

Structured Abstract
Objective
To test whether the mucus layer, luminal digestive enzymes, and intestinal mast cells are critical components in the pathogenesis of trauma-shock-induced gut and lung injury.
Summary Background Data
Gut-origin sepsis studies have highlighted the importance of the systemic component (ischemia-repefusion) of gut injury while the intraluminal component is less well studied.
Methods
In rats subjected to trauma-hemorrhagic shock (T/HS) or sham-shock (T/SS), the role of pancreatic enzymes in gut injury was tested by diversion of pancreatic enzymes via pancreatic duct exteriorization (PDE), while the role of the mucus layer was tested via the enteral administration of a mucus surrogate. Additionally the role of mast cells was assessed by measuring mast cell activation and the ability of pharmacologic inhibition of mast cells to abrogate gut and lung injury. Gut and mucus injury was characterized functionally, morphologically and chemically.
Results
PDE abrogated T/HS-induced gut barrier loss and limited chemical mucus changes. The mucus surrogate prevented both T/HS-induced gut and lung injury. Lastly, pancreatic enzyme-induced gut and lung injury appears to involve mast cell activation, since T/HS activates mast cells and pharmacologic inhibition of intestinal mast cells prevented T/HS-induced gut and lung injury.
Conclusions
These results indicate that gut and gut-induced lung injury after T/HS involves a complex process consisting of intraluminal digestive enzymes, the unstirred mucus layer, and a systemic ischemic-reperfusion injury. This suggests the possibility of intraluminal therapeutic strategies.
Introduction
The gut hypothesis of the multiple organ dysfunction syndrome (MODS) has been the subject of intensive investigation for almost three decades and has led to the recognition that systemic insults leading to splanchnic hypoperfusion cause subsequent gut injury, and ultimately result in a gut-induced systemic inflammatory state, acute lung injury, and MODS1. This work has contributed to specific gut-directed therapies such as early enteral nutrition, immunonutrition and selective digestive decontamination2. Mechanistic studies of gut-origin sepsis have highlighted the important role of the downstream intestinal ischemia-repefusion injury that results from decreased mesenteric perfusion as well as the global role of pancreatic digestive enzymes in shock3. However, the roles of many intraluminal non-bacterial factors remains less well researched and understood4,5. We and others have recently focused attention on these non-bacterial, luminal factors that modulate gut ischemia-reperfusion injury with a special emphasis on the protective role of the intestinal mucus layer6–8 and the potentially injurious effects of luminal digestive enzymes5,9,10. These results have led to the hypothesis that T/HS-induced splanchnic hypoperfusion causes oxidative damage to the mucus layer, thereby allowing pancreatic enzymes to reach and autodigest the otherwise protected intestinal mucosa, triggering the gut origin MODS response. However, much work is still required to understand the potential relationships between mucus oxidation, intestinal mucus loss, luminal digestive enzymes and the pathways by which pancreatic enzymes contribute to T/HS-induced gut injury and inflammation. Consequently, the overall goal of this study was to investigate the role of the intraluminal non-bacterial components of the intestine in T/HS-induced gut injury and gut-induced acute lung injury as well as potential interactions between the unstirred mucus layer, pancreatic proteases and mucosal mast cells. The results of this study support the protective role of the mucus layer, indicate that pancreatic proteases contribute to intestinal injury, and suggest an association with mast cell activation and degranulation.
Methods
Animals
Male Sprague-Dawley rats weighing 320 to 400 grams were housed under barrier-sustained conditions, at a temperature of 25°C, with 12-h light/dark cycles and acclimated for at least five days before experimentation. The rats had free access to water and chow (Teklan 22/5 Rodent Diet W-8640; Harlan Teklad; Madison, WI). All rats were maintained in accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals. All animal protocols were approved by the New Jersey Medical School Animal Care Committee.
Experimental Design
Recent studies suggest that T/HS-induced gut injury also involves luminal factors, especially the mucus layer and pancreatic digestive enzymes4–6. In this regard, the mucus layer appears to function as a key protective factor, limiting other factors, especially pancreatic digestive enzymes, from reaching the underlying epithelium and disrupting gut barrier function. The first set of experiments was therefore designed to determine the effect of pancreatic enzymes on the intestinal mucus layer and correlate these changes with gut morphology and barrier function. The goal of the second set of studies was to determine whether high molecular weight polyethelene glycol (HMW-PEG), which can function as a mucus surrogate11, would protect against T/HS-induced gut injury and gut-derived acute lung injury. HMW-PEG has previously demonstrated to adhere to the intestinal wall thereby altering surface electric charges, hydrophobicity, and bacterial/host interactions11. PEG is a known biologically inert material, is both non-toxic and non-immunogenic, and prevents adsorption of bioactive agents12. Furthermore, in prior in-vitro studies specifically evaluating barrier function, HMW PEG was shown to demonstrate a dose dependent increase in endothelial cell barrier function13. Lastly, as mast cells contain protease-activated receptors which could be activated by pancreatic proteases that cross the mucosal barrier, we tested the hypothesis that mast cell inhibition would limit T/HS-induced gut and lung injury. In all groups of animals, these studies were carried out at 3 hours after the end of the trauma hemorrhagic shock (T/HS) or trauma sham shock (T/SS) period, since both gut and lung injury is well established at this point14.
