

Abstract
Cell death has been extensively evaluated for decades and it is well recognized that pharmacological interventions directed to inhibit cell death can prevent significant cell loss and can thus improve an organ’s physiological function. For long, only apoptosis was considered as a sole form of programmed cell death. Recently necroptosis, a RIP1/RIP3-dependent programmed cell death, has been identified as an apoptotic backup cell death mechanism with necrotic morphology. The evidences of necroptosis and protective effects achieved by blocking necroptosis have been extensively reported in recent past. However, only a few studies reported the evidence of necroptosis and protective effects achieved by inhibiting necroptosis in liver related disease conditions. Although the number of necroptosis initiators is increasing; however, interestingly, it is still unclear that what actually triggers necroptosis in different liver diseases or if there is always a different necroptosis initiator in each specific disease condition followed by specific downstream signaling molecules. Understanding the precise mechanism of necroptosis as well as counteracting other cell death pathways in liver diseases could provide a useful insight towards achieving extensive therapeutic significance. By targeting necroptosis and/or other parallel death pathways, a significant cell loss and thus a decrement in an organ’s physiological function can be prevented.
Keywords: Necroptosis, Programmed necrosis, Apoptosis, Cell death, Liver disease
Core tip: Necroptosis has been identified as apoptotic “back up” cell death mechanism. The evidence of necroptosis and protective effects achieved by blocking necroptosis have been extensively reported in recent past such as in renal ischemic/reperfusion injury, myocardial infarction, and acute pancreatitis. However, only a limited number of studies reported necroptosis evidence and significance of key necroptosis molecules, RIP1 and RIP3, in liver related disease conditions. The current review focuses on evidence of necroptosis in liver related disease conditions as well as potential significance of other programmed necrosis pathways.
INTRODUCTION
The study of cell death and survival has been under extensive exploration. However, it is still not known how a cell critically decides to live or die. A cell can be labeled dead if it meets one of following criteria: loss of plasma membrane integrity and incorporation of vital dyes in vitro, cell fragmentation, and/or in vivo engulfment of cell fragments by neighboring cells[1]. Historically cell death mechanism can be broadly classified into regulated and unregulated. Apoptosis and necrosis are examples of regulated and unregulated types of cell deaths respectively, however; there are also evidences of other overlapping types of cell deaths with therapeutic potentials[2]. Moreover, even the exposure to the same death inducing substance at different concentrations can produce a mixed picture of cell deaths in which a diversity of different cell death types can be observed[3,4]. Regulated cell death is genetically controlled and unregulated cell death is due to failure of cell to overcome extreme stresses[2,4]. Among the various cell death types to be explored, apoptosis and necrosis are well-known morphologically distinct types of cell deaths[2,3]. Apoptosis, necrosis and autophagy are major distinct types of cell death each with a specific molecular, biochemical and morphological characteristics[1,4].
Necroptosis is a non-apoptotic backup, necrosis-like cell death mechanism, which is initiated when apoptosis is blocked[5]. Apoptotic pathway is a caspase-dependent dominant cell death pathway while necroptotic pathway is dependent on kinase cascade. A family of kinase activity containing proteins known as receptor interacting proteins (RIPs) are essential cell stress sensors[6]. The current review focuses on necroptosis, a caspase-independent programmed cell death, and potential protective effects achieved by intervening necroptosis in liver diseases.
