

Abstract
Decreased levels of cell cycle inhibitor p27Kip1 due to excessive degradation occur in a variety of aggressive human tumors. Since reduced p27Kip1 expression has been associated with a poor prognosis in many human cancers and resistance to certain antitumor therapies, elevation of p27Kip1 expression could improve prognosis and prevent excessive cell proliferation. SCFSkp2 is one of the major ubiquitin E3 ligases responsible for degradation of p27Kip1. Ubiquitination of p27Kip1 also requires a small adaptor protein, Cks1, which facilitates substrate recruitment by bridging the interaction between Skp2 and p27Kip1. It has been shown previously that a direct interaction between Cks1 and Skp2 is required for p27Kip1 degradation. Accordingly, perturbation of the Skp2-Cks1 interaction may represent an attractive target for pharmacological intervention. Here we describe a high-throughput AlphaScreen assay for discovering small-molecule inhibitors of the Skp2-Cks1 protein-protein interaction in vitro. Two compounds (NSC689857 and NSC681152) were identified and validated through a structure-activity relationship analysis. Both compounds were also shown to inhibit p27Kip1 ubiquitination in vitro. These studies demonstrate that disruption of the Skp2-Cks1 interaction provides a viable strategy to prevent p27Kip1 ubiquitination and may potentially be useful for the control of excessive degradation of this cell cycle inhibitor in tumor cells.
Keywords: E3 ligase, inhibitor, Skp2, Cks1, p27kip1, ubiquitin, proteolysis
Introduction
Eukaryotic cell proliferation requires sequential activation of cyclin-dependent kinases (CDKs).1 The activity of these enzymes is regulated positively and negatively by transcriptional, translational, and posttranslational mechanisms. CDKs are positively regulated by cyclins and negatively regulated by CDK inhibitory proteins (CKIs).1,2 There are at least two families of these small inhibitory proteins—namely, the p16 and the p21 families.1,2 Members of the p16 family, including p16INK4a, p15INK4b, p18INK4c, and p19INK4d, target CDK4 and CDK6 to prevent the association of the CDKs with cyclin D. Members of the p21 family of inhibitors (p21Cip, p27Kip1, and p57Kip2) bind to preassembled CDK/cyclin complexes and inhibit their activities. p27Kip1 (hereafter referred to as p27) is an inhibitor of both CDK2– cyclin E and CDK2–cyclin A, the activities of which are required for the G1 to S transition and triggering DNA replication.1,2 As cells enter the cell cycle and S phase, p27 levels must be decreased to allow Cdk activation. The rise and fall of p27 is primarily controlled by the ubiquitin-proteasome pathway.
Two ubiquitin E3 ligases are known to regulate p27 ubiquitination in vitro and in vivo.2 SCFSkp2 is the major ubiquitin E3 ligase that recognizes T187-phosphorylated p27 and targets it for destruction.2 SCF complexes are multiprotein machinery consisting of four proteins: the ring domain protein Rbx1 (or Roc1), Cullin-1, Skp1, and an F-box–containing substrate receptor.3 The p27 ubiquitination reaction has been fully reconstituted in vitro.4 Unlike other SCF substrates, p27 ubiquitination requires the accessory protein Cks1 for its recognition and ubiquitination by the SCFSkp2 E3 ligase.4–7 In addition, physical contact between the CDK2/cyclin E or the CDK2/cyclin A complex is also required for p27 ubiquitination.8,9 Recognition of p27 by the SCFSkp2 complex requires phosphorylation of p27 at T187 by either CDK2/cyclin E or CDK2/cyclin A (Fig. 1B). Cks1 functions as an adaptor protein that binds the CDK/cyclin complex, phosphorylated p27, and Skp2.6,7,10 Therefore, the complex consisting of phospho-p27/Cks1/CDK2/cyclin E can be considered the substrate for SCFSkp2.
Figure 1.
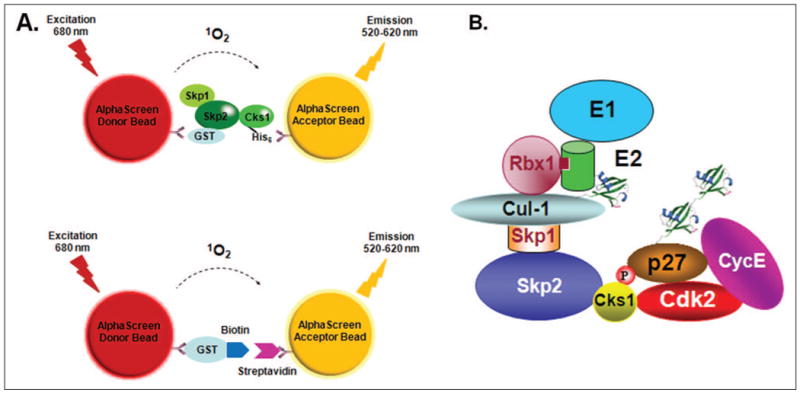
(A) Format of the AlphaScreen assay to detect Skp2-Cks1 interactions. Binding of the glutathione S-transferase (GST)– Skp2/Skp2 complex and His6-tagged Cks1 brings the beads together by immobilization to GST-coated donor beads and nickel-chelated acceptor beads, respectively. When excited at 680 nm, donor beads generate singlet oxygens, which diffuse to the acceptor beads, creating a chemiluminescent reaction that is measured as an AlphaScreen signal. The lower panel shows the format of the counterscreen AlphaScreen assay to eliminate false-positive hits. In this system, donor beads conjugated to a GST-biotin peptide served as a binding partner for streptavidin-conjugated acceptor beads. (B) A schematic of the SCFSkp2-dependent p27 ubiquitination pathway in vitro.