In the first set of studies, rats were divided into four groups. The animals were subjected to either trauma-hemorrhagic shock (T/HS) or trauma sham-shock (T/SS) with half of each group receiving either pancreatic duct ligation and cannulation (PDC) or sham PDC. The goal of PDC was to divert pancreatic enzymes from the intestinal lumen. Since it is known that the large sugar moieties within the mucin molecule act as physiologic free radical scavengers15 and that reactive oxygen and nitrogen species can damage and degrade mucin molecules leading to a loss of mucus viscosity, hydrophobicity and barrier function,15,16 we measured the extent of oxidant stress to the mucus layer as well as its anti-oxidant capacity in one set of animals. Mucus oxidant stress was assayed by measuring carbonyl derivates and nitrated tyrosine residues in the mucus, since these are markers of reactive oxidant and reactive nitrogen reactions17,18. Mucus function was evaluated by measuring the anti-oxidant activity of the mucus. In a second set of animals, in which mucus collection did not occur, gut barrier function was assessed functionally (intestinal permeability studies) as well as morphologically by quantifying villous injury and the extent of mucus loss.
In the second study, we tested the hypothesis that administration of a chemical mucin surrogate would ameliorate gut injury and prevent loss of gut barrier function. As the increase in gut permeability observed following trauma-hemorrhagic shock has been implicated in the development of the acute respiratory distress syndrome, our secondary aim was to determine if the potential gut protective effect of surrogate mucins would attenuate lung injury. Rats were divided into three groups: T/SS, T/HS + vehicle (sterile water), and T/HS + mucin surrogate (high molecular weight PEG). The HMW-PEG (15–20kD) was administered by injection into the duodenum as a 5% solution at a dose of 30ml/kg during the laparotomy as previously described11. Luminal contents were not disrupted or removed during the injection, and this volume of PEG was found in prior studies to be sufficient to reach throughout the duodenum without causing distention. Three hours following resuscitation, at the end of the shock or sham-shock period, gut barrier function was assayed functionally (intestinal permeability to FD4 (fluorescein isothiocyanate-dextran, a fluorescent labeled dextran molecule with an average molecular weight of 5,000 u) and bacterial translocation to mesenteric lymph nodes) as well as histologically by the measurement of villous injury. In a second group of rats, lung injury was measured by permeability to Evans blue dye and BALF leukoctye counts.
In the third study, we tested the hypothesis that T/HS-induced gut injury was associated with mast cell activation and that activated mast cells contribute to T/HS-induced gut and lung injury. Rats were divided into a T/HS group and a T/SS group as previously described. Following trauma shock or sham shock, serum and lymph was collected and analyzed for RMCP (rat mast cell protease) II levels, since RMCP II is a mast cell protease specific to enteric mast cells and is a marker of mast cell degranulation19. Terminal ileum samples were obtained and gut injury (villous damage) and the number of mast cells were measured. In subsequent experiments aimed at further testing the role of mast cell degranulation in T/HS-induced gut and lung injury, we measured both gut and lung injury in rats receiving either the mast cell stabilizer doxantrazole, which affects both mucosal and connective tissue mast cells20, or the mast cell stabilizer ketotifen, which only affects connective tissue mast cells21. This study tested the hypothesis that mucosal, but not the connective tissue, mast cell stabilization would be protective. We selected these two mast cell agents based on prior studies utilizing an LPS model of ischemia-reperfusion gut injury showing that pretreatment with doxantrazole, but not ketotifen, was effective in reducing gut mast cell activation and degranulation22. Doxantrazole (25 mg/kg ip) was administered at 2 and 20 hrs prior to T/HS (T/SS) as was ketotifen (20 mg/kg ip). These doses were based on previously published work19–21. Having demonstrated that doxantrazole was effective in decreasing T/HS-induced gut permeability, we then tested whether the protective effects of mast cell inhibition was associated with mucus layer preservation. In this experiment, rats were pretreated with doxantrazole (as previously described) or a normal saline vehicle and then underwent T/HS. Gut permeability was measured by FD4 permeability and a segment of the terminal ileum was obtained and mucus loss was measured.
Trauma-hemorrhagic shock and pancreatic duct diversion models
Rats were subjected to either T/HS or T/SS as previously described14. Briefly, rats were anesthetized with intraperitoneal sodium pentobarbital (50 mg/kg) and additional intraperitoneal sodium pentobarbital boluses (5 mg/kg) were given as needed throughout the post resuscitation experimental period. A 3 cm midline laparotomy (trauma) was performed, followed by evisceration of the small and large bowel for 15 minutes. In animals undergoing pancreatic duct ligation and exteriorization, the distal biliary-pancreatic duct was identified and cannulated with polyethylene (PE-10) tubing and externalized. The biliary-pancreatic tube was then secured with a 4–0 silk suture. The proximal duct was then ligated distal to the site of cannulation just proximal to its entrance into the duodenum. In all animals, the small and large intestines were then returned to the abdominal cavity and the abdomen was closed with a running 3–0 silk suture. Blood was withdrawn at a rate of 1 mL/minute until the mean arterial pressure reached 30 to 35 mmHg. The mean arterial pressure was maintained at 30 to 35 mmHg for 90 minutes by withdrawing or reinfusing the shed blood. At the end of the shock period, the animals were resuscitated by reinfusion of all the shed blood at a rate of 1 mL/min as previously described14. The mean arterial pressure returned to the normal level within a few minutes after the animals were resuscitated and remained at that level for the duration of the experiment. In the T/SS group, animals underwent cannulation of vessels and laparotomy but were not subjected to T/HS.
