

Abstract
In the age of systems biology, biologists seek to quantify the absolute number of molecules in experimentally treated samples. Immunoblotting remains a technique of choice for assessing the relative differences between the protein levels in different samples. Here we discuss how to exploit immunoblotting for estimating the number of Smad transcription factor molecules per cell. We focus on describing the calculations needed to analyze the data. Our methods are generally applicable to the quantification of other cellular proteins.
Keywords: Transforming Growth Factor-β, Smad, Cell signaling, Quantitative, Immunoblot, Western blot
1. Introduction
In the age of systems biology, we seek to prioritize the importance of putative molecular mechanisms and to determine the way in which they are integrated to give rise to phenotype. These goals depend on our ability to quantify and model the system of interest, which calls for experimental methods that can accurately quantify the absolute number of biological molecules in a sample. For example, the quantitative properties of Transforming Growth Factor-β (TGF-β)/Smad signaling determine the specific cell response (1). Understanding TGF-β biology therefore requires investigating these properties, such as kinetic parameter values and the cellular concentrations of relevant molecular species. The principal intracellular components of the TGF-β signaling pathway are the Smad transcription factors (2). Eight Smad isoforms exist in mammalian cells, which transduce signals from the TGF-β superfamily member ligands. Five of the isoforms (1, 2, 3, 5, and 8) are classified as the “receptor-regulated” Smads (R-Smads); one of the isoforms, Smad4, is classified as the “common-mediator” Smad (Co-Smad); and the two remaining isoforms, 6 and 7, are classified as the “inhibitory” Smads (I-Smads). Smads 2, 3, and 4 are the principal downstream mediators of TGF-β signaling proper, and we focus on quantifying Smad2 in the following discussion.
Ultimately, we hope to quantify the Smads in various cell states, as well as the amounts of Smads with different Post-translational modifications, and their abundance in different cellular locations (see the chapter by Chapnick and Liu in this volume). In this chapter, we describe our methods for quantifying the number of Smad transcription factor molecules per cell using quantitative immunoblotting. Normally, immunoblotting cannot be used to derive quantitative information by direct comparison of band optical densities because of the nonlinear relationship between protein abundance and optical density. However, by running a standard curve of proteins of known abundance alongside the experimental samples, immunoblotting can be used to estimate Smad transcription factor abundance in absolute units (e.g., moles per Liter or molecules per cell).
1.1. Relevant Background Information on Immunoblotting
Immunoblotting, otherwise known as “Western blotting”, is a protein analysis technique that involves separating proteins using polyacrylamide gel electrophoresis, transferring them to a nitrocellulose or polyvinylidene difluoride (PVDF) membrane, and then exposing the membrane to antibodies that specifically bind the protein of interest. Usually, a second round of antibody incubation follows in which the secondary antibody linked to an enzyme, a fluorescent moiety, or an otherwise detectable molecule, targets the first antibody. The bands are then detected, typically using chemiluminescence, fluorescence, or chemifluorescence. The signals are captured on autoradiography film or digitally and the bands are quantified by densitometry.
A key issue in immunoblotting is that the band intensity is nonlinearly related to the abundance of target protein in the sample. This is especially true for chemiluminescence detection using autoradiography film because the incident energy is related to the amount of energy passing through the film (the “emergent energy”) by Beer–Lambert's law, such that the band intensity is a power law function of the protein abundance (3). People often speak of the “linear range of detection” in immunoblotting and optimizing one's blotting conditions so that the band intensity of their sample falls in this range. In reality, there is no true linear range of detection – a linear range can be approximated, however, by employing blotting conditions that fall either on the steep initial rise of the power law curve or the more gradual rise that occurs more distally in the curve. Therefore, there are two options for quantifying immunoblot data using standard curves – (1) attempt to optimize the conditions to achieve an approximately linear standard curve that encompasses the sample signal intensity or (2) use a power law function to fit a broader standard curve. The first option is useful when you seek to quantify the band intensity of a single sample. For example, we discuss below how to use immunoblotting to quantify the abundance of endogenous Smad2 in the lysates of cells cultured in basal conditions (Fig. 1). The second option is likely better when you seek to quantify the band intensities of a number of samples in which the signal intensity is expected to vary over a broad dynamic range. We used this strategy to quantify the abundance of C-terminally phosphorylated Smad2 (phospho-Smad2) in cells exposed to a range of concentrations of TGF-β over time (Fig. 2).
Fig. 1.