NECROPTOSIS: EMERGING CONCEPT OF PROGRAMED CELL NECROSIS
Chan and colleagues introduced the term “programmed necrosis” for an alternative RIP-mediated form of cell death, which is morphologically distinct from apoptosis and is dependent on tumor necrosis factor receptor, Fas and tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) receptors activation[7]. RIPs are essential for necroptosis execution; for pro-necrotic complex formation, the kinase activities of RIP1 and RIP3 are essential and are tightly regulated within the necrosome[8]. There is an increasing number of necroptosis initiators; however, TNF-α induced necroptosis is extensively studied and reported[9,10] (Figure 1). Moreover, it is also still not known whether the different necroptosis inducers follow the same downstream signaling pathway[10]. The RIP1 and RIP3 interact with each other through homotypic interaction motif at their C terminus[11]. RIP1 is thought be a crucial kinase making the decision of cell survival or death[12]. RIP1 has three domains; a serine/threonine kinase domain essential for necroptosis, an intermediate domain, containing homotypic interaction motif, for nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and a death domain for apoptosis activation[13]. RIP1 ubiquitination promotes cell survival pathway while de-ubiquitination promotes kinase dependent cell death pathway[10]. As RIP1 has multiple domains, its activation can result in multiple outcomes such as NF-κB, mitogen-activated protein kinase (MAPK), apoptosis or necrosis[12]; however, only the kinase activity of RIP1 was reported to be essential for necroptosis execution but not for other pathways[8]. The necrostatin-1 (nec-1), a small potent molecule, blocks RIP1 kinase activity and thus blocks death receptors induced necroptosis[5]. Cho et al[8] reported that RIP3 controls programmed necrosis and found that RIP3 augments RIP1 recruitment to necrosome. Furthermore, apart from RIP1 and RIP3, several other kinases were also thought to be involved in phosphorylation of RIP1 and RIP3.
Figure 1.
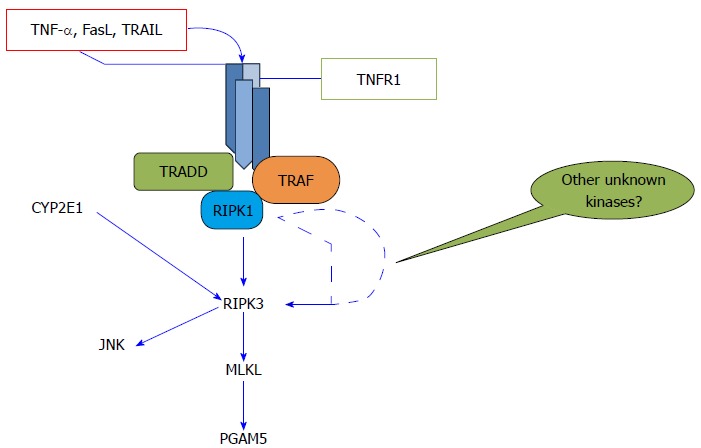
Diagrammatic representation of necroptosis pathway. Apoptosis and necroptosis can share the same death inducers molecules such as tumor necrosis factor-α (TNF-α), FasL, and TNF-related apoptosis-inducing ligand (TRAIL). The intracellular domain of TNF receptor (TNFR)1 further recruits many intracellular proteins including TNF receptor-associated death domain protein (TRADD), TNF receptor associated factor (TRAF), and receptor-interacting serine-threonine kinase 1 (RIP1). RIP1 phosphorylates and activates RIP3 which later forms the complex together with mixed lineage kinase domain-like (MLKL). C-Jun N-terminal kinase (JNK) and MLKL act downstream of RIP3. There is a possibility that some other unknown kinases might be acting between RIP1 and RIP3 which could explain why certain liver diseases do not show the protective effects of necrostatin-1.
Upon activation, RIP3 is reported to activate a number of different downstream signals such as contribution to necroptosis by formation of necrosome and later the activation of mixed-lineage kinase domain-like (MLKL), phosphoglycerate mutase 5 (PGAM5) and finally dynamin-related protein (Drp1) to induce reactive oxygen species (ROS) production and membrane permeability[14]. Moreover, through JNK activation, RIP1/RIP3 kinase cascade is also reported to regulate mitochondrial oxidative stress[15-17]. The increased RIP3 expression is also thought to be related to increased expression of pro-inflammatory cytokines[14,16] and inflammasome activation[14]. Furthermore, RIP3 is also reported to be linked with energy metabolism and to determine the switch between apoptosis and necrosis[18]. Recently, RIP3 inhibition has also been reported to increase allograft survival in renal ischemic reperfusion injury model[19,20].