Collection of mucus sample and measurement of protein concentration
The distal 30 cm segment of the terminal ileum was identified and transected from the intestinal tract and the underlying mesentery. The intestinal mucus from this sample was collected and processed as previously described6. The concentration of protein in the mucus samples were measured by the Thermo Scientific Pierce BCA (bicinchoninic acid) Assay (Thermo Fisher Scientific; Rockford, IL) according the manufacturer’s instructions. Protein concentrations are expressed as μg/ml.
Mucus structural changes assays
Nitration of tyrosine residues, a marker for (RNI)-mediated injury, was measured with a competitive ELISA (Nitrotyrosine ELISA; Millipore; Billerica, MA and 3-Nitrotyrosine ELISA; Abcam; Cambridge, MA) as previously described6. Nitrated tyrosine residues are expressed as nitro-BSA equivalents23. Carbonyl derivatives, a marker of ROS-mediated damage, were quantified by ELISA (OxiSelect Protein Carbonyl ELISA; Cell Biolabs; San Diego, CA) as previously described6. ROS-mediated damage was expressed as protein carbonyl nmol/mg.
Measurement of total antioxidant capacity
One measure of mucus function is its ability to function as an antioxidant14, thus mucus sample antioxidant capacity was measured via ELISA (Total Antioxidant Capacity Assay; Abcam; Cambridge, MA) as previously described6. Total antioxidant capacity is expressed as trolox equivalents (per nmol), the antioxidant capacity of 1 nmol of trolox, which is 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid, a standard measurement for antioxidant capacity24.
Villous injury and mucus coverage
After sacrifice, a 5 cm segment of the terminal ileum was collected for histologic studies. In order to visualize the mucus layer, the mucosal surface was then covered with 3% Alcian blue followed by fixation in Carnoy’s solution for two hours25. After processing, five, random fields containing 100–250 villi per animal were analyzed in a blinded fashion to quantitate morphologic villous damage and ileal mucus coverage is expressed as percent of the villi that were not covered with mucus as previously reported25.
In-vivo gut barrier permeability
Ileal permeability was measured in-vivo using the 4-kD dextran permeability probe FD-4 (fluorescein isothiocyanate-dextran, Sigma, St. Louis, Mo), a fluorescent labeled dextran molecule with an average molecular weight of 5,000 u as previously reported and was expressed as the amount of FD-4 found in systemic plasma in nanograms per milliliter14.
Bacterial Translocation
The mesenteric lymph node (MLN) complex was harvested and the level of translocating bacterial quantified as previously described26.
Lung Permeability and BALF leukocyte counts
Lung permeability was assessed by permeability to Evans blue dye as previously described27.
BALF volumes were noted and cell pellets were resuspended in fixed volume (100μL) of PBS. WBC counts were performed in resuspended cell pellets using an automated cell counter (Hematrue; Heska Corporation; Loveland, CO).
Lung injury score
Three hours following trauma-hemorrhagic shock or sham-shock, lungs were harvested by median sternotomy and immediately fixed in 10% buffered formalin solution. After fixation, lung tissue samples were dehydrated, embedded in paraffin blocks, cut to 4μm thickness and stained with hematoxylin-eosin. The slides were read under a standard light microscope, lung injury was scored by the histologic grading scale described by Claridge28, and the total number of neutrophils were counted. 30 random fields in each sample were analyzed in a blinded fashion.
Mucosal mast cell identification
Immunodetection of RMCP II was carried out on 10% buffered formalin-fixed section using a monoclonal antibody (Moredun Anima Health, Edinburgh, UK). Detection was performed with avidin/peroxidase (Vectastain ABC kit, Vector Lab., Burlingame, CA). Sections were counterstained with haematoxylin. Positively stained mast cells were counted and expressed as numbers in 100 villi.
Measurement of RMCP II levels
Serum samples were taken in all rats at 3 hours after resuscitation or sham resuscitation. RMCP II concentration in the serum and lymph was measured by enzyme-linked immunosorbent assay (ELISA) using commercial kits (Moredun Anima Health, Edinburgh, UK).
Statistical analysis: the results are expressed as mean ± SEM. Statistical analysis was conducted using Prism-4 software (Graphpad, USA). Unpaired student’s t-tests and ANOVA were used for single and group comparisons respectively. For data sets compared using ANOVA, the post-hoc Newman-Keuls method was used to determine significance between groups. The incidence of bacterial translocation was assessed by Fisher’s exact test. Statistical significance was considered achieved at p<0.05.
Results
Pancreatic duct cannulation with exclusion
T/HS was associated with an increase in gut injury as reflected by an increase in gut permeability and villous injury as compared to the T/SS groups (Figure 1A, 1B). In addition, the percent of villi covered with mucus was reduced in T/HS as compared to T/SS rats (Figure 1C). In contrast to the T/HS rats, the T/HS rats with PDC were protected and levels of gut permeability, villous injury and mucus coverage of the villi were similar to that of the T/SS groups (Figure 1).
