

Abstract
Summary
Survivin is expressed in tumor cells, including acute myeloid leukemia (AML), regulates mitosis, and prevents tumor cell death. The antisense oligonucleotide sodium LY2181308 (LY2181308) inhibits survivin expression and may cause cell cycle arrest and restore apoptosis in AML.
Methods
In this study, the safety, pharmacokinetics, and pharmacodynamics/efficacy of LY2181308 was examined in AML patients, first in a cohort with monotherapy (n=8) and then post-amendment in a cohort with the combination of cytarabine and idarubicin treatment (n=16). LY2181308 was administered with a loading dosage of 3 consecutive daily infusions of 750 mg followed by weekly intravenous (IV) maintenance doses of 750 mg. Cytarabine 1.5 g/m2 was administered as a 4-hour IV infusion on Days 3, 4, and 5 of Cycle 1, and idarubicin 12 mg/m2 was administered as a 30-minute IV infusion on Days 3, 4, and 5 of Cycle 1. Cytarabine and idarubicin were administered on Days 1, 2, and 3 of each subsequent 28-day cycle. Reduction of survivin was evaluated in peripheral blasts and bone marrow.
Results
Single-agent LY2181308 was well tolerated and survivin was reduced only in patients with a high survivin expression. In combination with chemotherapy, 4/16 patients had complete responses, 1/16 patients had incomplete responses, and 4/16 patients had cytoreduction. Nine patients died on study: 6 (monotherapy), 3 (combination).
Conclusions
LY2181308 alone is well tolerated in patients with AML. In combination with cytarabine and idarubicin, LY2181308 does not appear to cause additional toxicity, and has shown some clinical benefit needing confirmation in future clinical trials.
Keywords: antisense, acute myeloid leukemia, idarubicin, cytarabine
Introduction
Acute myeloid leukemia (AML) is a clonal disorder of hematopoiesis characterized by the accumulation of abnormal, non-functional myeloblasts. Due to the concomitant loss of normal hematopoiesis, AML has a high risk of mortality from disease- or treatment-related neutropenia and thrombocytopenia [1]. Classification and response to treatment in AML is currently being defined by World Health Organization (WHO) criteria [2,3].
Traditional induction chemotherapy (eg, anthracycline and cytarabine) produces a significant reduction in leukemic burden and a complete remission (CR) in a variable percentage of newly diagnosed patients (60% to 80% in younger patients and 40% to 55% in older patients) [4]. Unfortunately, the risk of relapse is considerable, particularly in patients with unfavorable AML cytogenetics and/or a history of an antecedent myelodysplastic syndrome. With the exception of patients with acute promyelocytic leukemia (APML), therapy for relapsed AML is often considered palliative, except for those patients for whom re-induction chemotherapy is a bridge to hematopoietic stem cell transplantation.
Given the relatively limited treatment options for patients with relapsed and refractory AML, there has been increasing interest in developing novel therapeutic agents targeting pathways important in leukemia biology. Survivin is a unique member of the mammalian inhibitor-of-apoptosis (IAP) protein family that is involved in mitotic regulation and suppresses apoptosis. Survivin is over-expressed during embryo-fetal development and in the vast majority of solid tumors and leukemias [5], but is not expressed in most normal, terminally-differentiated tissues [6]. In AML, it is hypothesized that constitutive expression of survivin leads to dysregulated cell proliferation and suppression of normal apoptotic signals [7]. Clinically, survivin-positive AML has been associated with a lower peripheral white blood cell count and is an independent negative prognostic factor for survival [8]. In chronic myeloid leukemia (CML), survivin is up-regulated during the transition from chronic phase to blast crisis, further implicating survivin in the pathophysiology of acute leukemic proliferation [9]. Consistent with this hypothesis, treatment of AML cells with the survivin antisense oligonucleotide (ASO) ISIS 23722 (clinically developed as the second-generation 2′-O-methoxyethyl, phosphorothioate antisense oligonucleotide, LY2181308) induced apoptosis by interfering with cell cycle progression [10]. In in vitro studies, LY2181308 has shown synergistic proapoptotic effect, with chemotherapies such as doxorubicin, gemcitabine, and taxanes [11]. This suggests that survivin ASO may restore the pro-apoptotic pathway in tumor cells, rendering tumor cells more susceptible to the subsequent apoptotic insult delivered by chemotherapy. Recently, encouraging evidence for LY2181308's activity in solid tumors suggests that targeting survivin expression in solid tumors is safe and potentially effective [12]. Taken together, these studies provide rationale for treating patients with relapsed or refractory survivin-positive AML with LY2181308.
Hence, for future development of LY2181308 in AML patients, it is important to establish the safety and PK profile of LY2181308 when combined with commonly used agents for the treatment of AML, such as idarubicin and cytarabine. The current study evaluated the safety of the monotherapy and combination in AML patients. In addition, the primary pharmacodynamic (PD) assessment in this analysis evaluated the reduction of survivin expression in AML cells of patients treated with LY2181308, as a single agent or in combination with idarubicin and cytarabine. Finally, the PD effect was related to the remission rates in patients with refractory or relapsed AML.
Materials and Methods
Eligibility criteria
Patients had to have a diagnosis of AML that was relapsed or refractory to at least 1 prior treatment for leukemia, or have CML in myeloid blast crisis which had failed at least 1 previous therapy with a tyrosine kinase inhibitor. For the monotherapy evaluation, only patients with high survivin expression levels (ie, greater than 2 for survivin molecules of equivalent fluorescence [MEFL]/isotype MEFL) in the leukemic blasts were eligible to participate in the trial (for details, see [13]). In the combination regimen, this eligibility criterion was removed, because survivin expression was based on gene expression rather than flow cytometry in AML blasts. A baseline bone marrow (BM) assessment was required ≤96 hours prior to the first dose of study drug. Patients had to have an Eastern Cooperative Oncology Group performance status of 0-2. Patients must have discontinued all previous therapies for cancer, including chemotherapy, radiotherapy, immunotherapy, cancer-related hormone therapy, or other investigational therapy for at least 21 days for myelosuppressive agents (ie, cytarabine, daunorubicin, and gemtuzumab ozogamicin) or 14 days for non-myelosuppressive agents prior to receiving study drug. Subjects must have recovered from the acute effects of prior therapy (ie, neurotoxicity, diarrhea, and mucositis) except for residual myelosuppression and alopecia. Hydroxyurea was permitted to control the peripheral blast cell count, but needed to be stopped at least 24 hours before study drug administration. In addition, patients had to have adequate organ function, including acceptable hepatic and renal function to allow safe administration of cytotoxic agents. Coagulation parameters also needed to be normal to evaluate potential toxicity for LY2181308.
Patients were excluded if they had been diagnosed with APML; had known hypersensitivity to oligonucleotides or any component of the formulation; or had leukemic involvement of the CNS by spinal fluid cytology or imaging. Patients with signs or symptoms of leukemic meningitis or a history of leukemic meningitis must have had a negative lumbar puncture within 2 weeks of study enrollment. Patients with a second primary malignancy and patients with serious pre-existing medical conditions were excluded. Patients with a known coagulopathy or bleeding disorder other than leukemia-related thrombocytopenia, or who were on concomitant anticoagulant therapy, were also ineligible.