Decreased levels of cell cycle inhibitor p27Kip1 due to excessive degradation occur in a variety of aggressive human tumors such that p27 is an independent prognostic marker in many types of human cancers.11–13 Low levels of p27 correlate with aggressive, high-grade tumors and a poor prognosis.11,14 Ectopic reexpression of p27 in brain, lung, or breast tumor cells often induces apoptosis,15,16 suggesting that elevating p27 expression levels may inhibit tumor growth and thus present a possible effective antitumor therapy. Because SCFSkp2 is largely responsible for excessive p27 proteolysis, perturbation of the Skp2-Cks1 interaction represents an attractive target for pharmacological intervention.
Here we describe the development, optimization, and implementation of a high-throughput screening (HTS) assay based on PerkinElmer (Waltham, MA) AlphaScreen technology to discover small organic molecules that disrupt interactions between Skp2 and Cks1. We demonstrate that the assay we developed is highly specific and can be automated in a 384-well format. Using this assay, we have discovered two compounds (NSC689857 and NSC681152) that perturb the Skp2-Cks1 interaction in vitro and stabilize green fluorescent protein (GFP)–p27 in cells. Our studies provide an experimental framework to systematically discover small-molecule inhibitors that can prevent excessive proteolysis of p27 in tumor cells.
Materials and Methods
Expression and Purification of GST-Skp2/Skp1 and Cks1 in Escherichia coli
A bicistronic pGEX vector containing Skp2 (lacking the N-terminal 100 residues) and Skp1 (lacking residues 38–43 and 71–82) was generously provided by Dr. Bing Hao (University of Connecticut). pGEX-4T-1 vector containing glutathione S-transferase (GST)–Thrombin-His6-TEV-Cks1 was used to purify recombinant Cks1.7,17 A detailed procedure for purifying GST-Skp2/Skp1 and 6xHis-Cks1 or untagged Cks1 can be found in the supplementary methods.
In Vitro Binding Assay of GST-Skp2/Skp1 and His6-Cks1
In total, 3 μg GST-Skp2/Skp1 or GST-Skp2(D331A)/Skp1 or GST alone was incubated with 3 μg His6-Cks1 in NETN buffer (150 mM NaCl, 1 mM EDTA, 0.5% NP40, 1 mM dithiothreitol [DTT], Tris-HCl [pH 7.5]) for 1 h at 4 °C. The binding reactions were then coupled to 20 μL of glutathione beads for 1 h at 4 °C. Beads were washed 3 times with NETN buffer. The bound proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and analyzed using Coomassie blue stain.
AlphaScreen Binding Assay between GST-Skp2/Skp1 and His6-Cks1
All AlphaScreen (PerkinElmer) assays were performed in triplicate using OptiPlate-384 (PerkinElmer) plates. A GST-Skp2/Skp1-His6-Cks1 concentration matrix was set up in the 384-well microplates in a total volume of 25 μL per well as follows: 5 μL of each protein solution (total 10 μL) was added to 5 μL of 1× AlphaScreen assay buffer. After mixing, the plate was incubated at room temperature while shaking for 1 h. Then, to each well, 5 μL of glutathione donor beads and 5 μL of nickel chelate acceptor beads (PerkinElmer) were added so that the working concentration of each bead was 0.02 mg/mL/well. All dilutions of proteins and reagents were made in 1× AlphaScreen assay buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 0.05% bovine serum albumin (BSA), 0.02% Tween, and 1 mM DTT. Additions of reagents and incubations were carried out in subdued lighting due to photosensitivity of the beads. Plates were sealed and incubated for 2.5 h at room temperature while shaking in the dark. After the incubation, laser excitations were carried out at 680 nm, and readings were performed at 520 to 620 nm using the EnVision 2102 Multilabel Reader (PerkinElmer).
To determine the binding constant (Kd), 5 μL of varying concentrations of GST-Skp2/Skp1 was mixed with 5 μL of a fixed concentration of Cks1 (125 nM). Readings were recorded at 520 to 620 nm using the EnVision 2102 Multilabel Reader. The sigmoidal curves were fit using GraphPad Prism software (GraphPad Software, La Jolla, CA) to obtain the apparent dissociation constant (Kd).
Untagged Cks1 of varying concentrations (0.69 nM–1.5 μM, 3-fold serial dilutions) was used to compete out His6-Cks1 in samples containing 78 nM His6-Cks1 and 50 nM GST-Skp2/Skp1. The AlphaScreen assay setup was identical to the one mentioned in the previous paragraph. The IC50 value was determined by GraphPad Prism software.
To determine the IC50 values of the hits from the screen for small-molecule inhibitors of Skp2-Cks1 binding, we added 5 μL GST-Skp2/Skp1 (20 nM) and 5 μL Cks1 (200 nM) to 5 μL of 1× AlphaScreen assay buffer. Then, 5 μL each of glutathione donor beads and nickel chelate acceptor beads was added (0.02 mg/mL/well working concentration) to the reaction mixture, following a 1-h incubation at room temperature. After incubating the reaction mixture for 2.5 h at room temperature in the dark, the plate was analyzed using an EnVision 2102 Multilabel Reader as described earlier. To obtain the IC50 values, the curves were fit using GraphPad Prism software.
Compound Library
The compounds tested in this study were provided by the National Cancer Institute (NCI)/National Institutes of Health (NIH) Developmental Therapeutics Program (DTP) (http://dtp.cancer.gov). We received 3125 compounds that were selected from the DTP Open Repository collection of 140,000 small molecules encompassing four focus sets: Challenge Set (57 compounds with unknown cell-killing mechanism), Natural Product Set (235 compounds), Diversity Set (1990 compounds), and Mechanistic Set (879 compounds). The compounds were supplied as 10- or 1-mM DMSO stocks in 96-well plates and stored at −20 °C.