Figure 1.

A) Pancreatic duct ligation (PDC) abrogates this ability of T/HS to increase gut permeability to intraluminally placed FD-4. B) PDC reduces the incidence of T/HS-induced villous injury and C) decreases the percentage of intestinal villi not covered with mucus. Data expressed as mean ± SEM with 6 animals per group. *p<0.05 vs. all other groups.
Since T/HS-induced mucus layer loss is related to an I/R-mediated oxidant injury6, and PDC limits T/HS-induced mucus loss, we tested the extent to which PDC would limit T/HS-induced mucus layer oxidation, nitration, and preservation of mucus anti-oxidant capacity. T/HS was associated with increased mucus oxidant injury as reflected in increased mucus protein carbonyl levels, while PDC was able to abrogate but not prevent mucus oxidation (Figure 2A). A similar pattern was observed for mucus protein nitration, with PDC abrogating but not totally preventing T/HS-induced mucus protein nitration (Figure 2B). Likewise, PDC was able to partially limit the loss of mucus anti-oxidant activity observed after T/HS (Figure 2C).
Figure 2.

T/HS induces structural and functional changes to the unstirred mucus layer. A) T/HS oxidatively modifies the unstirred mucus layer and this increase was partially prevented by pancreatic duct ligation. B) T/HS-induced RNI-mediated structural changes to the mucus layer was partially abrogated by PDC. C) T/HS induced a functional change in the mucus layer as evidenced by a decrease in the total antioxidant capacity, which was prevented by PDC. Data expressed as mean ± SEM with 6 animals per group. *p<0.01 vs. all other groups. ** p<0.05 vs. sham shock groups.
Administration of HMW-PEG
Since the mucus layer appears to function as a key protective barrier that limits T/HS-induced gut injury and subsequent lung injury4, we next tested whether the administration of HMW-PEG, which can function as a mucus surrogate11, would limit T/HS-induced gut and/or lung injury. As shown in Figure 3, the intraluminal administration of HW-PEG prevented the increase in gut permeability observed after T/HS as well as T/HS-induced morphologic villous injury and bacterial translocation. Since the bacterial intestinal population levels are an important variable in bacterial translocation, we measured the effect of HMW-PEG on cecal bacterial population levels and found that HMW-PEG’s ability to decrease bacterial translocation was not associated with changes in cecal bacterial population levels (Figure 3D). The administration of HMW-PEG was also able to prevent the T/HS-induced increase in lung permeability and bronchoalveolar WBC count (Figure 4). Based on in-vitro tests showing that HMW-PEG did not decrease trypsin activity, this protective effect of HMW-PEG on gut injury does not appear to be due its ability to interfere with enzymatic activities of luminal trypsin (data not shown).
Figure 3.

A) The addition of the mucus surrogate high-molecular weight polyethylene glycol (HMW-PEG) abrogated T/HS-induced increases in gut permeability and B) reduced morphologic evidence of illous injury. C) Following administration of HMW-PEG, bacterial translocation was reduced to sham values. D) There was no difference noted between cecal bacterial population levels amongst the groups. Data expressed as mean ± SEM with 7–8 animals per group. *p<0.001 vs. all other groups, **p<0.05 vs. T/SS groups.
Figure 4.
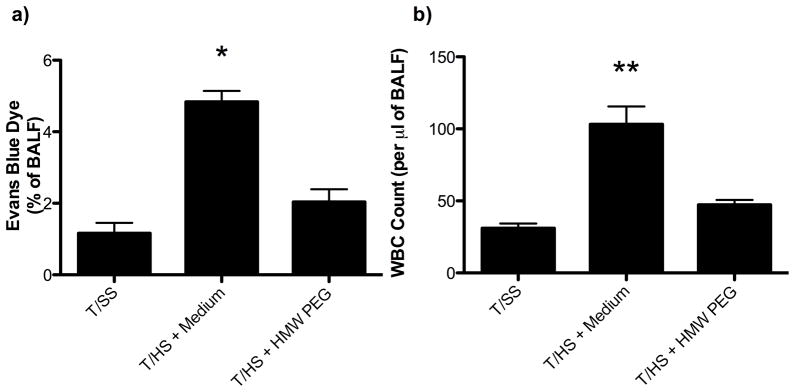
A) The T/HS-induced increase in the percentage of Evans blue dye leak was returned to sham levels following administration of HMW-PEG. B) The T/HS-induced increased number of WBCs seen in BALF fluid was returned to sham levels following administration of HMW-PEG. Data are expressed as mean ± SEM with 7–8 animals per group. *p<0.001 vs. all other groups, **p<0.01 vs. all other groups.
Intestinal mast cell activation
T/HS was associated with a 4-fold increase in intestinal mast cell numbers (Figure 5A, 5B) as well as an increase in serum and lymph rat mast cell protease II (Figure 5C, 5D). This indicates that T/HS is associated with mast cell activation three hours following reperfusion. Based on these results, we next tested and found that the mucosal mast cell inhibitor, doxantrazole, protected against T/HS-induced increased gut permeability and lung injury (Figure 6A, 6B). In contrast to doxantrazole, the connective tissue, non-mucosal mast cell stabilizer, ketotifen did not protect against T/HS-induced increased gut permeability or lung permeability (Figure 6C, 6D). While doxantrazole prevented increased T/HS-induced gut permeability, it did not prevent T/HS-induced loss of the intestinal mucus layer (Figure 7A, 7B). This observation suggests thats mucus loss is not dependent on mast cell activation.