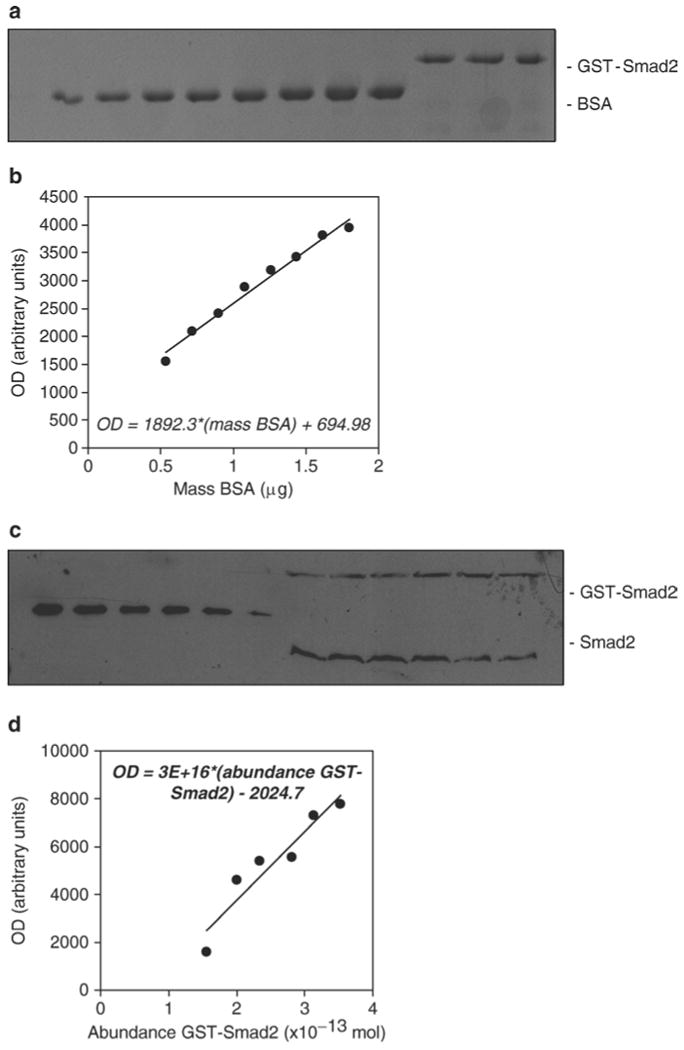
Quantification of the abundance of endogenous Smad2 molecules per cell in PE25 cells. (a) Measurement of the protein standard concentration. The recombinant GST-Smad2 protein standard was quantified using Coomassie staining with BSA standards (0.03–0.1 μg/μL in increments of 0.01 μg/muL in a loading volume of 18 μL sample + 6 μL 4× SDS buffer). Three replicates of the GST-Smad2 protein sample were run alongside the BSA standards. (b) A linear standard curve was generated using Excel's Trendline tool, from which the mass per band of each GST-Smad2 sample was interpolated. The mean mass per band was used to calculate the concentration of the stock GST-Smad2 protein standard. (c) Estimation of the number of Smad2 molecules per cell. Cells were seeded in 10 cm plates and grown to confluence, trypsinized and counted using a hemocytometer, followed by lysis with 500 μL of lysis buffer. Serial dilutions of the GST-Smad2 protein were separated by SDS-PAGE alongside the six independent lysates from untreated PE25 cells. The 12%, 15-well SDS-polyacrylamide gel was loaded with 24 μL of each sample. Proteins were subsequently transferred, immunoblotted with a Smad2-specific antibody, and detected using enhanced chemiluminescence. (d) The GST-Smad2 dilutions conferred a reasonably linear standard curve, which was fit using Excel's Trendline tool, and was then used to estimate the abundance of endogenous Smad2 molecules per band. These numbers were then divided by their respective number of cells, and the six estimates were used to calculate 95% confidence intervals for the number of Smad2 molecules per cell (we reported that 8.5–12 × 104 Smad2 molecules per cell exist in PE25 cells (14)).
Fig. 2.
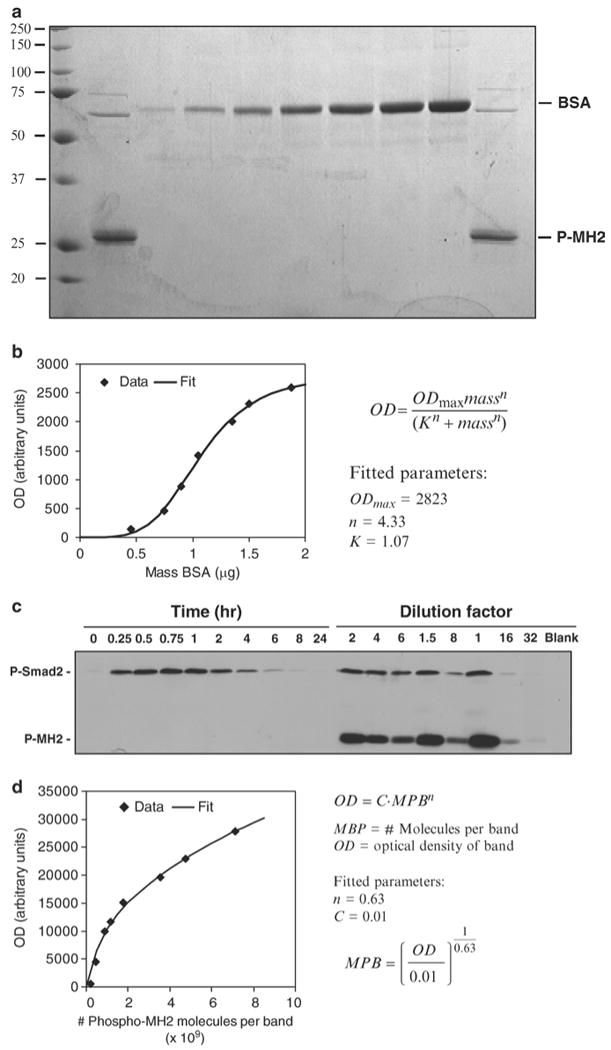
Quantification of phospho-Smad2 levels in whole cell lysates of PE25 cells exposed to 10 pM TGF-β over 8 h. (a) Quantification of the phospho-MH2 standards using Coomassie-staining.Two replicates of the phospho-MH2 (P-MH2) polypeptide were separated by SDS-PAGE alongside BSA standards (15 μL each of 0.03, 0.05, 0.06, 0.07, 0.09, 0.1, and 0.125 μg/μL serially diluted concentrations were loaded). The gel was then Coomassie-stained and quantified by densitometry. (b) A three-parameter logistic equation was used to fit the standard curve. Protein abundance per band of the phospho-MH2 peptides were numerically interpolated. The concentration of the standard was obtained by dividing the mass per band by the loading volume (15 μL). (c) Raw immunoblot data of a time series of lysates from cells treated with 10 pM TGF-β loaded alongside serial dilutions of cell lysate spiked with a known abundance of phospho-MH2 (P-MH2) polypeptide. An antibody specific to C-terminally phosphorylated Smad2 (P-Smad2) was used to probe the membrane. (d) Quantification of the immunoblot data. Phospho-MH2 band optical densities (OD) were quantified by densitometry and plotted versus the number of phospho-MH2 molecules per band. The data were fit to a power law equation in accordance with the chemiluminescence signal following the Beer-Lambert law (3). The fitted power law equation was used to calculate the estimated number of phospho-Smad2 molecules in the bands from the cell lysates. The number of molecules per cell was estimated by dividing the number of molecules per band by the estimated number of cells in the volume of lysate loaded on the gel. (1.5 × 106 cells were seeded in each well and, at the end of the experiment, were lysed in 200 μL of lysis buffer, giving a ratio of approximately 7.5 × 103cells/μL of lysate.) Specific loading volumes for each sample were determined to achieve equal total protein abundance across samples. From this type of experiment, we derived estimates for phospho-Smad2 molecules per cell over time in response to 10 pM TGF-β (15).