The protocol was approved by each participating institution's ethical review board, including the post-amendment rationale based on the safety profile of monotherapy LY2181308 in AML patients, in solid tumor patients [12], and the expected additive reduction in survivin expression after combining LY2181308 with chemotherapy. Written informed consent was obtained from each participant before enrollment. The study was conducted in accordance with the ethical principles of the Declaration of Helsinki and Good Clinical Practices.
Study design
This was a multicenter, single-arm, open-label study in patients with refractory or relapsed AML. The study was initiated as a monotherapy trial of LY2181308 in which 8 patients had received LY2181308 treatment at the 750-mg dose (monotherapy cohort). The primary endpoint for the monotherapy cohort (pre-amendment) was to describe the survivin reduction in AML blasts using different detection methods (see below).The study was amended as of February 2009 to include cytarabine and idarubicin in combination with LY2181308 (combination cohort). The primary endpoint for the combination cohort (post-amendment) was to characterize the safety/PK of LY2181308 in AML patients. In both cohorts, monotherapy (n=8) and combination (n=16), LY2181308 treatment consisted of a loading dosing regimen of 3 consecutive daily infusions (750 mg over 3 hours) on Days 1 through 3, followed by a maintenance dosing regimen consisting of weekly infusions (750 mg over 3 hours) starting on Day 8. For the combination cohort, the following chemotherapy treatment was added for a new 16 patient combination cohort: cytarabine was administered as 1.5 g/m2 on Days 3, 4, and 5 of Cycle 1 as a 4-hour infusion and idarubicin was administered as 12 mg/m2 on Days 3, 4, and 5 of Cycle 1 as a 30‐minute infusion. Cytarabine and idarubicin were administered on Days 1, 2, and 3 of each subsequent 28-day cycle. The cytarabine infusion was followed by the idarubicin infusion. When LY2181308 was administered on the same day as chemotherapy (Day 3 of Cycle 1 and Day 1 of Cycles 2-n), LY2181308 was administered prior to chemotherapy and chemotherapy did not start until at least 30 minutes after the end of LY2181308 infusion. For the monotherapy cohort, the cycle was defined as 7 days, with the first cycles defined as the loading dose of three consecutive days (750 mg over 3 hours) and all subsequent cycles as defined by the weekly maintenance dose. For the combination cohort, each cycle was defined by the chemotherapy as 28 days (Fig 1).
Fig 1.
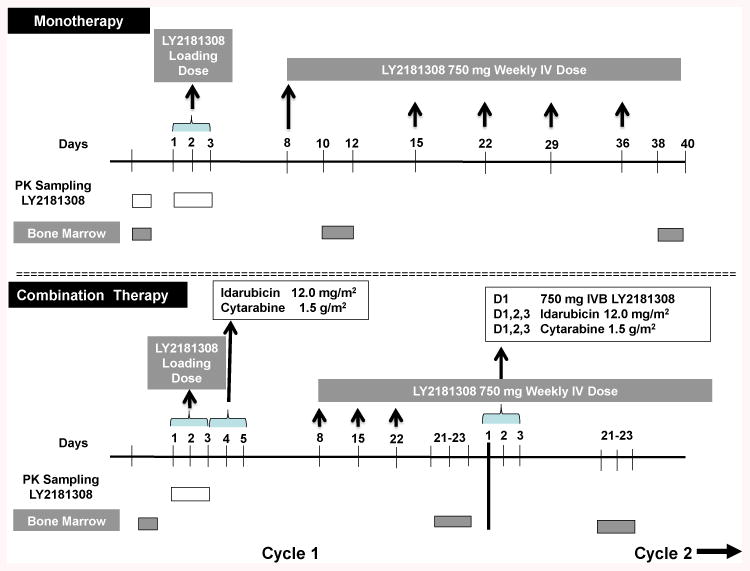
Study Design. Pre amendment- 750 milligrams intravenous bolus dose LY2181308 was administered in loading doses on days 1-3 and day 8. Weekly doses of 750 mg LY2181308 were administered intravenously on days 15, 22, 29, and day 36 thereafter. Post-amendment- 750 milligrams intravenous bolus dose LY2181308 was administered in loading doses on days 1-3 followed by idarubicin 12.0 mg/m2 plus cytarabine 1.5 mg/m2. Weekly doses of 750 mg LY2181308 were administered iv on days 8, 15, and 22 thereafter. Pharmacokinetic and bone marrow sampling are depicted as white and grey boxes, respectively. See methods for details.
There were assessments of survivin expression in peripheral blood leukemic blasts, and an assessment of anti-leukemic response was performed on a BM specimen obtained between Days 21 and 23 and compared to the pretreatment BM baseline assessment performed ≤96 hours prior to the first dose of LY2181308 (Figure 1). Patients with objective evidence of progression (defined as an increase in BM blast cell count of at least 50% from the baseline assessment) were taken off study drug treatment. For patients with baseline BM blast counts ≥70%, an absolute increase in blast percentage of 10 percentage points or more was considered progression (eg, from 70% to 80%). Patients without evidence of BM progression as determined by the cellularity/blast index were eligible to receive LY2181308 in combination with chemotherapy in Cycle 2 as long as discontinuation criteria had not been met. In patients with rapidly progressing disease, peripheral blood blast counts and clinical symptoms were sufficient to discontinue patients from the study.
Safety was assessed before each cycle using the National Cancer Institute's Common Terminology Criteria for Adverse Events, version 3.0.
Overview of Pharmacodynamic Assessments
The PD variables were:
Survivin evaluation, by the fluorescence-activated cell sorting (FACS) assay (data were expressed as mean equivalent fluorescence [MEFL])
Survivin messenger ribonucleic acid (mRNA) in whole blood and bone marrow, as measured by quantitative reverse transcriptase polymerase chain reaction (qRT-PCR)
Survivin protein in whole blood lysates, as measured by enzyme-linked immunosorbent assay (ELISA).
For each PD measurement, the longitudinal data was analyzed using a mixed effect model in order to account for any incomplete data and repeated measures on each patient. The models included patient as a random effect, baseline measurement and time as fixed effects. Time was modeled as a class effect. In addition, any additional variable collected for normalization purposes (e.g. house keeping genes) were included in the models as fixed variables. An unstructured covariance matrix was used unless it was deemed inappropriate for the dataset. The results are presented using Least-Squares Geometric Means (LSGM) for time categories and 90% confidence intervals for the key variables.