CROSSTALK BETWEEN NECROPTOSIS AND APOPTOSIS
Despite morphologically different and distinct molecular machinery involved in execution of different death pathways, there is a complex interaction and crosstalk between necroptosis and apoptosis[4]. In general, the blockage of single pathway does not block the cell death entirely; however, it prone the cell to choose an alternative death pathway[21]. Under apoptosis deficient conditions such as caspase inhibition by zVAD, cells can opt a type of programmed cell death pathway termed as programmed necrosis or necroptosis[5]. The existence of complex crosstalk and overlaps between different cell death pathways has indeed made it difficult to determine what actually defines cell’s priority to choose one pathway over another. Several studies pointed towards the involvement of different molecular mechanisms to determine this switch. For instance, the blockage of apoptotic pathway by pan-caspase inhibitors (e.g., zVAD) can lead to cell’s switch to necroptotic pathway, alternatively, the blockage of apoptosis is not necessary for necroptosis execution[5,22], and in neuronal subpopulation necroptosis might be a primary death mechanism[5]. RIP1 and RIP3 are thought to be critical molecules contributing in cell’s switching from apoptotic to necroptotic pathway[13,23,24]. Festjens et al[12] described that RIP1 integrates several upstream signals and is a critical decision maker in cell’s fate to live or die. Moreover, it was also suggested that the RIP1 kinase domain might have a role in shifts between cell survival and death[25]. Depending on the circumstances RIP1 can contribute to NF-κB, MAPK, apoptosis or necrosis activation[12]. Moreover, the cell shifts from apoptosis to necrosis when it has depleted energy stores such as during DNA repair by poly-ADP-ribose polymerase (PARP) activation[26]. Interestingly, the extracellular pH was also found to switch the TRAIL-induced apoptosis to necroptosis in human HT29 and HepG2 cells[27]. Despite the number of evidences reporting the switches between different pathways, it is still unclear what exactly determines the cell’s preference to choose one death pathway over another.
EVIDENCE OF NECROPTOSIS IN LIVER DISEASES
The evidence and evaluation of protective effects mediated by necroptosis inhibition have been reported in a number of disease conditions such as renal ischemic reperfusion injury[28], myocardial infarction[5], acute pancreatitis[24], retinal detachment[29], and traumatic brain injury[30]. However, recently, only a few studies reported the necroptosis evidences and protective effects achieved through inhibition of necroptosis in liver related disease conditions.
Roychowdhury et al[31] reported that ethanol feeding activates both apoptotic as well as non-apoptotic cell death pathways. Ethanol induced RIP3 expression which was independent of presence or absence of caspase inhibitor. Moreover, blocking apoptosis alone by using Bid deficient mice or VX166, a pan-caspase inhibitor, was not sufficient to prevent ethanol induced liver injury. Roychowdhury et al[16] also found that RIP3 deficiency did not have any effect on ethanol-induced apoptosis. Chronic ethanol fed mice showed an increased RIP3 expression; however, RIP1 expression remained the same. Moreover, the liver biopsies of alcoholic liver disease patients also showed an increased RIP3 expression, which signifies the execution of necroptosis in human hepatic pathologies as well. Furthermore, cytochrome P450 2E1 (CYP2E1)-deficient mice failed to induce RIP3, suggesting that CYP2E1 acts upstream of RIP3 in ethanol mediated liver injury. RIP3-deficient mice also showed reduced pJNK positive hepatocytes and were protected against ethanol medicated hepatocyte injury and pro-inflammatory cytokines expression[16].