1.2. Outline of a Quantitative Immunoblotting Experiment
Two sets of proteins are needed: (1) purified protein standards whose abundance or concentration is either already known or can be measured and (2) experimental samples for which the protein abundance is to be measured based on the standards. The stock concentration of the protein standards can be measured using gel-based methods such as Coomassie staining or the extinction coefficient method (4–6). Next, the specific dilutions to use for the immunoblot are determined by performing a series of immunoblots with different ranges of dilutions of the protein standard until a range of standards that encompasses the signal from the experimental samples is obtained. Once the conditions for the standard curve are determined, the immunoblot that includes both the standards and experimental samples is performed. Once a successful immunoblot is reproducibly obtained, the data is then analyzed to calculate the number of protein molecules contained in the experimental samples. In addition, the experimental design must include measurement or calculation of other variables, including the molecular weights of both the protein standard and of the target endogenous protein (these will often be different because the protein standard will likely be tagged) and the number of cells that comprise the experimental sample. The cell number can either be counted directly using the sample from which the proteins will be extracted or estimated by performing the experimental treatment in parallel on a separate group of cells that are subsequently counted. Deciding how to estimate cell number depends on whether the cells need to be rapidly processed at the end of the experiment.
2. Materials
2.1. Cell Culture and TGF-β Treatment
Cell culture consumables: 6-well plates (Sarstedt), 0.05% trypsin-EDTA (GIBCO), Dubecco's phosphate-buffered saline (D-PBS, GIBCO).
Cell culture medium: Dubecco's modified Eagle's Medium (DMEM) (GIBCO) supplemented with 100 IU/mL penicillin (ICN Biomedicals), 100 μg/mL streptomycin (Sigma), 2 mM l-glutamine (GIBCO), and 10% fetal bovine serum (SAFC Biosciences).
PE25 cells, a derivative of the mink lung epithelial cell line (Mv1Lu or CCL-64) (7, 8).
Reichert Bright-Line Hemocytometer.
Transforming growth factor-β, human recombinant isoform 1 (R & D Systems). Reconstitute in sterile 4 mM HCl containing 0.1% (w/v) fatty-acid-free bovine serum albumin (Sigma) and 75 mM NaCl (Sigma) to a final TGF-β1 concentration of no less than 1 μg/mL. Aliquot in single use amounts (e.g., 20–50 μL) in 0.7 mL microfuge tubes, snap freeze in N2, and store at −80°C.
2.2. Cell Lysis and Sample Preparation
Protein standards: We used recombinant glutathione-S-transferase (GST)-tagged Smad proteins for the quantification of Smad2 in whole-cell lysates (9). Phospho-Smad2 molecules in TGF-β-treated cells were quantified using recombinant phospho-MH2 domain phosphopeptide that was constructed by synthetic protein ligation. This polypeptide was a generous gift of Dr. Yigong Shi at Princeton University. General information on synthetic protein ligation is available in several excellent reviews (10, 11) while methods specific for the Smad MH2 domain are available in (12).
Cell lysis buffer: 50 mM Tris (Tris base, Research Products International) (pH 7.4), 400 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA, Fisher), 1% (v/v) NP-40 alternative (Calbiochem), 15% (v/v) glycerol (Sigma). The lysis buffer is kept at 4°C and lasts for months. Prior to use, supplement buffer with complete protease inhibitor cocktail (Roche) and 2 mM sodium fluoride (Fisher) and 1 mM sodium orthovanadate (Na3VO4, Sigma) phosphatase inhibitors. The Na3VO4 must first be activated in the following manner (13). First, prepare a 200 mM solution of Na3VO4. Adjust the pH to 10.0 using either 1 N NaOH or 1 N HCl (the starting pH of the Na3VO4 will depend on the source). At pH 10.0, the solution will be yellow. Boil the solution until it becomes colorless (approximately 10 min), followed by cooling it to room temperature, and then readjust its pH to 10.0. Repeat these steps until the solution remains colorless and the pH is stable at 10.0. Aliquot Na3VO4 and store at −20°C.
Cell lifter (Corning).
1.7 mL microfuge tubes (Life Science Products).
Bovine albumin standard (2 mg/mL, Pierce).
BCA protein assay kit (Pierce).
96-well microplate (Becton Dickinson).
Parafilm.
2.3. SDS-PAGE
Gel apparatus: For large gels, we used R Shadel Inc. Discovery 2040 Vertical Gel Unit; for mini-gels, we used Invitrogen Novex XCell SureLock gel boxes with Bio-Rad glass rear spacer plates (1.0 mm spacer) and custom-made glass front plates. Note that the methods are compatible with many gel systems.