Flow Cytometry and Survivin Index
Using a FACS assay specifically developed for this trial, the number of AML blasts (expressed as percent of circulating cells or as an absolute value) and the amount of survivin expression (protein) per cell (expressed as MEFL value) was measured using an anti-survivin polyclonal antibody (Clone 71G4, rabbit anti-survivin polyclonal antibody coupled with Alexa 647, Cell Signaling Technology, Danvers, MA) and a proprietary fixation/permabilization technique. To determine a high survivin expression level in AML cells, blasts from AML patients were used and compared to blood cells from healthy volunteers. The ratio of survivin MEFL over isotype MEFL was defined as the survivin index. A survivin index of at least 2.0 or more was considered high. For monotherapy patients, only patients with a survivin index of more than >2 were included. Apoptosis was evaluated by Annexin V immunofluorescence using a flow cytometry-based cell cycle assay. All blood samples were sent to a central laboratory for analysis using a standardized flow cytometry protocol for hematologic malignancies (Laboratory Corporation of America Holdings/LabCorp Clinical Trials, Cranford, NJ and Mechelen, Belgium) using a survivin flow analysis protocol validated prior to this trial (data on file) [13]. The primary PD endpoint was based on the change in survivin index in the total blast cell gate, where the survivin index was calculated as [(blast survivin MEFL – blast isotypic control MEFL)/blast isotypic control MEFL] using the FACS assay.
Survivin mRNA Expression
For evaluating the expression of survivin in peripheral cells, we used 2 analysis platforms for samples collected before and after treatment. We used target-directed cDNA amplification based qRT-PCR, as well as a non-amplification based target hybridization platform with signal amplification using branched DNA, Quantigene® 2.0 Assay (Affymetrix, Carlsbad, CA)[14]. Oligonucleotide probe sets for target and control genes were designed with standard probe design software for use in the QuantiGene® 2.0 Assay Reagent Systems (Panomics, Inc., Santa Clara, CA). The software algorithm identifies 1 or more continuous regions that serve as annealing templates for capture extenders (CEs) (5–10 per gene), label extenders (LEs) (10–20 per gene), or blocking probes (BLs). Each probe consisted of 3 types of oligonucleotides that trap target ribonucleic acid (RNA) to the surface of the plate and hybridize to DNA signal amplification molecules designed with partial homology to the target gene sequence. All reagents were used according to manufacturer's recommended protocols (Affymetrix, Carlsbad, CA). Briefly, probe set oligonucleotides (250 fmol CE, 500 fmol BL, and 1000 fmol LE) were mixed with the sample, and the mixture was added to each assay well in a 96-well plate covalently coated with capture probe oligonucleotide 5′-CACTTCACTTTCTTTCCAAGAG.
Target RNA was captured during an overnight incubation at 55°C. Unbound material was removed by washing with 200 to 300 ul of wash buffer (1/10 strength standard saline citrate containing 0.3 g/L lithium lauryl sulfate) followed by sequential hybridization with DNA amplifier molecules, then 3′-alkaline phosphatase-conjugated label Probe oligonucleotides, with 3 washes after each incubation. After the final wash, the dioxetane alkaline phosphatase substrate Lumiphos Plus (Lumingen Inc., Southfield, MI) was added to the wells, and after a short incubation, luminescent signal was measured in an LMax (Molecular Devices, Mountain View, CA).
Quantitative RT-PCR was performed using TaqMan® assay reagents (Roche Molecular Systems, Inc. provided by Life Technologies, Carlsbad CA) for baculoviral IAP repeat-containing 5 (survivin) BIRC5, assay id Hs00153353_m1, context sequence 5′-AGAACAAAATTGCAAAGGAAACCAA and GAPDH, assay id Hs99999905_m1, context sequence 5′-TTGGGCGCCTGGTCACCAGGGCTGC. The Quantigene® 2.0 reagent identifications for survivin and GAPDH were NM_001168, catalog number SA_50156 and NM_002046, catalog number SA_10001, respectively. The β-actin, RPL32, and RPS20 genes were used for normalization in branched DNA analysis.
ELISA for Survivin Protein
Survivin protein was measured in peripheral blood mononuclear cell (PBMC) lysates using an ELISA with an anti-survivin polyclonal antibody (clone #500-201) according to manufacturer's instructions (Novus Biologicals, Littleton, CO). Cell counts and total protein concentrations (Bio-Rad, Richmond, CA) were determined in order to normalize survivin expression levels.
Pharmacokinetic Assessment
Plasma samples were analyzed to determine the concentration of LY2181308. For LY2181308 PK assessment, samples were taken on Days 1 and 3 (loading dose regimen), on Days 4 and 5 (prior to and at the end of chemotherapy), and on Days 8, 15, 22, 29, and 36 (prior to the LY2181308 infusion). Thereafter, PK assessment samples were taken every fourth week starting on Visit 3, Day 57 (prior to LY2181308 infusion) and at the follow up visit 21 days after patients had taken their last dose of study drug. Mean population plasma LY2181308 PK parameters (clearance, exposure, volume of distribution, half-lives) and subject variability were computed using non-linear mixed effect modeling [15] (implemented in NONMEM®, ICON corporation, Ellicott City, MD, version VI).VI In addition, LY2181308 PK parameters (maximum concentration [Cmax], area under the curve [AUC], and clearance [Cl]) were determined by standard non-compartmental methods of analysis using WinNonlin® Professional Edition version 5.2 (Pharsight Corporation, Mountain View, CA).
Safety and Efficacy
Safety and efficacy analyses [16] were conducted on all patients receiving at least 1 dose of study drug according to the dose the patients were assigned. Patient data from all sites were pooled for the purposes of statistical analysis. Parameter estimates and their 90% confidence intervals were reported, unless otherwise stated. Summary statistics are provided separately for monotherapy and combination patients.
Results
Study Design and Patient Characteristics
The study design is depicted in Figure 1. A total of 42 patients were entered from the start of the study in March 2008 and its end in January 2010. Based on the survivin index for the flow cytometry assay, eighteen patients did not meet screening criteria for study entry at their first visit. In the monotherapy portion of the study, 8 patients received 750 mg LY2181308 as monotherapy (refer to Study Design in Materials and Methods for further details). In the monotherapy, 1 patient discontinued after receiving 1 cycle of therapy. In the combination therapy, an additional 16 patients received LY2181308 and chemotherapy (idarubicin and cytarabine). Thirteen patients discontinued after receiving 1 cycle of combination therapy. Of the 24 total patients treated in both study cohorts, 10 patients had previously received hematopoietic stem cell transplants (N=2 for LY2181308 and N=8 for LY2181308 plus cytarabine and idarubicin), and all 24 patients had received prior systemic therapy. Patient and baseline characteristics are described in Table 1.