In Vitro Ubiquitination of p27
Ubiquitination of p27 was carried out as described in Ungermannova et al.28 Mouse p27, cloned into pCS2, was translated in vitro in a rabbit reticulocyte lysate system (Promega, Madison, WI) in the presence of [35S]-labeled methionine. p27 was phosphorylated by purified recombinant CDK2–cyclin E as described previously.9 Then, 5 μL of the phosphorylation reaction was incubated with a ubiquitination mixture containing 100 nM ubiquitin E1, 200 nM His-Cdc34 (E2), 100 nM SCFSkp2 (E3), 50 nM Cks1, 10 μM ubiquitin (Sigma-Aldrich, St. Louis, MO), 10 μM methylated ubiquitin (Boston Biochem, Cambridge, MA), 1 μL of 20× energy regeneration system: (10 mM ATP, 20 mM HEPES [pH 7.4], 10 mM MgOAc, 300 mM creatine phosphate, and 0.5 mg/ml creatine phosphokinase), 1 μM ubiquitin aldehyde (Boston Biochem, Cambridge, MA), and 1 μM MG132 (Sigma-Aldrich, St. Louis, MO) in a total volume of 15 μL. The reaction was quenched after 30 min in a 30 °C water bath by addition of 4× SDS sample buffer. The products of ubiquitination were resolved on a 12% polyacrylamide gel that was destained in 45% methanol and 10% acetic acid, dried, and exposed overnight to a phosphoimager screen for imaging using a Typhoon scanner (Typhoon 9400 Variable Mode Imager; Amersham Biosciences, Pittsburgh, PA).
Results
Development of AlphaScreen Assay to Characterize Skp2-Cks1 Interactions
We searched for a homogeneous in vitro binding assay that would be adaptable to an HTS system where we could use a library of small molecules to identify Cks1-Skp2 inhibitor(s). To discover new leads through HTS, it is critical to find an assay that measures the interaction accurately. Among other criteria, the assay should be sensitive, reliable, fast, easy, homogeneous (mix-and-read, no wash steps), automatable, and cost-effective. Developed by Ullman and coworkers,18,19 AlphaScreen is a bead-based nonradioactive amplified luminescent proximity homogeneous assay that consists of donor and acceptor beads that are brought into close proximity upon biological interactions between two proteins that are immobilized on the beads. Upon excitation of the reaction mixture with a laser set at 680 nm, a photosensitizer within the donor bead converts the ambient oxygen into a more excited singlet state. The singlet oxygen diffuses up to 200 nm and makes contact with a thioxene derivative present in the acceptor bead that generates chemiluminescence at 370 nm (Fig. 1A). This is followed by activation of fluorophores in the same bead, which ultimately emit light in the 520- to 620-nm range that can be read by a number of available plate readers, such as the EnVision Multilabel Plate Reader from PerkinElmer (Fig. 1). One donor bead can generate about 60,000 singlet oxygens, resulting in exceptionally high signal amplification, permitting the AlphaScreen assay volume to be minimized and adapted to 96-, 384-, and 1536-well formats. Having a relatively long decay half-life of 0.3 s, the AlphaScreen system can perform time-resolved measurements. We adopted the AlphaScreen system to measure the binding of Skp2 and Cks1 (Fig. 1A).
Skp2 was cloned together with Skp1 using a bicistronic vector derived from the pGEX-4T-1 vector, which has been shown to improve Skp2 expression.20 Skp2/Skp1 was expressed as a GST-fusion complex for immobilization on anti-GST donor beads, whereas Cks1 had a His6 tag for recognition by anti-His acceptor beads. Both proteins were purified from bacteria through multistep purification protocols described under Materials and Methods (Fig. 2A). We tested the binding capacity of the two proteins using an in vitro binding assay and detected binding between wild-type GST-Skp2/Skp1 and His6-Cks1 (Fig. 2B, lane 1). We showed previously that a D→A mutation at position 331 of Skp2 completely abrogates Skp2-Cks1 interactions.17 Hence, we expected that GST-Skp2(D331A)/Skp1 would serve as a negative control during the development of the AlphaScreen assay. Indeed, in our in vitro binding assay, the D331A mutant did not interact with His6-Cks1 (Fig. 2B, lane 2).
Figure 2.
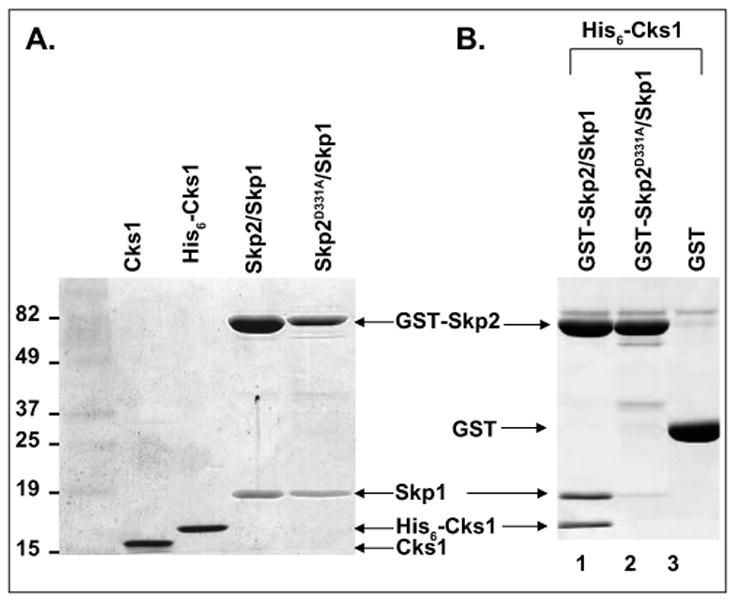
Purification and in vitro binding results of glutathione S-transferase (GST)–Skp2/Skp1 and His6-Cks1. (A) Coomassie-stained sodium dodecyl sulfate polyacrylamide gel electrophoresis gel of proteins to be used in the high-throughput screen. Lane 1: protein marker; lane 2: untagged Cks1; lane 3: His6-Cks1; lane 4: wild-type (WT) GST-Skp2/Skp1; lane 5: mutated GST-Skp2(D331A)/Skp1.(B) In vitro binding of GST-Skp2/Skp1 and His6-Cks1. WT GST-Skp2/Skp1 interacts with His6-Cks1 (lane 1). The binding between Cks1 and Skp2 is lost upon mutating D331 of Skp2 to A (lane 2). GST alone was used as a negative control (lane 3). The binding results were detected using Coomassie blue staining.