Polypropylene bags (VWR).
Heat Sealer: American International Electric Impulse Sealer.
Protogel (30% acrylamide-0.8% bis-acrylamide) (National Diagnostics). Store at room temperature. Note that this solution is toxic and proper safety precautions should be taken.
4× Resolving buffer: 1.5 M Tris (pH 8.8), 0.4% (w/v) sodium dodecyl sulfate (SDS, Sigma). Store at room temperature.
Stacking gel buffer: 1.0 M Tris (pH 6.8), 0.4% (w/v) SDS. Store at room temperature.
10% ammonium persulfate (Sigma), made fresh in H2O.
N,N,N′,N′-Tetramethylethylenediamine (TEMED, Research Products International Corp.).
4× SDS buffer: 200 mM Tris (pH 6.8), 40% (v/v) glycerol, 8% (w/v) SDS, 0.1% bromophenol blue (Sigma), and 400 mM β-mercaptoethanol (Sigma). Mix all components except β-mercaptoethanol, and store at room temperature (lasts for months). To make working reagent, aliquot 1.46 mL of solution into a 1.7 mL microfuge tube, and add 43 μL β-mercaptoethanol. Keep working reagent frozen at −20°C when not in use.
Electrophoresis buffer: 25 mM Tris, 192 mM glycine, 0.1% (w/v) SDS. Store at room temperature.
Protein marker: Spectra Multicolor Broad Range Protein Ladder (Fermentas).
2.4. Coomassie Staining
Destain solution: 10% (v/v) acetic acid (Mallinkrodt Chemicals), 45% (v/v) methanol, 45% (v/v) H2O. Store at room temperature.
Coomassie Brilliant Blue solution: 2.5 mg/mL Coomassie Brilliant Blue R-250 (Fisher) in destain solution. Filter with Whatman #1 filter paper. Store at room temperature.
2.5. Immunoblotting
Blotting membrane: Whatman Schleider Schuell BA83 Protran Nitrocellulose membrane.
Transfer buffer: 48 mM Tris, 39 mM glycine (Fisher), 0.37 g/LSDS, 20% (v/v) methanol (Mallinckrodt Chemicals). Store at room temperature.
Semi-dry transfer unit: Hoefer TE 70 connected to a Hoefer Scientific Instruments PS500XT DC power supply.
Blotting containers (Research Products International Corp).
Filter paper: 0.8 mm thick (Life Science Products).
Ponceau S staining solution: 0.2% (w/v) Ponceau S (Sigma) in 1% acetic acid. Store at room temperature.
Tris-buffered saline-Tween (TBS-T): 0.05 M Tris, 0.138 M NaCl, 0.0027 M KCl, 0.05–0.1% Tween (Sigma). Store at room temperature.
Blocking buffer: 4% (w/v) Safeway nonfat dried milk dissolved in TBS-T. Store at 4°C.
Primary antibodies: Smad2 (Zymed 51-1300), diluted to 1:1,000 in blocking buffer; phospho-Smad2 (Cell Signaling Technology #3108), diluted to 1:150 in blocking buffer.
Secondary antibody: Amersham Biosciences ECL Rabbit IgG, HRP-Linked Whole Ab from donkey.
Autoradiography cassette (Fisher Biotech).
Super-sensitive enhanced chemiluminescence substrate (Pierce SuperSignal West Dura Extended Duration Substrate).
Autoradiography film (ISC Bioexpress Blue Lite Autoradiography film F-9024-8×10).
3. Methods
3.1. Cell Culture and TGF-β Treatment
PE25 cells are cultured at 37°C/5% CO2 in cell culture medium in a humidified incubator.
Seed 1.5 × 106 PE25 cells per well in 6-well plates. Let the cells settle for at least 6 h or overnight.
If cells cultured under basal conditions (i.e., no TGF-β treatment) are to be analyzed, then the cell number in each well can be counted prior to lysis. Trypsinize the cells, collect them in a 15 mL conical tube, and store on ice. Dilute the cells with cell culture medium to a volume that confers approximately 50–75 cells per 1 mm2 (i.e., the area of one of the nine smaller squares in the hemocytometer counting area). Count the cells within at least five squares, calculate the average, divide the average by the volume above each square, and multiply by the dilution factor if necessary. Spin down the remaining cells at 300 × g for 5 min and proceed with lysis.
For experiments involving TGF-β stimulation, add the desired amount of TGF-β to sufficient culture medium to treat all the samples in the experiment. Warm the medium to 37°C in a water bath. Aspirate the old media, apply the medium containing the TGF-β to the cells, and incubate the cells for the desired time.
At the end of the experiment, aspirate the medium, wash the cells with 2 mL ice-cold D-PBS, aspirate D-PBS, and snap freeze the cells by carefully pouring N2 into each well. Store plates at −80°C until ready to lyse.
3.2. Cell Lysis and Sample Preparation
Thaw the frozen 6-well plates at 4°C for a few minutes to warm up.
Pipette 200 μL of lysis buffer onto the cells and scrape them using the cell lifter. Pipette the cells into a microfuge tube. Save an aliquot of lysis buffer to serve as a background control in the BCA assay.
Rotate the cells for 45 min at 4°C, followed by spinning down the insoluble material in a microfuge for 10 min at 16,100 × g at 4°C. Transfer lysate into a fresh microfuge tube and either store at −80°C for later use or proceed directly to the following steps, keeping tubes on ice. Avoid repeated freeze-thaw cycles, because this can reduce the signal on subsequent immunoblots.
Measure the protein concentration of the lysates using the microplate version of the bicinchoninic acid (BCA) method following the manufacturer's instructions (see Note 1).