Table 1. Demographic and Baseline Characteristics by Treatment Group.
| LY2181308 (Monotherapy Cohort) N=8 |
LY2181308 + idarubicin + cytarabine (Combination Cohort) N=16 |
Total N=24 |
|
|---|---|---|---|
| Sex: female/male, n | 5/3 | 6/10 | 11/13 |
| Age: mean ± SD, years | 60.3 (15.9) | 48.2 (14.7) | 52.2 (15.9) |
| Race: n (%) | |||
| African | 0 | 2 (12.5) | 2 (8.3) |
| Caucasian | 7 (87.5) | 11 (68.8) | 18 (75.0) |
| East Asian | 1 (12.5) | 0 | 1 (4.2) |
| Hispanic | 0 | 1 (6.3) | 1 (4.2) |
| West Asian | 0 | 2 (12.5) | 2 (8.3) |
| ECOG PS: n (%) | |||
| 0 | 1 (12.5) | 1 (6.3) | 2 (8.3) |
| 1 | 7 (87.5) | 11 (68.8) | 18 (75.0) |
| 2 | 0 | 4 (25.0) | 4 (16.7) |
| AML WHO Criteria: n (%) | 7 (87.5) | 15 (93.8) | 22 (91.7) |
| De novo: n (%) | 6 (75.0) | 14 (87.5) | 20 (83.3) |
| Not specified | 0 | 1 (6.3) | 1 (4.2) |
| Genetic abberations | 0 | 2 (12.5) | 2 (8.3) |
| Erythroblastic | 0 | 2 (12.5) | 2 (8.3) |
| Myeloblastic | |||
| Minimal differentiation | 3 (37.5) | 3 (18.8) | 6 (25.0) |
| Differentiated | 1 (12.5) | 3 (18.8) | 4 (16.7) |
| Monocytic | 1 (12.5) | 0 | 1 (4.2) |
| Myelomonocytic | 1 (12.5) | 3 (18.8) | 4 (16.7) |
| Secondary AML: n (%) | |||
| Not specified | 1 (12.5) | 2 | 3 (12.5) |
| Multi-lineage | 0 | 0 | 0 |
| Myeloblastic with minimal differentiation | 1 (12.5) | 0 | 1 (4.2) |
| With FLT3 mutation: n (%) | 0 | 2 (12.5) | 2 (8.3) |
| Prior history | |||
| Prior remissions: n (%) | |||
| 1 | 1 (25.0) | 4 (25.0) | 5 (20.8) |
| 2 | 3 (37.5) | 4 (25.0) | 7 (29.2) |
| 3 | 1 (12.5) | 1 (6.3) | 2 (8.3) |
| ≥4 | 1 (12.5) | 2 (25.0) | 3 (12.5) |
| Primary refractory disease: n (%) | 2 (25.0) | 5 (31.3) | 7 (29.2) |
| WBCs at first visit actual, ×109/L: n (%) | |||
| <4.5 | 3 (37.5) | 8 (50.0) | 11 (45.8) |
| ≥4.5 and ≤11 | 2 (25.0) | 3 (18.8) | 5 (20.8) |
| >11 | 3 (37.5) | 5 (31.3) | 8 (33.3) |
Abbreviations: AML = acute myeloid leukemia; ECOG = Eastern Cooperative Oncology Group; n = number of patients in each category; N = number of enrolled patients; PS = performance status; SD = standard deviation; WBC = white blood cell; WHO = World Health Organization.
Pharmacokinetics
LY2181308 PK properties in AML patients are characterized by a rapid and extensive distribution of LY2181308 from plasma to tissue: high volume of distribution and rapid distribution clearance from plasma to tissue (Table 2) [15]. Thus, within 1 to 2 hours after the end of LY2181308 infusion, the drug administered is distributed into tissues. The moderate elimination clearance in tissues (>80 L/h per the PK model) associated with the extensive volume of distribution leads to a long terminal elimination half-life of ∼17 days (based on late sampling of PK data up to 80 days after the last LY2181308 dose [n=7 patients]). The individual PK data, Cmax and trough-predose LY2181308 concentrations, do not indicate accumulation of LY2181308 plasma exposure over the first month of treatment following repeated weekly LY2181308 dosing (Figure 2). From the observed data, the ratio of LY2181308 Cmax Day 3/Day 1 ranged from 0.67 to 1.72 (with 20th -80th percentile of 0.85 and 1.09, respectively; data on file) [15]. The average plasma concentration and exposure for LY2181308 are shown in Table 2. LY2181308 exposure is similar overall between the monotherapy LY2181308 and the LY2181308/chemotherapy arms in AML patients. LY2181308 PK characteristics in AML patients are similar to those described in a conventional Phase 1 cancer patient population [12] but with a slightly higher volume of distribution and elimination clearance [15]. Therefore the average plasma concentration and exposure were about 33% lower in patients with AML compared to a conventional Phase 1 cancer patient population [data on file, 11]. This lower exposure in AML patients should be put in perspective of the slightly greater variability in overall LY2181308 exposure in that population. The coefficient of variation of LY2181308 exposure in a conventional Phase 1 cancer population was 31.5% (n=24 patients at 750-mg dose) [15] and 45.7% (n=15) in this AML patient population (Table 2). LY2181308 concentration in BM and PBMCs was measured using similar techniques to those described in patients with solid tumors (Figure 3) [12]. The LY2181308 concentration levels observed were predicted to be sufficient to inhibit survivin protein.
Table 2. LY2181308 Plasma PK Parameters Following Day 1 and Day 3 Dosing (750-mg 3-hour Infusion).
| Day 1 | Day 3 | ||||||
|---|---|---|---|---|---|---|---|
| Patient Na |
Cmax (ng/mL) |
Patient Na |
Cmax (ng/mL) |
Plasma CLss (L/h) |
AUCτ (ng*h/mL) |
Vz (L) |
|
| Group 1c | N=8 | 50503 (51.0) | N=8 | 46822 (57.0) | 3.45 (60.0) | 216947 (60.0) | 654 (43.4) |
| 26830 - 88137b | 20586 - 90339b | 1.56 - 7.88b | 95189 - 480259b | 376 - 1315 | |||
| Group 2c | N=15d | 45678 (28.7) | N=16 | 45565 (35.1) | 4.04 (45.7) | 185734 (45.7) | 1149 (71.9) |
| 27740 - 77519b | 26497 - 80711b | 1.63 - 8.1b | 92621 - 460082b | 300 - 3430 | |||
Abbreviations: AUCτ = area under the curve over the dosing interval; CLss = steady state clearance; Cmax = maximum concentration; N = number of enrolled patients; PS = performance status; SD = standard deviation; Vz = Volume of Distribution (calculated plasmaCLss/(0.693/t1/2), t1/2 from the model).
Number of patients.
Range.
Group 1 represents LY2181308 monotherapy; Group 2 represents LY2181308 monotherapy on Days 1 and 2, and together with chemotherapy on Day 3.
No PK data collected for 1 patient on Day 1.
Fig 2.
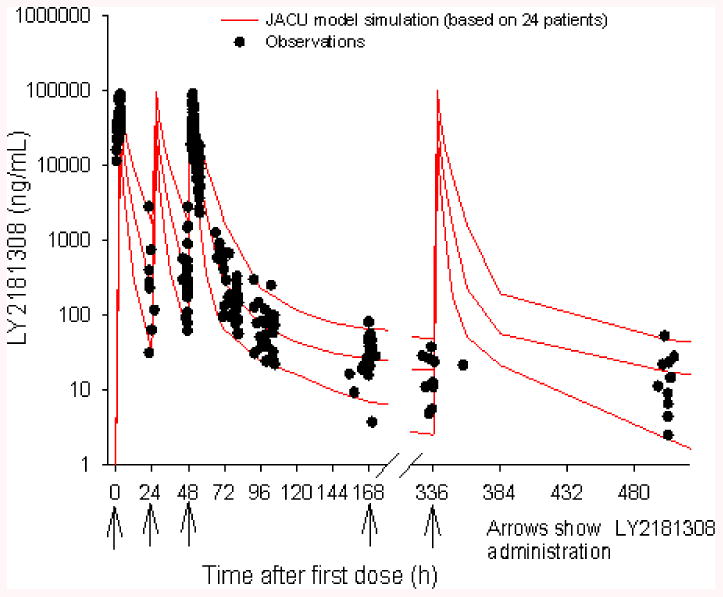
LY2181308 plasma pharmacokinetic profile following Day 3 dosing (LY2181308 750 mg 3-hour infusion). Black circles represent observed data-- red lines represent simulated profile from a four compartment PK model (median, 5th, and 95th percentiles) LY2181308 (15). Data were analyzed on the log scale using a mixed effect model in order to account for any incomplete data and repeated measures on each patient
Fig 3.