Figure 5.

A) Three hours following T/HS, the number of intestinal mast cells were increased. B) Representative photomicrographs (100x) illustrating the increase in mast cell number (brown cells) in the T/HS as compared to T/SS rats. C) RMCP-II levels were increased in the serum and D) lymph following T/HS indicating mast cell activation. Data are expressed as mean ± SEM with 6 animals per group. *p<0.001 vs. trauma sham shock (T/SS), ** p<0.01 vs. trauma sham shock (T/SS).)
Figure 6.

Administration of the intestinal specific mast cell degranulation inhibitor, doxantrazole, decreasesd gut (A) and lung (B) injury following T/HS. Data are expressed as mean ± SEM with 6–8 animals per group. In contrast, the administration of the connective tissue mast cell degranulation inhibitor, ketotifen, did not decrease gut (C) or lung (D) injury following T/HS. Data are expressed as mean ± SEM with 5–6 animals per group. *p<0.05 vs. all other groups.
Figure 7.

Administration of the intestinal specific mast cell degranulation inhibitor doxantrazole decreased gut permeability (7A) following T/HS, but did not alter the extent of mucus loss (7B). Data are expressed as mean ± SEM with 5–6 animals per group. *p<0.05 vs. T/HS + vehicle.
Discussion
The ischemia-reperfusion paradigm has helped clarify the mechanisms through which shock states, including hypoperfusion, are transduced into tissue injury and systemic inflammation. During times of stress or shock, blood is preferentially shunted from the splanchnic circulation into the central circulation to sustain perfusion of vital organs29, this makes the gastrointestinal tract quite susceptible to I/R induced-injury. However, unlike most organs, the gut serves as a barrier to the potentially hostile external environment. Not only does the lumen of the gut contain a large number of bacteria and bacterial products, but it also contains high concentrations of potentially injurious digestive enzymes. Thus, the normal intestine must be capable of digesting and absorbing nutrients while simultaneously preventing the escape and systemic spread of intestinal bacteria, bacterial products, and digestive enzymes. An important, yet relatively under appreciated, component of the gut barrier is the mucus layer. The mucus layer facilitates absorption of nutrients and other molecules, while at the same time, it prevents bacteria, their products as well as digestive enzymes from coming into direct contact with the underlying enterocytes4 where they could induce injury and inflammation. In fact, basic pharmacologic and physiologic studies of the intestinal barrier have documented that the mucus layer is more important in preventing the escape of luminal factors from the gut than the tight junctions between enterocytes5,30. Consequently, we previously tested the hypothesis that the loss of the mucus layer significantly contributes to gut barrier failure in conditions associated with splanchnic hypoperfusion and ischemia-reperfusion-mediated gut injuries. These prior studies demonstrate that mucus layer loss was associated with increased gut permeability and that anatomic sites of mucus disruption directly correlate with areas of morphologic gut injury7,8,14,25. Subsequent studies indicate that the T/HS-induced gut ischemia-reperfusion insult leads to oxidative modification and extrusion of the mucus layer into the intestinal lumen, further contributing to loss of gut barrier function6. Early pioneering studies performed by Schmid-Schonbein’s laboratory5,31,32 and the subsequent work by other groups, including our own9–10, illustrate the important role that intraluminal pancreatic digestive enzymes play in the pathogenesis of shock-induced gut injury and the MODS response. The culmination of such studies led Schmid-Schonbein to propose the autodigestion hypothesis, which postulates that the products of pancreatic enzymatic digestion, rather than the enzymes themselves, lead to systemic inflammation and organ injury33. Consequently, the goal of the current work was to integrate these two lines of work and test the basic hypothesis that oxidative injury to the mucus layer during ischemia-reperfusion is a key initiating factor in mucosal barrier disruption that allows digestive enzymes access into the intestinal wall thereby initiating an autodigestive response.
Our results documenting that diversion of pancreatic enzymes from the intestinal lumen by PDC prevents T/HS-induced increases in gut permeability and intestinal villous injury validates earlier studies10. However, the results from this set of experiments also show that pancreatic enzyme diversion only partially protected against T/HS-induced oxidative and physiologic modifications of the mucus layer. These results have several implications. First, PDC is able to completely prevent gut injury and mucus loss despite the presence of oxidative and nitrosylated modifications to the mucus layer. Second, an intestinal ischemia-reperfusion insult is directly responsible for the chemical changes observed in the mucus layer following T/HS. Third, the T/HS-induced systemic ischemia-reperfusion injury to the gut is not sufficient to induce demonstrable gut injury and dysfunction in the absence of intraluminal pancreatic proteases. The potential explanation of the synergistic effects of a systemic ischemia-reperfusion injury and the presence of luminal digestive enzymes in the pathogenesis of T/HS-induced gut injury will need further study. The pancreatic proteases already present in the intestine of the PDL-treated rats subjected to T/HS do not appear to be sufficient by themselves to cause gut injury. The reason for this observation is unclear but may represent a dose-response effect, where a certain minimal amount of pancreatic proteases need to be present during and after the shock state to injure the mucosa. In the absence of additional secretion of pancreatic proteases in the pancreatic duct ligated animals, the gut luminal level of pancreatic proteases might not have reached sufficiently high concentrations to contribute to gut injury. It is also known that reactive oxygen and nitrogen species damage and degrade mucin molecules resulting in a loss of viscosity and a decrease in hydrophobicity15,34. The oxidant-mediated damage to the mucus layer not only compromises the gut barrier, but increases the susceptibility of the mucins to pancreatic protease cleavage35,36. As a result of these modifications, mucins are less able to prevent translocation of digestive enzymes into the underlying mucosa37. Consequently, it appears that the pathogenesis of T/HS-induced gut injury is a multi-hit phenomenon whereby the initial insult is an intestinal ischemia-reperfusion insult to the mucus layer6 and the second hit is the intraluminal pancreatic proteases reaching and crossing the layer of enterocytes lining the villi38.