To determine the optimal protein concentration in the AlphaScreen binding reaction, we performed cross-titration of both binding partners. The 1:1.5 serial dilutions of GST-Skp2/Skp1 and His6-Cks1 were mixed together with 20 μg/mL of both beads, and the luminescence signal was measured on an EnVision plate reader. Using this approach, we observed a robust signal when we combined 50 nM GST-Skp2/Skp1 with 125 nM His6-Cks1 (Suppl. Fig. S1, bar 1). Since Skp1 is a part of the protein complex, it is possible that Skp1 contributes to Cks1 binding. To validate specificity of Skp2 binding to Cks1, we added 50 nM of untagged Skp1 and noted that the association between Skp2 and Cks1 was not affected (bar 2), which implies that it is unlikely that Skp2-Cks1 interactions are mediated by Skp1. As expected, the AlphaScreen signal was substantially reduced using the following reaction conditions: GST-Skp2/Skp1 was replaced with GST alone (bar 3), donor or acceptor beads were left out (bars 4 and 5), reactions were incubated without His6-Cks1 or GST-Skp2/Skp1 (bars 6 and 7), or GST-Skp2 was replaced by a point mutation (D331A, within the Cks1 binding region [bar 8]). On the basis of these results, we selected assay concentrations of 50 nM GST-Skp2/Skp1 and 125 nM His6-Cks1 for further optimization and validation.
Skp2-Cks1 Kd Determination and Competition with Untagged Cks1
To determine the binding affinity for the Skp2-Cks1 interactions, we added 5 μL GST-Skp2/Skp1 of varying concentrations ranging from 2 to 180 nM in a 384-well Optiplate to His6-Cks1 (at a fixed concentration of 125 nM), which was incubated at room temperature for 1 h. Next, 10 μL of glutathione donor/His acceptor beads was added, bringing the total volume to 25 μL per well, and the reaction was incubated with shaking for 2 h before reading. Triplicates were used to demonstrate assay reproducibility. The apparent Kd of 15.7 nM for Skp2-Cks1 binding was determined by applying a sigmoidal dose-response model for curve fitting using GraphPad software (Fig. 3A). The obtained Kd correlates well with a Kd of 14 nM acquired by isothermal titration calorimetry4,10 but deviates from the 140 nM reported by Xu et al.,21 who used a homogeneous time-resolved fluorescence energy transfer assay. To show the specificity of GST-Skp2/Skp1 binding to His6-Cks1, we performed competitive displacement measurements with untagged Cks1. In this experiment, 125 nM His6-Cks1 was incubated with 50 nM GST-Skp2/Skp1 complex in the presence of increasing concentrations of unbound, untagged Cks1. Untagged Cks1 effectively competed with His6-Cks1 with an IC50 of 17.7 nM (Fig. 3B).
Figure 3.

Measuring the Skp2-Cks1 interaction using the AlphaScreen assay. (A, B) Skp2-Cks1 Kd determination and competition with untagged Cks1, measured by the AlphaScreeen assay. (A) Apparent Kd for Skp2 binding to Cks1. In total, 62.5 nM His6-Cks1 was incubated with glutathione S-transferase (GST)–Skp2/Skp1 ranging from 2 to 180 nM for 1 h at room temperature, followed by addition of donor/acceptor beads for 2 h and reading the results in the plate reader. The experiment was set up in triplicates, and Kd was extracted from binding curves generated using GraphPad Prism software (GraphPad Software, La Jolla, CA). (B) Competition with untagged Cks1. In total, 50 nM GST-Skp2/Skp1 was bound to 62.5 nM His6-Cks1 in the presence of increasing concentrations of free Cks1. IC50 for the competition was determined by a seven-point dose ranging from 0.3 to 300 nM of Cks1 and analyzed by GraphPad Prism.
Optimization of GST-Skp2/Skp1-His6-Cks1 AlphaScreen Assay for HTS
To evaluate the suitability of the Skp2-Cks1 AlphaScreen assay for HTS, we tested the following parameters: (1) stability of the reagents in DMSO, (2) duration of the AlphaScreen signal, (3) order of addition of the reagents, and (4) effect of reaction volume on the AlphaScreen signal. Because HTS compounds are typically dissolved in DMSO, it was necessary to examine whether Skp2 and Cks1 can retain activity in its presence. We tested DMSO concentrations up to 20% and found that it had no significant effect on Skp2-Cks1 binding activity (Fig. 4A). Considering that it takes about 5 min for the plate reader to scan one 384-well plate, we checked the stability of the signal over the course of 24 h. Five identical reactions were tested for each time point (2, 3, 4, 5, 6, and 24 h) where, after incubating Skp2 and Cks1 for 1 h and after the addition of both beads (A/D, acceptor and donor beads), the plate was sealed and time points were taken after unsealing the plate. The plate was resealed between readings to avoid photobleaching. We found that the signal was stable up to at least 6 h, whereas incubation overnight resulted in a significant signal decrease, leading to a lower signal-to-background (S/B) ratio (Fig. 4B). This indicated that we can batch process many plates for reading using a stacker-equipped microplate reader.
Figure 4.

Optimization of the glutathione S-transferase (GST)–Skp2/Skp1-His6-Cks1 AlphaScreen assay for high-throughput screening. (A) Stability of AlphaScreen reaction components in DMSO. (B) Time dependence of the AlphaScreen signal (in hours). For each time point, five wells were measured and plotted as the average and standard deviation. The background was negligible when acceptor or donor beads were omitted (±A/D). (C) Order of addition. When compared with the AlphaScreen reaction where the two proteins were incubated together without adding any DMSO (bar 1), the signal was unaffected when DMSO was incubated with His6-Cks1 for 1 h prior to adding Skp2 (bar 2). There was a reduction of signal when Skp2 was first incubated with DMSO followed by addition of Cks1 one hour later (bar 3). After each incubation, donor/acceptor beads were mixed in and the results were read after 2 h using an EnVision plate reader (PerkinElmer, Waltham, MA). The reactions were set up in triplicate. (D) The effect of reaction volume on AlphaScreen signal. The signal decreased with decreasing reaction volume while keeping the ratio of all reaction components constant. In each case, the concentration of beads was maintained at 20 μg/mL.