LY2181308 detection in peripheral blood mononuclear cells (PBMCs) after monotherapy. Enzyme-linked immunosorbent assay was used to measure LY2181308 abundance in peripheral blood monocyte lysates before and 10-12 days after initial bolus dose administration (2-4 days after Day 3 dose). The red circle denotes the median concentration value predose on Day 8. The black lines denote the model simulated median, 5th, and 95th percentile concentrations at the end of LY2181308 infusion on Day 3 in combination patients (ie, prior to administration of the first dose of chemotherapy – idarubicin and cytarabine). ID = identification; PK = pharmacokinetic
Pharmacodynamics
Consistent with these PK findings, it was observed that on average, the target, as measured by the survivin protein index in AML blast, was reduced significantly on Days 1 to 4, as indicated by the 90% confidence interval of the ratio, which excludes 1 (Figure 4 and Table 3). The maximum effect was observed on Day 2 (Figure 4 and Table 3) with a ratio of LSGM (post dose/baseline predose) estimated to be 0.43, with a 90% confidence interval of 0.28 to 0.67. This corresponds to a 57% reduction from baseline (mean value).
Fig 4.

Survivin expression before and up to 5 days after LY2181308 administration as monotherapy (n=8 pts). The survivin index was calculated as [(blast survivin mean equivalent fluorescence (MEFL) – blast isotypic control MEFL)/blast isotypic control MEFL] using the fluorescence-activated cell sorting (FACS) assay. The results are presented using least-squares geometric means (LSGM) for time categories and 90% confidence intervals for the key variables
Table 3. Survivin Index from Baseline to Day 5 or After in Monotherapy Patients.
| Day | LSMeans (90% CI) | Ratio of LSMeansa (90% CI) |
|---|---|---|
| Baseline | 7.98 (4.64-13.70) | |
| Day 1 | 4.84 (2.81-8.33) | 0.61 (0.40-0.92) |
| Day 2 | 3.43 (1.96-6.00) | 0.43 (0.28-0.67) |
| Day 3 or 4 | 5.44 (3.31-8.96) | 0.68 (0.48-0.97) |
| Day 5 or after | 6.50 (3.93-10.75) | 0.82 (0.57-1.16) |
Abbreviations: CI = confidence interval; LSMeans = Least Square Geometric Means.
Relative to baseline.
Survivin gene expression was measured in whole blood and BM using qRT‐PCR. In the whole blood, 2 patients had a reduction in survivin during the loading dose and 1 patient had a rebound after the loading dose. In addition, 1 patient had a large rebound during the loading dose, and had a reduction in survivin after completing the loading dose compared to baseline levels. Overall, when modeling the longitudinal data, there was no evidence suggesting changes to the survivin expression levels post-treatment compared to pre-treatment in the whole blood. No patterns were observed in the bone marrow either. Lastly, Survivin protein expression was measured in PBMC lysates using an ELISA assay. According to a mixed model used to analyze and normalize the data, there were no changes in survivin protein expression post treatment.
Efficacy
Although monotherapy patients stayed on treatment longer than combination patients, little evidence of efficacy with monotherapy LY2181308 was observed. The reduced time in treatment for combination patients is explained by investigator decisions to treat these patients with alternative therapies. Four of 16 combination patients had morphologic complete responses, 1/16 had an incomplete response, and 4/16 had cytoreductions. Overall, at least 9/16 combination patients had a clinically significant response. An additional 2 patients had clinical benefit, defined as stable disease.
Safety and Tolerability
The majority of patients (7/8 patients; 87.5%) who received LY2181308 as a single agent completed Cycle 2 (1 cycle defined as 7 days), while only 3/16 patients (18.8%) who received LY2181308 plus cytarabine and idarubicin completed Cycle 2 (1 cycle defined as 28 days). In the combination cohort, none of the 16 patients completed Cycle 3 or higher. Of the 8 monotherapy patients, 3 patients (37.5%) completed Cycle 3, 3 patients (37.5%) completed Cycle 4, 1 patient (12.5%) completed Cycle 5, and 1 patient (12.5%) completed Cycle 6. There were no dose reductions of LY2181308 either pre- or post-treatment.
A total of 19/24 patients (79.2%) experienced 1 or more serious adverse events (SAEs). All the SAEs were ultimately determined by investigators to be unrelated to LY2181308. The most frequently reported SAEs in the study overall were febrile neutropenia (8/24 patients; 33.3%) and sepsis (5/24 patients; 20.8%). A greater percentage of combination patients experienced 1 or more SAEs (13/16 patients; 81.3%) compared with patients on monotherapy (6/8 patients; 75%). SAEs that were reported in both monotherapy and combination patients were febrile neutropenia, sepsis, hypotension, pneumonia, and clostridial infection. Febrile neutropenia was the most frequently reported SAE in combination patients (7/16 patients; 43.8%), while it was reported only for 1/8 monotherapy patients (12.5%). Sepsis was the most frequently reported SAE in monotherapy patients (4/8 patients; 50%), but was reported in only 1/16 combination patient (6.3%). There were several SAEs reported in combination patients but not in monotherapy patients, such as respiratory failure (2/16 patients; 12.5%). Similarly, there were some SAEs reported in monotherapy patients but not in combination patients, such as dyspnea (2/8 patients; 25%).
In the study overall, there were 13 patients (54.2%) who experienced at least 1 TEAE that was deemed to be possibly study drug related by the investigator (primarily related to chemotherapy), which included 2 of the 8 patients (25%) in the monotherapy group, and 11 of the 16 patients (69%) in the combination therapy group. The possibly drug-related TEAEs that were reported most frequently overall in the study were nausea (5 of 24 patients; 20.8%), diarrhea (3 of 24 patients; 12.5%), fatigue (3 of 24 patients; 12.5%), and leukopenia (3 of 24patients; 12.5%). In the monotherapy group, no grade 3 or 4 drug related events were observed. The maximum CTCAE severity was grade 1: one event of dyspepsia, one event of nausea, and one event of insomnia. In the combination group, there were six grade 3 AEs (one report each of hyperbilirubinemia, esophagitis, febrile neutropenia, blood infection, respiratory infection, and tumor lysis syndrome), five grade 4 AEs (one report each of diarrhea, and platelets, and three reports of leukopenia), and two grade 5 AEs (death related to the AEs in one patient of CNS cerebrovascular ischemia and hypoxia).