Combine the volume of lysate necessary for the desired amount of total protein loaded per sample (we typically load 30–36 μg of protein per well), dilute to the desired volume with H2O, followed by adding 4× SDS buffer to the final volume (we typically load each well with 38 μL, such that 27.75 μL of diluted sample is combined with 9.25 μL of 4× SDS buffer). Cap the samples tightly and boil them in a heat-block at 90°C for 5 min, let them cool at room temperature, followed by centrifuging the samples at 3,300 × g for 3 min in a microfuge at room temperature (see Note 2).
Use samples for SDS-PAGE or store at −80°C for future use.
3.3. SDS-PAGE
Determine the percentage acrylamide, the size, and the number of wells needed for the gel. For quantification of the protein standards, we run mini-gels with the BSA standards and 2-3 replicates of the protein standard (GST-Smad2 or phospho-MH2 polypeptide) (14, 15). For quantification of the Smad2 transcription factor under basal conditions, we use 15-well minigels loaded with the molecular weight marker, 6 standards, and 6 independent cell lysates (14). For quantification of phospho-Smad2 molecules per cell during TGF-β signaling, we run a large 20-well gel (15). For mini-gels, follow the manufacturer's instructions. We provide instructions for making and running large gels below.
Wash the glass plates and combs with dish soap and water, followed by wiping down with a Kimwipe and glass cleaning solution or ethanol.
Prepare gel-casting bags by heat-sealing the polypropylene bags using a heat sealer. The bag dimensions should just be big enough to snugly fit the front and rear glass plates and spacers for two gels. Check for leaks by filling with water and looking for drips. Empty the bag.
Very carefully, insert the glass plates for two gels into the gel-casting bag. In particular, be careful not to cause any small tears in the bag, because this will cause leaks upon pouring the gels (see Note 3). Insert two spacers in between each set of front and rear glass plates at their lateral edges.
Using large office supply binder clamps, sandwich an additional glass plate to the front and back of the gel-casting bag.
Insert one of the combs, measure approximately 1 cm below the teeth, and mark this spot on the glass plate used to sandwich the gel plates. This represents the line to which the running gel will be poured. Remove the comb.
In a 125 mL filtering flask, combine 21 mL H2O, 15.6 mL resolving gel buffer, 25 mL Protogel, and 625 μL 10% APS. Degas gel for about 1 min by capping flask with a rubber stopper and attaching to a vacuum pump. Add 23 μL TEMED to solution, swirl gently, and immediately pour the gel (see Note 4).
Pipette 500 μL butanol slowly and evenly onto gel. Avoid disturbing the gel otherwise during polymerization (∼15–30 min). Let the gel solution remaining in the flask polymerize. When that gel is polymerized, so should be the poured gel. Another sign is the presence of excluded H2O in a separate layer under the butanol at the top of the polymerized gel.
Soak up the butanol with filter paper. Remove the polymerized gel from the filter flask and wash. Rinse the flask with distilled H2O, and dry it with paper towels.
Prepare the stacking gel by combining 18.75 mL H2O, 7.5 mL stacking gel buffer, 4.5 mL protogel, and 300 μL 10% APS. Degas the solution for about 1 min. Add 23 μL TEMED, and pour the gel up to the top of the glass plates. Insert combs, ensuring that no bubble gets between the teeth (small bubbles below the teeth are acceptable). Let the gel polymerize for 15-30 min, and use the gel immediately or store in electrophoresis buffer at 4°C for up to 3 weeks.
Remove the combs carefully by clamping the glass plates at the bottom with an office supply binder clamp and gently and evenly pulling out the comb. The vacuum created by removing the combs will distort the well walls. Use a narrow strip of 1 mm thick Teflon to straighten the well walls.
Set up the gel apparatus, and fill the upper and lower reservoirs with electrophoresis buffer. Wash out the gel wells with electrophoresis buffer using a 5 mL syringe fitted with a 20 g1 needle.
Load gels using pipettor fitted with gel-loading pipette tips (see Note 5). Load 3–5 μL of the molecular weight marker, and load any empty wells with the same volume of 4× SDS buffer that is included in each sample.
Attach the gel apparatus to the power supply, and run on constant current mode at ∼35 mA per gel. If desired, the gel can be run at lower currents to prolong the run time (e.g., overnight).
Run gel until the dye line has passed 5.5 cm from the border of the stacking and resolving gels. This is to ensure that all the proteins from the sample will be transferred onto a membrane that fits in the blotting containers.
3.4. Coomassie Staining
Carefully remove the resolving gel from the plates by cutting the stacking gel with a gel tool and by gently lifting the gel off the plates and placing it into the stain solution. Incubate the gel at room temperature for 1 h on a rocking shaker at a low speed (e.g., 3 out of 10).
Destain the gel by replacing the stain solution with destain solution. Replace the destain solution periodically (e.g., every 10–30 min) until the unbound stain has exited the gel and the stained bands are left behind (Figs. 1a and 2a). Document the gel, and perform densitometry (see Subheading 3.7).
3.5. Immunoblotting
Cut a piece of nitrocellulose membrane and four pieces of 0.8 mm thick filter paper to dimensions that match the intended size of the section of gel to be transferred. Cut a small notch in the upper left-hand corner to orient the membrane. Wet nitrocellulose membrane with H2O, and store in transfer buffer until use. Keep filter paper dry until just prior to use.
When the gel is finished running, lay the plates flat and gently remove the front plate using a gel tool. Cut off the stacking gel, and cut the unused portion of the resolving gel such that the dimensions of the resolving gel containing the separated proteins are the same as those of the membrane and filter paper. Equilibrate the gel in transfer buffer for about 5 min.