We next examined the most favorable order of addition of the two interacting proteins. In this experiment, we preincubated DMSO with His6-Cks1 or GST-Skp2/Skp1 for 1 h before addition of the second interacting protein (GST-Skp2/Skp1 and His6-Cks1, respectively) for another hour, followed by incubating the reaction with beads for 2 h. We determined that the signal is more robust if His6-Cks1 is incubated with the compound first (Fig. 4C, bar 2 vs. bar 3); hence, we decided to incubate it with the compounds first and mix in GST-Skp2/Skp1 one hour later.
Finally, we explored the possibility of lowering the reaction volume to bring down the assay cost. The effect of different reaction volumes (25 to 5 μL) on the assay signal was tested with a bead concentration of 20 μL/mL. The signal decreased linearly with decreasing reaction volume with a maximum signal at 25 μL and a minimum signal at 5 μL (Fig. 4D). On the basis of the results of the experiments in this section, we determined that a suitable HTS assay signal can be obtained by incubating Cks1 with the compounds first, then adding GST-Skp2 and incubating the reaction with donor/acceptor beads for 2 h in a reaction volume of 20 μL, while being highly tolerant to DMSO.
Pilot Screen for Small-Molecule Inhibitors of Skp2-Cks1 Binding
A pilot screen of potential inhibitors of Skp2-Cks1 interactions (totaling 1600 compounds supplied by the NCI) was performed at final concentrations of 4 or 40 μM, depending on whether the stock concentration of the compound was 1 or 10 mM. The compounds were shipped in a 96-well format and were transferred to 384-well screening plates. The compounds were dispensed by the pipetting robot and were diluted in DMSO to give a final concentration of 400 or 40 μM, resulting in 10% DMSO content. The data were analyzed and Z′ factors and S/B were extracted for each plate (Suppl. Fig. S2A). With the average Z′ value of 0.7 and S/B ratio of 19, the assay performance and robustness were deemed acceptable. Two independent screening runs were executed to assess the run-to-run screening variability since replicate testing improves hit confirmation and reduces false positives and false negatives.22 Duplicate runs of the AlphaScreen assay showed a high degree of correlation (Suppl. Fig. S2B), suggesting that this assay is a reliable HTS method. Duplicate experiments of the AlphaScreen runs detected 45 compounds with a hit rate of 2.8% when the hit limit was set as a 75% decrease in signal (three standard deviations away from the sample mean).
Hit Confirmation and Counterscreen
Suppression in AlphaScreen signal could be attributed to either on-target events or false positives due to off-target events. Two strategies were used to validate the hits from the primary screen. First, we retested all 45 compounds in a counterscreening assay using an AlphaScreen assay kit in which GST-biotin conjugated to donor beads operated as a binding partner for streptavidin-bound acceptor beads (Fig. 1A). Small molecules that inhibit this control reaction interfere with the generation of the signal and not with protein-protein interactions tested in the primary assay. Second, we tested the hits (40 μM) in an in vitro p27 ubiquitination assay. With these two independent assays, we were able to reduce the list to 12 compounds (Fig. 5A,B). The level of polyubiquitination was compared with the reaction that was supplemented with DMSO, and at least 50% inhibition of ubiquitin ligation quantified by ImageJ (NIH, Bethesda, MD) was required in order for the compound to be considered a confirmed hit. Among the hits identified, NSC689857 and NSC681152 are particularly intriguing as these two hits are structurally related compounds. In addition, both compounds are known to have antitumor activities, but their mechanism(s) of action remains elusive23 (Suppl. Fig. S4A,B). Moreover, both compounds are hydroquinones, which are commonly found in anticancer drugs but are often filtered out due to potential toxicity and poor development potential. Finally, in the crystal structure of the Skp2-Cks1 complex, a benzamidine molecule was found in the interface of these two proteins, which suggests the possibility of targeting the binding interface with small molecules. For these reasons, we decided to further characterize this class of compounds.
Figure 5.
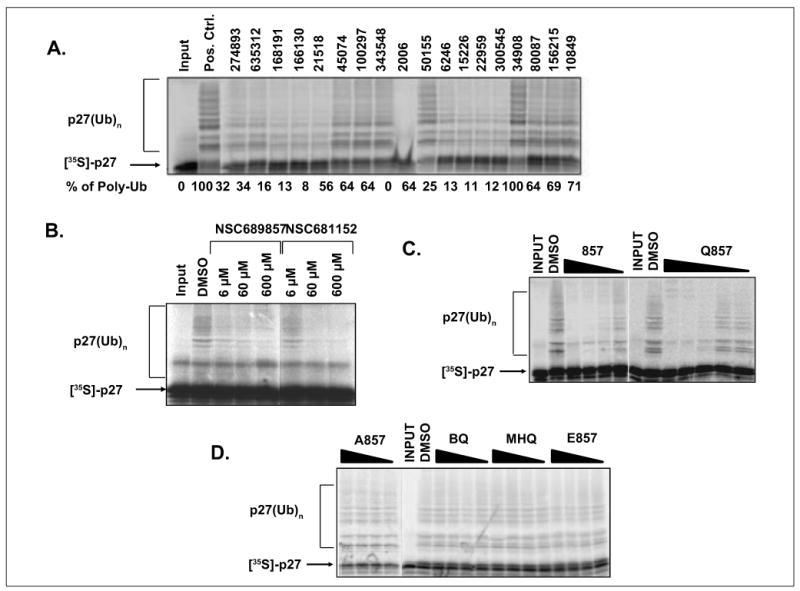
Secondary screen evaluated by in vitro p27 ubiquitination. (A) Representative compounds identified in the primary AlphaScreen assay were tested in the p27 ubiquitination reaction at 40 μM each. A total of 15 compounds were found to be inhibitory. Partial results are shown. (B) NSC689857 and NSC681152 impeded p27 polyubiquitination in a dose-dependent manner. DMSO was used as a positive control in all sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels. (C, D) The effect of NSC689857 and its analogues on in vitro p27 ubiquitination. Compounds 857, A857, E857, methylhydroquinone (MHQ), and 1,4-benzoquinone (BQ) were added at 250, 125, 62.5, and 31 μM. The concentrations of Q857 were 100, 50, 25, 12.5, and 6.25 μM. The indicated compounds were incubated for 20 min with Skp2/Skp1 complex at room temperature. Subsequently, Cul1/Rbx1, E1, Cdc34, energy regeneration system, ubiquitin, Cks1, and CDK2–cyclin E–prephosphorylated p27 were mixed in to initiate the transfer of ubiquitin onto p27. The reactions were incubated for 1 h at 30 °C, run on an SDS-PAGE gel, and developed by autoradiography.