In total, there were 9 deaths reported during the study, including 6 deaths due to progression of AML in monotherapy patients (single‐agent LY2181308) and 3 deaths in combination patients (LY2181308 plus cytarabine and idarubicin). One of the 3 patients in the combination group was reported to have died due to study drug toxicity (reason: hemorrhagic stroke and respiratory failure) while receiving LY2181308 plus cytarabine and idarubicin in Cycle 1; the investigator found this death to be likely related to treatment with cytarabine and idarubicin rather than LY2181308.
Discussion
In addition to safety, the objective of this non-randomized open-label, proof-of-concept study was to evaluate whether LY2181308 reduces measurable survivin levels in AML patients similar to that observed in solid tumors [12]. We first evaluated the effect of LY2181308 as a monotherapy and later as part of a combination with pro-apoptotic chemotherapy. During the monotherapy treatment, only patients with a pre-defined survivin expression index using flow cytometry were enrolled. The assay determining a pre-specified level of survivin protein expression (survivin expression index) was validated prior to the start of the study [13, 17]. Reduction in survivin protein expression was observed in monotherapy patients with high survivin protein expression in AML blast cells. In the combination cohort we evaluated survivin expression based on both gene and protein expression. However, protein expression changes were not readily observed, as patients with low survivin expression were allowed to be enrolled in this combination cohort. Consequently, it was difficult to detect a change in survivin protein expression by flow cytometry.
In contrast to the survivin protein expression measurements, other studies used survivin gene expression to determine the PD changes induced by ASOs. At a LY2181308 dose of 750 mg, the inhibition of both survivin mRNA and protein was on average 20% in patients with solid tumors [12]. In contrast to the solid tumor patients, a consistent inhibition pattern based on mRNA measurements using qRT-PCR or branched DNA assays was not observed in this study. The observed changes in survivin gene expression (qRT-PCR) and survivin protein expression (flow cytometry) were not correlated.
The monotherapy activity of 750 mg LY2181308 to decrease survivin protein expression appeared moderate in this study and was less than anticipated. When monotherapy patients with relatively high survivin expression were enrolled, changes in survivin protein expression levels were detected. In the cohort with the combination, this threshold for high survivin expression upon study entry was lowered and may have compromised the ability to reliably distinguish changes in survivin expression.
The apparent discordance in antisense presence/absence and in survivin target diminution or lack thereof can have several explanations. First, it is possible that the flat dosing used for LY2181308 was not achieving the required tissue concentrations compared to an alternative dosing regimen. For example, the ASO against the X-linked inhibitor of apoptosis (XIAP) AEG35156 used body-surface area (BSA)-based dosing and reported that the XIAP mRNA levels were reduced in AML patients [18]. At the 350 mg/m2 dose of antisense XIAP, the inhibition of the target mRNA ranged from 45% to 90% in AML patients. By contrast, LY2181308 inhibited survivin mRNA expression at an average of 20% with flat dosing in solid tumors [15]. When we retrospectively applied BSA-based dose calculations to the patients treated in the study, we found that the flat dosing had not underestimated the BSA-based administration of LY2181308, but rather would have increased the risk of LY2181308-associated toxicity. While higher doses of LY2181308 may be required in AML patients due to their tumor burden, the ASO-associated off-target toxicity in the liver and complement-activation limited the option to increase the dose above levels observed to be tolerable in solid tumor patients [12].
Second, ASOs may be differentially eliminated in patients with AML, and thus the time on target may be altered. Compared to solid tumor patients, LY2181308 in AML patients has a similar PK profile, such as a multiphasic rapid distribution, moderate clearance, and high volume of distribution associated with the long terminal half-life [15]. While the range of LY2181308 exposure is comparable between the 2 patient populations in these studies, the average plasma concentration and exposure for LY2181308 was observed to be approximately 33% lower in AML patients compared with solid tumor patients. This lower average plasma exposure for LY2181308 in AML patients is associated with higher clearance. The higher clearance and lower exposure and half-life in AML patients compared with conventional Phase 1 cancer patients could be explained hypothetically by the presence of blast cells in AML. These blast cells could constitute a tissue into which LY2181308 could be distributed and eliminated. Saleem et al. [19] indicated that LY2181308 distributes into blood cells with a 1.5 plasma-to-blood concentration ratio. This may also explain the variability in levels of survivin reduction observed in AML patients. Based on the PK measurements, the survivin inhibition we observed may also have been of too short a duration to produce a substantial inhibitory effect on tumor cell proliferation. Such low cellular exposure levels have not been reported for other ASOs, and may explain why LY2181308 may not have achieved sufficient levels for inhibition of survivin in AML patients [12, 18]. LY2181308 concentration data were available for 10 combination therapy patients of BM pre- and post-treatment. In all patients, LY2181308 concentration in BM following 3 weeks of treatment was below the limit of quantitation. In some cases, low content of cells in BM aspirates due to the chemotherapy made the assessment inconclusive. While our observations imply the possibility that LY2181308 behaves differently in AML patients, such observations have not been reported about other ASOs targeting IAPs. For instance, the Phase 1 PK data of AEG35156 in patients with refractory AML and in patients with advanced cancer [18,20] did not show a greater clearance of the ASO in patients with AML.
Third, it has been observed that survivin and XIAP can form heteromeric binding partners [21]. It is possible that 1 confounding element of these studies may be the potential for survivin protein levels to be altered in the face of antisense inhibition by this interaction with XIAP. Some other potential binding partners include the XIAP-associated factor 1 (XAF1) [22] and the oncogenic transcription factor STAT3, which was identified as a binding partner of nuclear survivin [23]. Many tumor cells express reduced levels of XAF1 which might be involved in survivin stabilization [21]. Recently, aberrant STAT3 expression has been demonstrated in AML cells and primary cell lines from pediatric patients. A small molecule STAT3 inhibitor induced apoptosis of these cells and inhibited AML blast colony formation [24]. Alternatively, AML patients may not have sufficient levels of either RNase H1 and/or H2, which is required to digest the ASO/mRNA hybridization product [25]. While we did not specifically investigate the levels or the activity of RNase H1 and H2, gene array analyses suggest that many tumor cells have sufficient levels of RNase H2 for drug targeting [26], as do the above results described for AEG35156 [18, 20]. Also, the LY2181308 binding site can be identified in survivin alpha, survivin DeltaEx3, survivin 2B β, and survivin 3B variants, but not in survivin 2-α and survivin 3-α mRNA splice variants. The possibility exists that the survivin-2-α or survivin-3-α splice variants may play a more important role in AML than in solid tumors. It could also be that some of the other splice variants of survivin mRNA are more poorly inhibited by LY2181308 than the survivin mRNA it was designed and tested against and therefore may be able to compensate, in some circumstances, for the loss of survivin. As noted by others, difficulties in correlating changes in survivin expression and clinical outcome are not uncommon [28]. Clearly, further understanding of survivin biology in AML and other tumor types will be necessary to better address these issues.