The protective effect of the mucus layer was further validated by our observation that the enteral administration of HMW-PEG was able to limit T/HS-induced gut injury and loss of barrier function. PEG with a molecular weight of 15–20kD was tested, since it has been previously shown to act as a mucus surrogate and to be protective in a lethal gut-origin sepsis model while low molecular weight PEG (3–5kD) found in golytely was not effective11, 13. While the mechanisms by which HMW-PEG limited gut injury, bacterial translocation and increased gut permeability following T/HS remains to be determined, certain facts about HMW-PEG are known that could have contributed to its protective effects. First, HMW-PEG has been shown to adhere to enterocytes as globules and decrease bacterial contact and internalization by enterocytes39. Second, HMW-PEG can directly bind to bacteria and further decrease their ability to reach and translocate across the gut barrier11. Although not explicitly tested, it appears that the ability of HMW-PEG to bind luminal bacteria was likely not responsible for the protective effects seen in the current studies as the intestinal bacterial population levels were similar in both arms. Third, HMW-PEG has been shown to up-regulate various components of the barrier-enhancing signal transduction pathway13. Furthermore, by modulating the intraluminal compartment, HMW-PEG was also efficacious in preventing T/HS-induced acute lung injury. This suggests that the strategy of limiting acute gut injury after T/HS by the oral administration of a mucus surrogate also has potential systemic and clinical benefits. Since these results are consistent with a large body of evidence on gut-origin sepsis1, they are thus not surprising.
Our results also highlight the important role that mast cell activation and degranulation play in T/HS-induced gut injury. T/HS was associated with mast cell degranulation and the release of the mucosal mast cell specific tryptase RMCP-II. Furthermore, the observation that the mast cell stabilizer doxantrazole, but not ketotifen, prevented both T/HS-induced gut and lung injury has physiologic implications since doxantrazole’s major action is on both mucosal and connective tissue mast cells20–21 while ketotifen’s actions affect only connective tissue mast cells20. Thus, the differential effects of these two mast cell stabilizers are important in implicating intestinal mucosal mast cells as being active participants in T/HS-induced gut injury and gut-induced MODS. Taken together our results complement previous studies documenting that chemical mast cell stabilizers limit gut injury in isolated gut ischemia-reperfusion and in LPS models of gut injury and that mast cell-deficient rats and mice are relatively resistant to ischemia-reperfusion-induced gut injury and the downstream SIRS response20,21,40, 41. Furthermore, mucosal mast cell activation under conditions associated with gut barrier failure is consistent with their position at host-environment interfaces where they can serve as first responders to invading pathogens and act as cell stress signals. While additional research is needed to further investigate the protective effect of gut specific mast cell inhibition in the pathogenesis of gut mediated sepsis, this study clearly documents an association between T/HS-mediated I/R intestinal injury and mast cell activation.
In conclusion, we hypothesize that T/HS, or other conditions associated with splanchnic hypoperfusion, induces an intestinal ischemia-reperfusion injury that results in a mild enterocyte injury and an oxidant-mediated injury to the intestinal mucus layer. Although the ischemia-reperfusion injury is not independently sufficient to completely disrupt the gut barrier, the chemically-modified mucus is sufficiently modified to allow further mucin degradation by intraluminal, water soluble pancreatic proteases. This allows the pancreatic proteases, and other luminal factors (such as bacteria and bacterial products), to cross the enterocyte barrier, reach the submucosal space, and initiate an injurious response that is likely mediated, in part, by mucosal mast cells. Thus, translocated luminal factors appear to exacerbate the magnitude of gut injury and contribute to the development of MODS.