Load and run the transfer unit according to manufacturer's instructions. Briefly, create a sandwich (in order from bottom to top) of two 0.8 mm thick transfer-buffer-soaked pieces of filter paper, the wetted nitrocellulose membrane, the gel, and two more pieces of soaked filter paper. As each layer is added, gently roll a test tube along the layer to press out any bubbles.
Upon completion of the transfer, check its effectiveness by soaking the membrane in Ponceau S solution for a few seconds, and then rinse with H2O until the bands are clearly visible.
Perform the following steps at room temperature on a rocking shaker set at low speed. Block the membrane by incubating it for 1 h in ∼10 mL blocking buffer.
Replace the blocking buffer with primary antibody, and incubate the membrane for ∼2 h (see Note 6). Afterwards, collect primary antibody back into its tube and refreeze. Wash the membrane 3 × 5 min in TBS-T (see Note 7).
Incubate the blot in secondary antibody (5–6 mL for a small membrane and 8 mL for a large membrane) for 45–60 min (see Note 8) followed by washing the membrane 3 × 10 min in TBS-T. Discard the used secondary antibody.
Detect the blot by enhanced chemiluminescence (see Note 9) and visualize using autoradiography film. Perform exposures of several durations (typically between 30 s and 5 min) to optimize the signal relative to the background (e.g., Fig. 1c and Fig. 2c). Afterwards, align the developed film with the membrane, and mark the location of the molecular weight markers. Dry the membrane, label, and store (see Note 10).
3.6. Densitometry Using ImageJ
For Coomassie-stained gels, use a gel documentation system (e.g., AlphaImager, AlphaInnotech Corp.) to obtain a digital image of the gel. For immunoblots, scan the film using a digital scanner (e.g., we use a UMAX PowerLook III with MagicScan v4.5 software) in transmissive mode, grayscale, and at a resolution of 300–600 dots per inch. Save as TIFF files.
Perform densitometry using ImageJ (Scion Corp.). Open the file containing the gel image, and ensure that the corresponding bands in each lane are as horizontal as possible by rotating the image (see Note 11).
Define boundaries for each band by tracing a vertical line and pressing “Backspace” in the space between each band and at the outer edges of the first and last bands. This deletes pixel density between the bands, which demarcates the bands in subsequent steps.
Trace a box around the entire set of bands and press <Ctrl-1>. If bands from different molecular weights must be quantified (which is typically the case if tagged recombinant proteins or protein fragments are used as standards), then move the box around those bands and press <Ctrl-2>. Press <Ctrl-3> to display the plotted optical densities (see Note 12).
In the plotted optical densities window, trace a horizontal line across the bands at the level of the optical densities at the outer edges of the gel. This removes the background optical density from each lane.
Quantify each band density using the magic wand tool by clicking within the space defined by each band and the background line. A yellow tracing should appear with each click showing that the band has been quantified. The results window will appear automatically with the densitometric measurements (arbitrary units).
Select “Copy All” from the Edit menu, and paste the results into a spreadsheet.
3.7. Data Analysis and Calculations for Coomassie-Stained Gels Using Microsoft Excel
Plot the mass of BSA in each lane versus the corresponding OD. Examine the shape of the curve – if the curve appears reasonably linear, go to step 2 (Fig. 1a). If the standard curve resembles an S-shape (Fig. 2a), a three-parameter logistic equation (or Hill equation) will be required, and its use is described in steps 4–9 of this subheading.
In the case of a linear standard curve, use Excel's Add Trendline feature by right-clicking on the data points within the plot. Select “Add Trendline,” and then select the “Linear” type. Click the “Options” tab, and check the “Display Equation on Chart” option and the “Display the R2-value on chart” if desired (as one way to evaluate the goodness of fit). The equation will be of the form y = mx + b, where y = OD, m = slope of the line, x = mass of BSA per lane, and b = the y-intercept (Fig. 1b).
Interpolate the mass of protein per lane for the protein standards by solving the equation with respect to x, substituting the OD values of the protein standards into the equation and calculating the mass of protein per lane. Compute the mean and standard deviation if multiple replicates of the protein standard were measured.
-
If an S-shaped curve results, then the following equation can be used to fit the data:
where OD is the optical density of the band, ODmax is the asymptotic maximum OD, K is the protein abundance that confers the 50% of ODmax, and n is a parameter that determines the steepness of the S-curve. This equation is most easily solved using a numerical approach implemented using a spreadsheet.
First, generate the standard curve equation by estimating the values for the parameters ODmax, K, and n. This equation will then be used to estimate protein abundances that correspond to the experimentally observed OD values. Start by entering the model parameters in three cells just below the data columns. Enter guess values for ODmax, K, and n. Sensible guess values include the maximum observed OD value for ODmax, the mass of BSA that approximately corresponds to the OD that is half of ODmax for K, and between 0.5 and 2 for n.
In the column next to the data columns, enter the model equation for each BSA mass using the parameters entered above (use absolute cell references or cell names).
In the following column, calculate the “error” or “residuals” by subtracting the modeled OD value by the observed OD value. At the bottom of this column, use the SUMSQ function applied to residuals to calculate the sum of squared errors (SSE).
Use Excel's Solver tool to find the parameter values that best fit the data. From the menu bar, select Tools, and from the dropdown menu, select Solver (see Note 13). In the Solver dialog box, set the “Target Cell” as the cell containing the SSE value, choose the “Min” radio button for the “Equal to” option, enter the cells containing the model parameters in the “By Changing Cells” box, and press “Solve” (see Note 14). The SSE value should change to a smaller value, the model parameter values should change, and, in the standard curve plot, the model line should run through the data points (Fig. 2b). The Solver step should be repeated several times to ensure convergence (i.e., the solution ceases to change).