Fourth, differential turnover of the target protein requires an altered dosing regimen for AML patients. Since survivin is often found in distinct subcellular compartments including the cytoplasm, nucleus and mitochondria in different cell types, it may again be that differential turnover of the protein within these locations may differ within tumor cells. Heat Shock Protein-90 (Hsp90), Hsp60, and the aryl hydrocarbon receptor-interacting protein (AIP) [21] have also been shown to physically interact with survivin in vivo, and may play a role in subcellular localization. Recently, BMI-1 has been suggested to be a survivin-interacting protein in B-cell lymphoma cells that may stabilize survivin levels post-translationally without a change in survivin mRNA level [29]. If such interactions stabilized survivin in protein complexes, turnover of the survivin protein may not closely follow that of its coding mRNA and may help to explain why survivin activity was not impacted as predicted. However, such interactions do not readily explain the lack of clear direction in our mRNA-based assays to detect survivin gene expression. As several pre-and post-dose samples exhibited a spike upward in survivin protein expression post-dose in our assays (data not shown), one speculative possibility may be that survivin expression itself is self-limiting and diminution of mRNA for survivin induces survivin expression through disruption of a negative feedback loop that ordinarily keeps survivin protein levels low. Evidence for positive transcriptional regulatory signaling between p53 and survivin in nasopharyngeal carcinoma without inducing apoptosis has recently been reported [30]. Further analysis of survivin regulation in tumor cells should help answer these questions.
Eleven of sixteen patients in the combination therapy (69%) had a clinical benefit (9 with clinical response and 2 with stable disease). The goal of salvage therapy for patients with relapsed and refractory acute myeloid leukemia has been to achieve response. It is not clear that response leads to improvement in survival in this patient population, but may allow patients to proceed to other potentially curative options (such as allogeneic hematopoietic stem cell transplantation). However, therapy is not continued indefinitely due to the toxicity of the chemotherapy regimen and the lack of benefit of prolonged chemotherapy treatment. The observation of the clinical benefit in the combination arm )raises the question of whether this response is due to the fact that LY2181308 sensitized the AML cells to subsequent chemotherapy. In this combination therapy portion of the study, the effect was not tied to a reduction of survivin expression in blast cells (data not shown). This pattern of response is more likely to be a result of the chemotherapy. Further discrimination of the ability of LY2181308 to enhance subsequent chemotherapy in AML patients will require randomized Phase 2 trials with assays to better discriminate low levels of survivin expression at the gene and protein level. The PK profile of LY2181308 in AML patients is consistent with that observed in patients with solid tumors (with a potential shorter on target time), and this profile does not appear to be affected by chemotherapy.
In general, the combination of LY2181308 with idarubicin and cytarabine is consistent with the profile of idarubicin and cytarabine. All SAEs and AEs were either attributed to the chemotherapies or associated with progressive disease. There was no obvious contribution of LY2181308 to the observed and known toxicity profile of the chemotherapies.
Acknowledgments
The authors would like to thank all patients and site personnel for supporting the study. They would also like to thank David Ohannesian PhD and Joseph Durrant (Pharmanet/i3, Indianapolis, IN; an InVentiv Health company) for writing and editorial support and Bret Monia PhD (ISIS Pharmaceuticals, Carlsbad, CA) for his review of the manuscript. This study was sponsored by Eli Lilly and Company, Indianapolis, IN, USA.
Supported in part by grants from NIH CA 55164, CA 049639, and CA 016672 (to MA).
Footnotes
Ethical Standards: This study was conducted in compliance with the current laws of the United States.
Conflict of Interest: M.L., V.A., S.C., S.S., S.K., and J.T.B. are employees and minor shareholders of Eli Lilly and Company, Indianapolis, IN. The other authors have no financial relationship to declare.
References
- 1.Stone RM, DeAngelo DJ, Klimek V, Galinsky I, Estey E, Nimer SD, Grandin W, Lebwohl D, Wang Y, Cohen P, Fox EA, Neuberg D, Clark J, Gilliand DG, Griffin JD. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005;105:54–60. doi: 10.1182/blood-2004-03-0891. [DOI] [PubMed] [Google Scholar]
- 2.Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, Harris NL, Le Beau MM, Hellström-Lindberg E, Tefferi A, Bloomfield CD. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114:937–951. doi: 10.1182/blood-2009-03-209262. [DOI] [PubMed] [Google Scholar]
- 3.Schlenk RF, Dohner K, Krauter J, Frohling S, Corbacioglu A, Bullinger L, Habdank M, Spath D, Morgan M, Benner A, Schlegelberger B, Heil G, Ganser A, Dohner H German-Austrian Acute Myeloid Leukemia Study Group. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358:1909–1918. doi: 10.1056/NEJMoa074306. [DOI] [PubMed] [Google Scholar]
- 4.Tallman MS. New strategies for the treatment of acute myeloid leukemia including antibodies and other novel agents. Hematology Am Soc Hematol Educ Program. 2005:143–150. doi: 10.1182/asheducation-2005.1.143. [DOI] [PubMed] [Google Scholar]
- 5.Cong XL, Han ZC. Survivin and leukemia. Int J Hematol. 2004;80:232–238. doi: 10.1532/IJH97.A10408. [DOI] [PubMed] [Google Scholar]
- 6.Liu K, He X, Lei XZ, Zhao LS, Tang H, Liu L, Lei BJ. Pathomorphological study on location and distribution of Kupffer cells in hepatocellular carcinoma. World J Gastroenterol. 2003;9:1946–1949. doi: 10.3748/wjg.v9.i9.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carter BZ, Milella M, Altieri DC, Andreeff M. Cytokine-regulated expression of survivin in myeloid leukemia. Blood. 2001;97:2784–2790. doi: 10.1182/blood.V97.9.2784. [DOI] [PubMed] [Google Scholar]
- 8.Adida C, Recher C, Raffoux E, Daniel MT, Taksin AL, Rousselot P, Sigaux F, Degos L, Altieri DC, Dombret H. Expression and prognostic significance of survivin in de novo acute myeloid leukaemia. Br J Haematol. 2000;111:196–203. doi: 10.1111/j.1365-2141.2000.02328.x. [DOI] [PubMed] [Google Scholar]
- 9.Hernandez-Boluda JC, Bellosillo B, Vela MC, Colomer D, Alvarez-Larran A, Cervantes F. Survivin expression in the progression of chronic myeloid leukemia: a sequential study in 16 patients. Leuk Lymphoma. 2005;46:717–722. doi: 10.1080/10428190500052131. [DOI] [PubMed] [Google Scholar]