Footnotes
Disclosure: Supported by NIH grants GM 59841 (EAD) and T32 069330 (VA, SUS)
References
- 1.Deitch EA. Gut-origin sepsis: evolution of a concept. Surgeon. 2012;10:350–356. doi: 10.1016/j.surge.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deitch EA, Fishman JE, Levy G. Microbial translocation, gut origin sepsis, probiotics, prebiotics, selective gut decontamination. In: Fry D, editor. Surgical Infections. 2. London, UK: JP Medical Ltd; 2013. pp. 261–274. [Google Scholar]
- 3.DeLano FA, Hoyt DB, Schmid-Schonbein GW. Pancreatic digestive enzyme blockade in the intestine increases survival after experimental shock. Sci Transl Med. 2013;5:169ra11. doi: 10.1126/scitranslmed.3005046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharpe SM, Doucet DR, Qin X, et al. Role of intestinal mucus and pancreatic proteases in the pathogenesis of trauma-hemorrhagic shock-induced gut barrier failure and multiple organ dysfunction syndrome. J Organ Dysfunct. 2008;4:168–176. [Google Scholar]
- 5.Mitsuoka H, Kistler EB, Schmid-Schonbein GW. Generation of in vivo activating factors in the ischemic intestine by pancreatic enzymes. Proc Natl Acad Sci U S A. 2000;97:1772–1777. doi: 10.1073/pnas.97.4.1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fishman JE, Levy G, Alli V, et al. Oxidative modification of the intestinal mucus layer is a critical but unrecognized component of trauma hemorrhagic shock-induced gut barrier failure. Am J Physiol Gastrointest Liver Physiol. 2013;304:G57–G63. doi: 10.1152/ajpgi.00170.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qin X, Sheth SU, Sharpe SM, et al. The mucus layer is critical in protecting against ischemia-reperfusion-mediated gut injury and in the restitution of gut barrier function. Shock. 2011;35:275–281. doi: 10.1097/SHK.0b013e3181f6aaf1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sharpe SM, Qin X, Lu Q, et al. Loss of the intestinal mucus layer in the normal rat causes gut injury but not toxic mesenteric lymph nor lung injury. Shock. 2010;34:475–481. doi: 10.1097/SHK.0b013e3181dc3ff5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deitch EA, Shi HP, Lu Q, et al. Serine proteases are involved in the pathogenesis of trauma-hemorrhagic shock-induced gut and lung injury. Shock. 2003;19:452–456. doi: 10.1097/01.shk.0000048899.46342.f6. [DOI] [PubMed] [Google Scholar]
- 10.Caputo FJ, Rupani B, Watkins AC, et al. Pancreatic duct ligation abrogates the trauma hemorrhage-induced gut barrier failure and the subsequent production of biologically active intestinal lymph. Shock. 2007;28:441–446. doi: 10.1097/shk.0b013e31804858f2. [DOI] [PubMed] [Google Scholar]
- 11.Wu L, Zaborina O, Zaborin A, et al. High-molecular-weight polyethylene glycol prevents lethal sepsis due to intestinal Pseudomonas aeruginosa. Gastroenterology. 2004;126:488–498. doi: 10.1053/j.gastro.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 12.Sheth SR, Leckband D. Measurements of attractive forces between proteins and end-grafted poly(ethylene glycol) chains. Proc Natl Acad Sci U S A. 1997;94:8399–404. doi: 10.1073/pnas.94.16.8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chiang ET, Camp SM, Dudek SM, et al. Protective effects of high-molecular weight polyethylene glycol (PEG) in human lung endothelial cell barrier regulation: role of actin cytoskeleton rearrangement. Microvasc Res. 2009;77:174–186. doi: 10.1016/j.mvr.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rupani B, Caputo FJ, Watkins AC, et al. Relationship between disruption of the unstirred mucus layer and intestinal restitution in loss of gut barrier function after trauma hemorrhagic shock. Sugery. 2007;141:481–489. doi: 10.1016/j.surg.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 15.Cross CE, Halliwell B, Allen A. Antioxidant protection: a function of tracheobronchial and gastrointestinal mucus. Lancet. 1984;8390:1328–1330. doi: 10.1016/s0140-6736(84)91822-1. [DOI] [PubMed] [Google Scholar]
- 16.Brownlee IA, Knight J, Dettmar PW, et al. Action of reactive oxygen species on colonic mucus secretions. Free Radic Biol Med. 2007;43:800–808. doi: 10.1016/j.freeradbiomed.2007.05.023. [DOI] [PubMed] [Google Scholar]
- 17.Stadtman ER, Levine RL. Protein oxidation. Ann N Y Acad Sci. 2000;899:191–208. doi: 10.1111/j.1749-6632.2000.tb06187.x. [DOI] [PubMed] [Google Scholar]
- 18.Rubbo H, Radi R. Protein and lipid nitration: role in redox signaling and injury. Biochim Biophys Acta. 2008;1780:1318–1324. doi: 10.1016/j.bbagen.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 19.Moriez R, Leveque M, Salvador-Cartier C, et al. Mucosal mast cell proteases are involved in colonic permeability alterations and subsequent bacterial translocation in endotoxemic rats. Shock. 2007;28:118–124. doi: 10.1097/SHK.0b013e3180315ba9. [DOI] [PubMed] [Google Scholar]
