompson SA, Thomas D, et al. Mol Pharmacol. 1996;50:670–678. [PubMed] [Google Scholar]
- 11.Sundström I, Backstrom T. Psychoneuroendocrinology. 1998;23:73–88. doi: 10.1016/s0306-4530(97)00064-4. [DOI] [PubMed] [Google Scholar]
- 12.Adkins CE, Pillai GV, Kerby J, et al. J Biol Chem. 2001;276:38934–38939. doi: 10.1074/jbc.M104318200. [DOI] [PubMed] [Google Scholar]
- 13.Brown N, Kerby J, Bonnert TP, et al. Br J Pharmacol. 2002;136:965–974. doi: 10.1038/sj.bjp.0704795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ebert B, Thompson SA, Saounatsou K, et al. Mol Pharmacol. 1997;52:1150–1156. [PubMed] [Google Scholar]
- 15.Moran M, Smith S. Brain Res. 1998;807:84–90. doi: 10.1016/s0006-8993(98)00782-3. [DOI] [PubMed] [Google Scholar]
- 16.Peng Z, Hauer B, Michalek RM, et al. J Comp Neurol. 2002;446:179–197. doi: 10.1002/cne.10210. [DOI] [PubMed] [Google Scholar]
- 17.Saxena NC, Macdonald RL. Mol Pharmacol. 1996;49:567–579. [PubMed] [Google Scholar]
- 18.Corbett R, Fielding S, Cornfeldt M, et al. Psychopharmacology. 1991;104:312–316. doi: 10.1007/BF02246029. [DOI] [PubMed] [Google Scholar]
- 19.Nusser Z, Sieghart W, Somogyi P. J Neurosci. 1998;18:1693–1703. doi: 10.1523/JNEUROSCI.18-05-01693.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nusser Z, Mody I. J Neurophysiol. 2002;87:2624–2628. doi: 10.1152/jn.2002.87.5.2624. [DOI] [PubMed] [Google Scholar]
- 21.Brickley SG, Revilla V, Cull-Candy SG, et al. Nature. 2001;409:88–92. doi: 10.1038/35051086. [DOI] [PubMed] [Google Scholar]
- 22.Crestani F, Martin JR, Mohler H, et al. Proc Natl Acad Sci USA. 2002;99:8980–8985. doi: 10.1073/pnas.142288699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bai D, Zhu G, Pennefather P, et al. Mol Pharmacol. 2001;59:814–824. doi: 10.1124/mol.59.4.814. [DOI] [PubMed] [Google Scholar]