- 10.Carter BZ, Wang RY, Schober WD, Milella M, Chism D, Andreeff M. Targeting survivin expression induces cell proliferation defect and subsequent cell death involving mitochondrial pathway in myeloid leukemic cells. Cell Cycle. 2003;2:488–493. doi: 10.4161/cc.2.5.500. [DOI] [PubMed] [Google Scholar]
- 11.Carrasco RA, Stamm NB, Marcusson E, Sandusky G, Iversen P, Patel BK. Antisense inhibition of survivin expression as a cancer therapeutic. Mol Cancer Ther. 2011;10:221–232. doi: 10.1158/1535-7163.MCT-10-0756. [DOI] [PubMed] [Google Scholar]
- 12.Talbot DC, Ranson M, Davies J, Lahn M, Callies S, André V, Kadam S, Burgess M, Slapak C, Olsen AL, McHugh PJ, de Bono JS, Matthews J, Saleem A, Price P. Tumor survivin is downregulated by the antisense oligonucleotide LY2181308: a proof-of-concept, first-in-human dose study. Clin Cancer Res. 2010;16:6150–6158. doi: 10.1158/1078-0432.CCR-10-1932. [DOI] [PubMed] [Google Scholar]
- 13.Wacheck V, Lahn M, Dickinson G, Füreder W, Meyer R, Herndlhofer S, Füreder T, Dorfner G, Pillay S, André V, Burkholder TP, Akunda JK, Flye-Blakemore L, Van Bockstaele D, Schlenk RF, Sperr WR, Valent P. Dose study of the multikinase inhibitor, LY2457546, in patients with relapsed acute myeloid leukemia to assess safety, pharmacokinetics, and pharmacodynamics. Cancer Manag Res. 2011;3:157–175. doi: 10.2147/CMAR.S19341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang W, Maqsodi B, Ma Y, Bui S, Crawford KL, McMaster GK, Witney F, Luo Y. Direct quantification of gene expression in homogenates of formalin-fixed, paraffin-embedded tissues. Biotechniques. 2006;40:481–486. doi: 10.2144/000112133. [DOI] [PubMed] [Google Scholar]
- 15.Callies S, André V, Patel B, Waters D, Francis P, Burgess M, Lahn M. Integrated analysis of preclinical data to support the design of the first in man study of LY2181308, a second generation antisense oligonucleotide. Br J Clin Pharmacol. 2011;71:416–428. doi: 10.1111/j.1365-2125.2010.03836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheson BD, Bennett JM, Kopecky KJ, Buchner T, Willman CL, Estey EH, Schiffer CA, Doehner H, Tallman MS, Lister TA, LoCocco F, Willemze R, Biondi A, Hiddemann W, Larson RA, Lowenberg B, Sanz MA, Head DR, Ohno Ryuzo, Bloomfield CD. Revised Recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol. 2003;21:4642–4649. doi: 10.1200/JCO.2003.04.036. [DOI] [PubMed] [Google Scholar]
- 17.Shults K, Flye L, Green L, Daly T, Manro JR, Lahn M. Patient-derived acute myeloid leukemia (AML) bone marrow cells display distinct intracellular kinase phosphorylation patterns. Cancer Manag Res. 2009;1:49–59. doi: 10.2147/CMAR.S5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schimmer AD, Estey EH, Borthakur G, Carter BZ, Schiller GJ, Tallman MS, Altman JK, Karp JE, Kassis J, Hedley DW, Brandwein J, Xu W, Mak DH, LaCasse E, Jacob C, Morris SJ, Jolivet J, Andreeff M. Phase I/II trial of AEG35156 X-linked inhibitor of apoptosis protein antisense oligonucleotide combined with idarubicin and cytarabine in patients with relapsed or primary refractory acute myeloid leukemia. J Clin Oncol. 2009;27:4741–4746. doi: 10.1200/JCO.2009.21.8172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saleem A, Matthews JC, Ranson M, Callies S, Andre V, Lahn M, Dickinson C, Prenant C, Brown G, McMahon A, Talbot DC, Jones T, Price PM. Molecular imaging and pharmacokinetic analysis of carbon-11 labeled antisense oligonucleotide LY2181308 in cancer patients. Theranostics. 2011;1:290–301. doi: 10.7150/thno/v01p0290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dean E, Jodrell D, Connolly K, Danson S, Jolivet J, Durkin J, Morris S, Jowle D, Ward T, Cummings J, Dickinson G, Aarons L, Lacasse E, Robson L, Dive C, Ranson M. Phase I trial of AEG35156 administered as a 7-day and 3-day continuous intravenous infusion in patients with advanced refractory cancer. J Clin Oncol. 2009;27:1660–1666. doi: 10.1200/JCO.2008.19.5677. [DOI] [PubMed] [Google Scholar]
- 21.Altieri DC. Survivin and IAP proteins in cell death mechanisms. Biochem J. 2010;430:199–205. doi: 10.1042/BJ20100814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arora V, Cheung HH, Plenchette S, Micali OC, Liston P, Korneluk RG. Degradation of survivin by the XIAP-XAF1 complex. J Biol Chem. 2007;282:26202–26209. doi: 10.1074/jbc.M700776200. [DOI] [PubMed] [Google Scholar]
- 23.Wang H, Holloway MP, Ma L, Cooper ZA, Riolo M, Samkari A, Elenitoba-Johnson KS, Chin YE, Altura RA. Acetylation directs survivin nuclear localization to repress STAT3 oncogenic activity. J Biol Chem. 2010;285:36129–36137. doi: 10.1074/jbc.M110.152777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Redell MS, Ruiz MJ, Alonzo TA, Gerbing RB, Tweardy DJ. Stat3 signaling in acute myeloid leukemia: ligand-dependent and -independent activation and induction of apoptosis by a novel small-molecule Stat3 inhibitor. Blood. 2011;117:5701–5709. doi: 10.1182/blood-2010-04-280123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crooke S. Antisense Drug Technology: principles, strategies, and application. 2nd. CRC Press (Taylor&Francis Group); Boca Raton, FL: 2008. [Google Scholar]
- 26.Flanagan JM, Funes JM, Henderson S, Wild L, Carey N, Boshoff C. Genomics screen in transformed stem cells reveals RNASEH2A, PPAP2C, and ADARB1 as putative anticancer drug targets. Mol Cancer Ther. 2009;8:249–260. doi: 10.1158/1535-7163.MCT-08-0636. [DOI] [PubMed] [Google Scholar]
- 27.Carter BZ, Mak DH, Morris SJ, Borthakur G, Estey E, Byrd AL, Konopleva M, Kantarjian H, Andreeff M. XIAP antisense oligonucleotide (AEG35156) achieves target knockdown and induces apoptosis preferentially in CD34+38- cells in a phase 1/2 study of patients with relapsed/refractory AML. Apoptosis. 2011;16:67–74. doi: 10.1007/s10495-010-0545-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kelly RJ, Lopez-Chavez A, Citrin D, Janik JE, Morris JC. Impacting tumor cell-fate by targeting the inhibitor of apoptosis protein survivin. Mol Cancer. 2011;10:35. doi: 10.1186/1476-4598-10-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhattacharyya J, Mihara K, Ohtsubo M, Yasunaga S, Takei Y, Yanagihara K, Sakai A, Hoshi M, Takihara Y, Kimura A. Overexpression of BMI-1 correlates with drug resistance in B-cell lymphoma cells through the stabilization of survivin expression. Cancer Sci. 2012;103:34–41. doi: 10.1111/j.1349-7006.2011.02121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo L, Tang M, Yang L, Xiao L, Bode AM, Li L, Dong Z, Cao Y. Epstein-Barr virus oncoprotein LMP1 mediates survivin upregulation by p53 contributing to G1/S cell cycle progression in nasopharyngeal carcinoma. Int J Mol Med. 2012;29:574–580. doi: 10.3892/ijmm.2012.889. [DOI] [PMC free article] [PubMed] [Google Scholar]