Use the standard curve to interpolate the protein masses per band for the protein standards. In the column adjacent to the OD values corresponding to the experimental samples, enter guess values for the protein abundances of the protein standards using the standard curve data as a guide.
In the adjacent column, write the model equation with the fitted parameters to calculate an OD value corresponding to the guess value.
Subtract the calculated OD value from the observed OD value (the residual) and square it.
For each protein standard replicate, use Excel's Solver tool to minimize the squared error value by changing the guess value for protein abundance. Repeat the Solver step until convergence is reached (i.e., until the calculated OD and observed OD values are equal through at least three decimal places, which should correspond to a very low squared error value). Calculate the mean and standard deviation of the protein standard masses if more than one replicate was run on the gel.
Calculate the concentration of the protein standard by dividing the mass by the volume of protein standard loaded on the gel.
3.8. Data Analysis and Calculations for Immunoblot Data Using Microsoft Excel
Estimate protein molecular weights using the ExPASy “Compute pI/MW” tool (http://www.expasy.ch/tools/ pi_tool.html), entering the primary sequence of the proteins as the input. Using this method, we found that Smad2 has a predicted molecular weight (MW) of 52,306 Da, GST-Smad2 has a predicted MW of ∼78,306 Da, and phospho-MH2 polypeptide has a predicted MW of 25,517 Da.
Create the standard curve by fitting an appropriate function to the data. First, express the standard curve in the correct units. Because antibody staining is proportional to the number of epitopes present in the sample, express the concentration of the protein standard in units of moles per microliter by dividing the concentration (in units of g/μL) by the molecular weight of the protein standard.
Calculate the protein abundance per band (in units of mol) of the standards by multiplying the protein standard concentrations by the volume of standard loaded. (If desired, the protein abundance can also be expressed as the number of molecules per band simply by multiplying by Avogadro's number, instead of performing the same operation in step 9.)
Plot the known protein abundances versus the OD values. The shape of the curve will likely be either reasonably linear (Fig. 22.1d) or resemble the power law form (Fig. 2d).
If the curve is linear, use the same procedure outlined in Subheading 3.7 steps 2–3 to generate the standard curve and to interpolate the protein abundances per band from the experimental samples.
-
If the curve resembles a power-law function, enter the following equation adjacent to the observed OD values:
where C and n are the model parameters and Protein abundance is the abundance of protein per band calculated in step 3. Choose initial guess values for C and n. Plot the calculated values alongside the experimental data, and manually adjust the parameters until the model curve is reasonably close to the data points. A reasonable initial guess value for n should be between 0.3 and 0.7, as we and others (3) typically observe fitted values of n from this range. Choose C such that the plot of the model curve scales well with the data.
Follow the procedure described in Subheading 3.7 steps 4–8 to fit the C and n parameters to the data. Briefly, calculate the residuals for the modeled and observed OD values, calculate the SSE, and minimize the SSE by using Excel's Solver tool to change the values of C and n until the minimum SSE is found (Fig. 2d).
- Use the equation with fitted parameter values to estimate the protein abundances in the experimental samples from their corresponding optical densities. The solution to the power law equation with respect to Protein abundance is
Convert the protein abundance per band into molecules of protein per band by multiplying Protein abundance by Avogadro's number (6.02 × 1023 molecules/mol) (if not already done in step 3 for the standard).
-
Calculate the cell concentration:
For example, we estimated 1.5 × 106 cells in the lysates of the phospho-Smad2 kinetics experiment (Fig. 2), and each well was lysed with 200 μL of lysis buffer. Therefore, we estimate a cell concentration of 7.5 × 103 cells/μL. The result is then multiplied by the volume of lysate loaded to estimate the number of cells that contributed to the signal of the corresponding band in the immunoblot.
Calculate the number of molecules per cell by dividing the number of molecules per band by the number of cells.
4. Notes
To bring the protein concentration of the lysate within the range of the standard curve, we dilute the lysate 10× in water, e.g., 3 μL of lysate+ 27 μL of H2O, followed by loading 10 μL of the diluted lysate and combining with 200 μL of the BCA working reagent in two separate replicates. Cover the microplate with parafilm, and incubate at 37°C for 30 min. It is also possible to use the Bradford method, but our lysis buffer reacts with the Bradford reagent, resulting in high background, and the standard curve is not as linear as that for the BCA assay.
Loading the optimal (not necessarily the most) amount of sample is imperative for achieving clear separation between the bands in adjacent lanes. Our experience has been that gels loaded with too much protein tend to cause the adjacent bands to overlap.
We line up the glass plates upside down and slowly slide the gel-casting bag down over the plates, taking care to avoid cutting the bag with the leading corners of the plates.
Pour the resolving gel at a consistent rate until the gel line approaches the marked line. Let it settle for a few seconds, and then pour slowly until the gel line matches up with the marked line. Watch gel for leaks. Minor slow leaks can be replenished as needed until leaks cease as the gel polymerizes. Note that the use of properly poured, relatively fresh gels is crucial for obtaining sharp, straight bands. Distorted or smeared bands usually result from imperfections in the gel and are unusable for quantification purposes.
Insert tip as far into well as possible and slowly pipette sample into well until the sample lies above the tip orifice. Continue pipetting while slowly drawing tip upward to the first stop of the pipette. Purge remaining sample with tip orifice well above the sample (to avoid blowing out sample from well).