- 20.Kanwar S, Wallace JL, Befus D, et al. Nitric oxide synthesis inhibition increases epithelial permeability via mast cells. Am J Physiol. 1994;266:G222–G229. doi: 10.1152/ajpgi.1994.266.2.G222. [DOI] [PubMed] [Google Scholar]
- 21.Kanwar S, Kubes P. Mast cells contribute to ischemia-reperfusion-induced granulocyte infiltration and intestinal dysfunction. Am J Physiol. 1994;267:G316–G321. doi: 10.1152/ajpgi.1994.267.2.G316. [DOI] [PubMed] [Google Scholar]
- 22.Brown JF, Chafee KA, Tepperman BL. Role of mast cells, neutrophils and nitric oxide in endotoxin-induced damage to the neonatal rat colon. Br J Pharmacol. 1998;123:31–38. doi: 10.1038/sj.bjp.0701576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuhn DM, Sakowski SA, Sadidi M, et al. Nitrotyrosine as a marker for peroxynitrite-induced neurotoxicity: the beginning or the end of the end of dopamine neurons? J Neurochem. 2004;89:529–536. doi: 10.1111/j.1471-4159.2004.02346.x. [DOI] [PubMed] [Google Scholar]
- 24.Prior RL, Wu X, Schaich K. Standardized methods for the determination of antioxidant capacity and phenolics in food and dietary supplements. J Agric Food Chem. 2005;53:4290–4302. doi: 10.1021/jf0502698. [DOI] [PubMed] [Google Scholar]
- 25.Lu Q, Xu DZ, Sharpe S, et al. The anatomic sites of disruption of the mucus layer directly correlate with areas of trauma/hemorrhagic shock-induced gut injury. J Trauma. 2011;70:630–635. doi: 10.1097/TA.0b013e3181e1221b. [DOI] [PubMed] [Google Scholar]
- 26.Deitch EA, Bridges W, Berg R, et al. Hemorrhagic shock-induced bacterial translocation: the role of neutrophils and hydroxyl radicals. J Trauma. 1990;30:942–951. doi: 10.1097/00005373-199008000-00002. [DOI] [PubMed] [Google Scholar]
- 27.Levy G, Fishman JE, Xu DZ, et al. Vagal nerve stimulation modulates gut injury and lung permeability in trauma-hemorrhagic shock. J Trauma Acute Care Surg. 2012;73:338–342. doi: 10.1097/TA.0b013e31825debd3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Claridge JA, Enelow RI, Young JS. Hemorrhage and resuscitation induce delayed inflammation and pulmonary dysfunction in mice. J Surg Res. 2000;92:206–213. doi: 10.1006/jsre.2000.5899. [DOI] [PubMed] [Google Scholar]
- 29.Ceppa EP, Fuh KC, Bulkley GB. Mesenteric hemodynamic response to circulatory shock. Curr Opin Crit Care. 2003;9:127–32. doi: 10.1097/00075198-200304000-00008. [DOI] [PubMed] [Google Scholar]
- 30.Bounous G, McArdle AH, Hodges DM, et al. Biosynthesis of intestinal mucin in shock: relationship to tryptic hemorrhagic enteritis and permeability to curare. Ann Surg. 1966;164:13–22. doi: 10.1097/00000658-196607000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kistler EB, Hugli TE, Schmid-Schonbein GW. The pancreas as a source of cardiovascular cell activating factors. Microcirculation. 2000;7:183–192. [PubMed] [Google Scholar]
- 32.Schmid-Schonbein GW, Hugli TE. A new hypothesis for microvascular inflammation in shock and multiorgan failure: self-digestion by pancreatic enzymes. Microcirculation. 2005;12:71–82. doi: 10.1080/10739680590896009. [DOI] [PubMed] [Google Scholar]
- 33.Schmid-Schonbein GW. Inflammation and the autodigestion hypothesis. Microcirculation. 2009;16:289–306. doi: 10.1080/10739680902801949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grisham MB, Von Ritter C, Smith BF, et al. Interaction between oxygen radicals and gastric mucin. Am J Physiol. 1987;253:G93–96. doi: 10.1152/ajpgi.1987.253.1.G93. [DOI] [PubMed] [Google Scholar]
- 35.Kemper AC, Specian RD. Rat small intestinal mucins: a quantitative analysis. Anat Rec. 1991;229:219–226. doi: 10.1002/ar.1092290209. [DOI] [PubMed] [Google Scholar]
- 36.Forstner JF. Intestinal mucins in health and disease. Digestion. 1978;17:234–263. doi: 10.1159/000198115. [DOI] [PubMed] [Google Scholar]
- 37.Chang M, Alsaigh T, Kistler EB, et al. Breakdown of mucin as barrier to digestive enzymes in the ischemic rat small intestine. PLoS One. 2012;7:e40087. doi: 10.1371/journal.pone.0040087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang M, Kistler EB, Schmid-Schonbein GW. Disruption of the mucosal barrier during gut ischemia allows entry of digestive enzymes into the intestinal wall. Shock. 2012;37:297–305. doi: 10.1097/SHK.0b013e318240b59b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Henry-Stanley MJ, Wells CL. Polyethylene glycol influences microbial interactions with intestinal epithelium. Shock. 2009;31:390–396. doi: 10.1097/SHK.0b013e31818348a5. [DOI] [PubMed] [Google Scholar]
- 40.Andoh A, Fujiyama Y, Araki Y, et al. Role of complement activation and mast cell degranulation in the pathogenesis of rapid intestinal ischemia/reperfusion injury in rats. Digestion. 2001;63 (Suppl 1):103–107. doi: 10.1159/000051920. [DOI] [PubMed] [Google Scholar]
- 41.Cai C, Cao Z, Loughran PA, et al. Mast cells play a critical role in the systemic inflammatory response and end-organ injury resulting from trauma. J Am Coll Surg. 2011;213:604–615. doi: 10.1016/j.jamcollsurg.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]