Structure-Activity Relationship of Skp2-Cks1 Inhibitor NSC689857
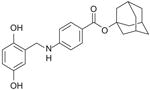
To derive a preliminary structure-activity relationship (SAR), we generated several analogues of NSC689857 (Table 1) to investigate the role of the phenyl ring and the hydroquinone in an effort to identify the pharmacophore of the hit compound. The original hydroquinone molecule, designated as 857 (abbreviated for NSC689857), and the following analogues—quinone (Q857), two oxidation-blocking analogues that were generated by nonselective methylation (E857, A857), methylhydroquinone (MHQ), and 1,4-benzoquinone (BQ)—were tested in an AlphaScreen assay for Skp2-Cks1 interaction, counterscreen (Suppl. Fig. S5), and p27 ubiquitin ligase reaction (Table 1). We found that NSC689857 and its quinone derivative (Q857) as well as NSC681152 blocked the Skp2-Cks1 interaction at low μM concentrations (Table 1). Similarly, all three compounds also inhibited p27 ubiquitination in vitro (Fig. 5C,D). Modifications of either hydroxyl by methylation destroyed activity, whereas quinone modification slightly improved activity in the ubiquitination assay but not in the Skp2-Cks1 binding assay (Table 1). However, quinone alone (BQ) or MHQ had no effect on disrupting the Skp2-Cks1 interaction or the conjugation of ubiquitin onto p27, suggesting that the specific structure rather than quinone functionality is necessary for the inhibitory activity (Table 1). The dihydrophenol ring configuration and the linker are likely critical elements of the pharmacophores.
Table 1. Structure-Activity Relationship of Skp2-Cks1 Inhibitors.
| Compound | Structure | Inhibition of Skp2-Cks1 IC50 (μM) | p27 Ub IC50 (μM) |
|---|---|---|---|
| 857 |
|
36 ± 6.2 | ~30 |
| Q857 |

|
71 ± 4.4 | ~15 |
| A857 |

|
1090 ± 189 | >300 |
| E857 |
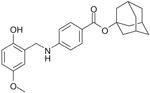
|
8311 ± 1458 | >300 |
| MHQ |
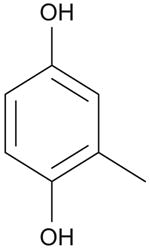
|
434 ± 39.5 | >300 |
| BQ |

|
626 ± 65.7 | >300 |
| NSC681152 |

|
75.6 ± 6.8 | ~80 |
BQ, 1,4-benzoquinone; MHQ, methylhydroquinone.
Discussion
We employed AlphaScreen technology to identify compounds that disrupt binding between the recognition subunit of SCFSkp2-Cks1 E3 ligase. As the interaction between Skp2 and Cks1 is categorically required for p27 ubiquitination by SCFSkp2 in vitro and in vivo, blocking the interaction between the two proteins by a small molecule is expected to prevent p27 ubiquitination, thereby inhibiting proteasomal destruction. We show that the AlphaScreen assay provides a solid method that measures Skp2-Cks1 interactions. The assay is homogeneous, very robust, and reproducible, and it can be miniaturized, which makes it very cost-effective for large-scale screening projects. We demonstrated the feasibility of this HTS assay by screening 1600 compounds from an NIH library. Using a combination of a counterscreen and an independent functional in vitro p27 ubiquitination assay, we were able to validate some of hits that emerged from the primary screen.
Interestingly, two hits (NSC689857 and NSC681152) are structurally related and were originally made for the SAR studies of AG957, which is a known inhibitor of epidermal growth factor receptor or tyrosine kinases such as p210bcr-abl.23 Neither AG957 nor its two analogues bear any resemblance to classical adenosine triphosphate competitive kinase inhibitors. The mechanism of kinase inhibition by this class of molecules is still elusive. The IC50 values of NSC689857 and NSC681152 against p210bcr-abl in vitro are 12.62 μM and 2.64 μM, respectively.23 In cell line testing, the GI50 values against K562 leukemia cell lines for both compounds are 12.84 μM and 18.47 μM. Both compounds have been tested in NCI 60 cell lines with average GI50s over all cell lines of 1.25 μM and 7.97 μM, respectively (Suppl. Fig. S4A,B). Although it is quite likely that both compounds target tyrosine kinases in tumor cells, our results suggest that they may also be involved in perturbing the Skp2-Cks1 interaction and consequently p27 degradation. We treated a mink lung epithelial cell line stably expressing GFP-p27 with these two compounds. Both compounds can cause dose-dependent stabilization of GFP-p27 (Suppl. Fig. S3), although the endogenous p27 increases only marginally. The dual action of these inhibitors may be a mechanism for their antitumor activity in a variety of cell lines. Future studies are necessary to further elucidate their activity in vivo.