Using concentrated primary antibody exceeding the usual recommended dilution is typically needed for endogenous protein detection. Our antibodies are often diluted to 1:150–1:250. We also emphasize the need to have sufficient volume of antibody solutions to maximize the contact time of the antibody solution with the entire surface of the membrane. For mini-gels (5 × 8 cm), we typically use >6 mL of antibody solution, and for large gels (14 × 5.5 cm), we use 10–15 mL of antibody solution. While this might seem exhorbitant, we save reagent and costs by reusing the antibody stock multiple times and freezing between uses at −20°C (we often use the same antibody stock ten or more times this way). In addition, we have observed that antibody incubations performed at room temperature for as long as feasibly possible (we typically do 2 h incubations) confer better signal than incubations performed overnight at 4°C.
We shorten the wash times from the manufacturer's recommendation and adjust the Tween concentration in the Tris-buffered saline-Tween (TBS-T) wash solution to achieve a blot with good signal and low background. Typical wash times are 3 × 5 min after the primary antibody and 3 × 10 min after the secondary antibody incubation, and the Tween concentration is 0.05% (v/v) and increased to 0.1% if there is too much background.
We keep the secondary antibody incubation to less than 1 h. Intuitively, one might think that prolonged secondary antibody incubations would confer stronger signal. This is not true, because in standard immunoblotting technique, the secondary antibody solution contains no primary antibody. Since antibody binding is reversible, primary antibodies bound to the membrane will experience a driving force to dissociate according to the laws of equilibrium, thus reducing the overall signal if left too long.
To detect endogenous proteins (which are often expressed at levels that are difficult to detect), we use Thermo Scientific (formerly Pierce) SuperSignal West Dura Extended Duration Substrate (34076). This chemiluminescent substrate delivers a much stronger and longer-lasting signal than standard chemiluminescent reagents.
Nitrocellulose (and presumably PVDF) membranes can be readily dried and rewetted at a later time for subsequent use with no loss of signal. Therefore, whenever a membrane is not in use, we always dry and store it in a plastic sheet protector in a binder. One caveat is that membranes become brittle over time, and great care is needed to avoid tearing the membrane until rewetted.
We do not use ImageJ's “Subtract background” function, because the optical densities of the bands can be unevenly affected. Instead, we process the background as part of our quantification procedure.
The box should be just large enough to encompass all the bands, with some space at the outer edges to measure the background levels. If multiple ranges of bands are to be quantified, the same box must be used to ensure compatibility between sampled ranges. Therefore, the box dimensions must be sufficiently large to incorporate the bands with the largest area and all the bands within each range.
If the Solver option does not appear, it may need to be installed.
This step should be repeated several times until convergence is reached. In addition, different initial guesses for the parameter values should also be tried to enhance the probability of finding the best-fit parameter values.
Acknowledgments
We thank Scott Dixon, Dana Ungermannova, and Meredith Brown for critical reading of the manuscript. This work was supported by a National Institutes of Health grant (GM083172) to X. L.
References
- 1.Clarke DC, Liu X. Decoding the quantitative nature of TGF-β signaling. Trends Cell Biol. 2008;18:430–442. doi: 10.1016/j.tcb.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- 3.Pitre A, Pan Y, Pruett S, Skalli O. On the use of ratio standard curves to accurately quantitate relative changes in protein levels by Western blot. Anal Biochem. 2007;361:305–307. doi: 10.1016/j.ab.2006.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 6.Pierce. Extinction coefficients. Rockford, IL: 2006. [Google Scholar]
- 7.Hua X, Liu X, Ansari DO, Lodish HF. Synergistic cooperation of TFE3 and smad proteins in TGF-beta-induced transcription of the plasminogen activator inhibitor-1 gene. Genes Dev. 1998;12:3084–3095. doi: 10.1101/gad.12.19.3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Macdonald M, Wan Y, Wang W, et al. Control of cell cycle-dependent degradation of c-Ski proto-oncoprotein by Cdc34. Oncogene. 2004;23:5643–5653. doi: 10.1038/sj.onc.1207733. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y, Feng X, We R, Derynck R. Receptor-associated Mad homologues synergize as effectors of the TGF-beta response. Nature. 1996;383:168–172. doi: 10.1038/383168a0. [DOI] [PubMed] [Google Scholar]
- 10.Muralidharan V, Muir TW. Protein ligation: an enabling technology for the biophysical analysis of proteins. Nat Methods. 2006;3:429–438. doi: 10.1038/nmeth886. [DOI] [PubMed] [Google Scholar]
- 11.Muir TW. Semisynthesis of proteins by expressed protein ligation. Annu Rev Biochem. 2003;72:249–289. doi: 10.1146/annurev.biochem.72.121801.161900. [DOI] [PubMed] [Google Scholar]
- 12.Wu JW, Hu M, Chai J, et al. Crystal structure of a phosphorylated Smad2. Recognition of phosphoserine by the MH2 domain and insights on Smad function in TGF-beta signaling. Mol Cell. 2001;8:1277–1289. doi: 10.1016/s1097-2765(01)00421-x. [DOI] [PubMed] [Google Scholar]
- 13.Gordon JA. Use of vanadate as protein-phosphotyrosine phosphatase inhibitor. Methods Enzymol. 1991;201:477–482. doi: 10.1016/0076-6879(91)01043-2. [DOI] [PubMed] [Google Scholar]
- 14.Clarke DC, Betterton MB, Liu X. Systems theory of Smad signalling. Systems Biology (Stevenage) 2006;153:412–424. doi: 10.1049/ip-syb:20050055. [DOI] [PubMed] [Google Scholar]
- 15.Clarke D, Brown M, Erickson R, Shi Y, Liu X. Transforming growth factor-beta depletion is the primary determinant of Smad signaling kinetics. Mol Cell Biol. 2009;29:2443–2455. doi: 10.1128/MCB.01443-08. [DOI] [PMC free article] [PubMed] [Google Scholar]