We show here that it is possible to implement an HTS assay for Skp2-Cks1 interactions using the AlphaScreen platform and identify hits that may perturb this interaction. However, AlphaScreen tends to generate a higher rate of hits, and a large fraction of them may not be subsequently validated. These observations heighten the need to develop secondary screens to filter false positives. Some of the false positives can be attributed to compounds that interfere in binding interactions by forming aggregates, those that are protein reactive, or those that directly interfere in assay signaling.24,25 It has been recognized that a subset of substructural features appear as frequent hitters in many biochemical high-throughput screens.24–26 These are known as pan-assay interference compounds (PAINS), and it is generally advised to exclude them from the library.26 We also noted that colored compounds are generally incompatible with the AlphaScreen assay, and these were excluded from our assay. Quinones have been classified as PAINS. Since both NSC689857 and NSC681152 are hydroquinones, we further tested whether hydroquinones and quinones indiscriminately interfere with the AlphaScreen assay. When all the compounds tested at the same concentration (10 μM) (Suppl. Fig. S5), we did not observe a dramatic reduction of AlphaScreen signal under this condition. Modest reduction was seen in isolated cases, but there does not appear to be a general effect, and suppression of the AlphaScreen signal in the counterscreen assay does not correlate with the effectiveness of these compounds in our functional assays. Although we should be cautious about interpreting hits with a hydroquinone and quinone substructure, our results suggest that there is also a need to analyze them individually with medicinal chemistry optimization. Some of the natural or synthetic quinones are known for their pharmacological activities in anticancer and anticardiovascular diseases. As long as specific activity can be demonstrated in a biological context, their utilities as potential drug candidates should not be categorically ignored.
Previously, a homogeneous time-resolved fluorescence (HTRF) assay for inhibitors of the Cks1-Skp2 interaction was described.27 The advantage of the HTRF assay lies in very low background as lanthanides have a very long, excited state relative to most fluorophores. By that time, all of the fluorescence of the acceptor and contaminants in the buffers and the plate have decayed, and the only source of a signal is sensitized emission from the acceptor being excited by nonradiative energy transfer from the donor. However, disadvantages of the HTRF assay include its limited dynamic range (<10-fold) and significant constraints on the range of detection of interacting binding partners. In contrast, the AlphaScreen assay format, like the one described in this report, is that the assay described here has a very high dynamic range (>200-fold; Suppl. Fig. S1) and is relatively easy to set up. An HTRF assay has been developed for quantitative analysis of the Cks1-Skp2 interaction,21,27 but the actual performance of this HTRF assay for the HTS screen of inhibitors of Skp2-Cks1 has not been reported in detail. It will be interesting to compare these two HTS assays side-by-side in future studies.
In summary, we developed a high-throughput screen based on AlphaScreen technology and applied it to screen a small compound library for potential inhibitors of p27 proteolysis. We identified NSC689857 and NSC681152 as validated potential leads to disrupt the Skp2-Cks1 interaction. Future screening efforts will be focused on identifying more potent inhibitors with a much larger collections of compounds and testing the leads identified in this report in animal models.
Supplementary Material
Acknowledgments
We thank Craig Manahan for excellent technical help. We thank Dr. Kristen Barthel and Edward Kee for editing of the manuscript. We thank Drs. Bing Hao, Wei Wang, and Robert Kuchta for discussions.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by a grant from the National Institutes of Health (CA107089) and a BDEG grant from University of Colorado Tech Transfer Office to Xuedong Liu.
Footnotes
Supplementary material for this article is available on the Journal of Biomolecular Screening Web site at http://jbx.sagepub.com/supplemental.
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Sherr CJ, Roberts JM. CDK Inhibitors: Positive and Negative Regulators of G1-phase Progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 2.Nakayama KI, Nakayama K. Ubiquitin Ligases: Cell-Cycle Control and Cancer. Nat Rev Cancer. 2006;6:369–381. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- 3.Jin J, Cardozo T, Lovering RC, Elledge SJ, Pagano M, Harper JW. Systematic Analysis and Nomenclature of Mammalian F-box Proteins. Genes Dev. 2004;18:2573–2580. doi: 10.1101/gad.1255304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ganoth D, Bornstein G, Ko TK, Larsen B, Tyers M, Pagano M, Hershko A. The Cell-Cycle Regulatory Protein Cks1 Is Required for SCF(Skp2)–Mediated Ubiquitination of p27. Nat Cell Biol. 2001;3:321–324. doi: 10.1038/35060126. [DOI] [PubMed] [Google Scholar]
- 5.Spruck C, Strohmaier H, Watson M, Smith AP, Ryan A, Krek TW, Reed SI. A CDK-Independent Function of Mammalian Cks1: Targeting of SCF(Skp2) to the CDK Inhibitor p27Kip1. Mol Cell. 2001;7:639–650. doi: 10.1016/s1097-2765(01)00210-6. [DOI] [PubMed] [Google Scholar]
- 6.Sitry D, Seeliger MA, Ko TK, Ganoth D, Breward SE, Itzhaki LS, Pagano M, Hershko A. Three Different Binding Sites of Cks1 Are Required for p27-Ubiquitin Ligation. J Biol Chem. 2002;277:42233–42240. doi: 10.1074/jbc.M205254200. [DOI] [PubMed] [Google Scholar]
- 7.Wang W, Ungermannova D, Chen L, Liu X. Molecular and Biochemical Characterization of the Skp2-Cks1 Binding Interface. J Biol Chem. 2004;279:51362–51369. doi: 10.1074/jbc.M405944200. [DOI] [PubMed] [Google Scholar]
- 8.Montagnoli A, Fiore F, Eytan E, Carrano AC, Draetta GF, Hershko A, Pagano M. Ubiquitination of p27 Is Regulated by Cdk-Dependent Phosphorylation and Trimeric Complex Formation. Genes Dev. 1999;13:1181–1189. doi: 10.1101/gad.13.9.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ungermannova D, Gao Y, Liu X. Ubiquitination of p27Kip1 Requires Physical Interaction with Cyclin E and Probable Phosphate Recognition by SKP2. J Biol Chem. 2005;280:30301–30309. doi: 10.1074/jbc.M411103200. [DOI] [PubMed] [Google Scholar]
- 10.Hao B, Zheng N, Schulman BA, Wu G, Miller JJ, Pagano M, Pavletich NP. Structural Basis of the Cks1-Dependent Recognition of p27(Kip1) by the SCF(Skp2) Ubiquitin Ligase. Mol Cell. 2005;20:9–19. doi: 10.1016/j.molcel.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 11.Chu IM, Hengst L, Slingerland JM. The Cdk Inhibitor p27 in Human Cancer: Prognostic Potential and Relevance to Anticancer Therapy. Nat Rev Cancer. 2008;8:253–267. doi: 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
- 12.Pagano M, Benmaamar R. When Protein Destruction Runs Amok, Malignancy Is on the Loose. Cancer Cell. 2003;4:251–256. doi: 10.1016/s1535-6108(03)00243-5. [DOI] [PubMed] [Google Scholar]
- 13.Hiramatsu Y, Kitagawa K, Suzuki T, Uchida C, Hattori T, Kikuchi H, Oda T, Hatakeyama S, Nakayama KI, Yamamoto T, et al. Degradation of Tob1 Mediated by SCFSkp2-Dependent Ubiquitination. Cancer Res. 2006;66:8477–8483. doi: 10.1158/0008-5472.CAN-06-1603. [DOI] [PubMed] [Google Scholar]
- 14.Slingerland J, Pagano M. Regulation of the Cdk Inhibitor p27 and Its Deregulation in Cancer. J Cell Physiol. 2000;183:10–17. doi: 10.1002/(SICI)1097-4652(200004)183:1<10::AID-JCP2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 15.Craig C, Wersto R, Kim M, Ohri E, Li Z, Katayose D, Lee SJ, Trepel J, Cowan K, Seth P. A Recombinant Adenovirus Expressing p27Kip1 Induces Cell Cycle Arrest and Loss of Cyclin-Cdk Activity in Human Breast Cancer Cells. Oncogene. 1997;14:2283–2289. doi: 10.1038/sj.onc.1201064. [DOI] [PubMed] [Google Scholar]
- 16.Chen J, Willingham T, Shuford M, Nisen PD. Tumor Suppression and Inhibition of Aneuploid Cell Accumulation in Human Brain Tumor Cells by Ectopic Overexpression of the Cyclin-Dependent Kinase Inhibitor p27KIP1. J Clin Invest. 1996;97:1983–1988. doi: 10.1172/JCI118631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang W, Ungermannova D, Chen L, Liu X. A Negatively Charged Amino Acid in Skp2 Is Required for Skp2-Cks1 Interaction and Ubiquitination of p27Kip1. J Biol Chem. 2003;278:32390–32396. doi: 10.1074/jbc.M305241200. [DOI] [PubMed] [Google Scholar]
- 18.Ullman EF, Kirakossian H, Singh S, Wu ZP, Irvin BR, Pease J, Switchenko AC, Irvine JD, Dafforn A, Skold CN, et al. Luminescent Oxygen Channeling Immunoassay: Measurement of Particle Binding Kinetics by Chemiluminescence. Proc Natl Acad Sci U S A. 1994;91:5426–5430. doi: 10.1073/pnas.91.12.5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ullman EF, Kirakossian H, Switchenko AC, Ishkanian J, Ericson M, Wartchow CA, Pirio M, Pease J, Irvin BR, Singh S, et al. Luminescent Oxygen Channeling Assay (LOCI): Sensitive, Broadly Applicable Homogeneous Immunoassay Method. Clin Chem. 1996;42:1518–1526. [PubMed] [Google Scholar]
- 20.Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P, Chu C, Koepp DM, Elledge SJ, Pagano M, et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF Ubiquitin Ligase Complex. Nature. 2002;416:703–709. doi: 10.1038/416703a. [DOI] [PubMed] [Google Scholar]
- 21.Xu K, Belunis C, Chu W, Weber D, Podlaski F, Huang KS, Reed SI, Vassilev LT. Protein-Protein Interactions Involved in the Recognition of p27 by E3 Ubiquitin Ligase. Biochem J. 2003;371:957–964. doi: 10.1042/BJ20021722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ozdamar B, Bose R, Barrios-Rodiles M, Wang HR, Zhang Y, Wrana JL. Regulation of the Polarity Protein Par6 by TGFbeta Receptors Controls Epithelial Cell Plasticity. Science. 2005;307:1603–1609. doi: 10.1126/science.1105718. [DOI] [PubMed] [Google Scholar]
- 23.Kaur G, Narayanan VL, Risbood PA, Hollingshead MG, Stinson SF, Varma RK, Sausville EA. Synthesis, Structure-Activity Relationship, and p210(bcr-abl) Protein Tyrosine Kinase Activity of Novel AG 957 Analogs. Bioorg Med Chem. 2005;13:1749–1761. doi: 10.1016/j.bmc.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 24.McGovern SL, Caselli E, Grigorieff N, Shoichet BK. A Common Mechanism Underlying Promiscuous Inhibitors from Virtual and High-Throughput Screening. J Med Chem. 2002;45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 25.Feng BY, Shelat A, Doman TN, Guy RK, Shoichet BK. High-Throughput Assays for Promiscuous Inhibitors. Nat Chem Biol. 2005;1:146–148. doi: 10.1038/nchembio718. [DOI] [PubMed] [Google Scholar]
- 26.Baell JB, Holloway GA. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 27.Huang KS, Vassilev LT. High-Throughput Screening for Inhibitors of the Cks1-Skp2 Interaction. Methods Enzymol. 2005;399:717–728. doi: 10.1016/S0076-6879(05)99047-2. [DOI] [PubMed] [Google Scholar]
- 28.Ungermannova D, Parker SJ, Nasveschuk CG, Chapnick DA, Phillips AJ, Kuchta RD, Liu X. Identification and Mechanistic Studies of a Novel Ubiquitin E1 Inhibitor. J Biomol Screen. 2012;17:421–434. doi: 10.1177/1087057111433843. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.