In another study, acetaminophen (APAP) toxicity induced RIP3 expression, increased alanine aminotransferase (ALT) levels, and led to extensive necrosis. RIP3 morpholinos treatment targeting RIP3 and use of RIP3 deficient mice not only decreased ALT levels and oxidative stress but also altered mitochondrial function. The reduced RIP3 expression reduced JNK and Drp1 activation, which later reduced the mitochondrial oxidative stress, fission and finally prevented the necrosis. Moreover, RIP3 deficient mice and RIP3 anti-sense morpholinos also attenuated necrotic cell death in vivo as well as in vitro by decreasing the activation and translocation of JNK and Drp1[17]. Similar results were also reported in two other studies that nec-1 is protective against APAP-induced hepatocytes toxicity and RIP1/RIP3 act upstream of JNK activation[15,32].
In concanavalin A (ConA) induced murine hepatitis model nec-1 was found to protect against ConA-induced hepatitis and decreased serum aspartate aminotransferase and ALT and PARP-1 expression[33]. Another study also reported similar results that nec-1 not only reduced ConA induced acute liver injury and but also improved liver histology, reduced liver enzymes, inflammatory cytokines, and improved animal survival[34]. Moreover, nec-1 decreased RIP1, TNF-α, interferon-γ, interleukin (IL)-2 and IL-6 expressions; however, interestingly, nec-1 also inhibited autophagy in vitro[34]. In another study, treatment of septic mice with nec-1 was found to be damaging for hepatocytes. Nec-1 treatment altered liver glycogen contents and increased serum liver injury markers, pro-inflammatory cytokines, and caspase-3 activity[25].
RIP1 DEPENDENT AND RIP1 INDEPENDENT NECROPTOSIS
Although, RIP1 kinase is thought to be the key molecule regulating necroptosis; however, the current reports regarding protective effects achieved through RIP1 inhibition by nec-1 in liver related disease conditions are inconsistent. For instance, in Con-A induced hepatitis[33,34] and in APAP toxicity[17,32] models, RIP1 inhibition by nec-1 is reported to be protective while in alcoholic liver disease model[16], only RIP3 inhibition was protective and RIP1 inhibition by nec-1 had no protective effects at all (Table 1). Furthermore, interestingly, in septic mouse model nec-1 not only increased caspase-3 activity but was also hepatotoxic[25]. As there are a numbers of potential necroptosis executers, the disparity can be resolved by noticing first, that in each disease model, the initiator of necroptosis might be different. For example, in Con-A induced hepatitis model[33] TRAIL was reported to induce necroptosis while in alcoholic liver disease model[16] TNF-α was thought to induce necroptosis. However, in APAP toxicity model, the initiator of necroptosis is still not known[17]. Moreover, there are also evidences supporting that in certain conditions necroptosis is only RIP3 dependent and RIP1-independent[10,16,35,36]. Second, the RIP1/RIP3-dependent necroptosis and RIP3 alone dependent necroptosis could be representing the splitting of necroptosis pathway at the level of RIP3. There is a possibility that in RIP3 alone dependent necroptosis, the upstream kinase activating RIP3 could not be RIP1 but some other unknown kinase. Therefore, depending on disease condition necroptosis signal might be bypassing the kinase activity of RIP1. Cho et al[8] also pointed towards the possible existence of several other kinases to be involved in necroptosis kinase cascade and the possibility that RIP3 might be acting upstream of RIP1 and that some other unknown kinase might also be activating RIP3 downstream of RIP1. With recent evidences indicating the existence of distinct pathways of regulated necrosis[37,38], it is fascinating to speculate that both RIP1 and RIP3 mediated and RIP3 alone mediated necroptosis might also be representing the splitting of necroptosis pathway. It is yet to be answered which necroptosis executers promote the involvement of both RIP1 and RIP3 and which executers only promote the involvement of RIP3 alone. Furthermore, it should also be noticed that nec-1 is reported to depict a number of diverse effects which are sometimes independent of RIP1[22,30,39-41].
Table 1.
Brief summary of necroptosis and necrostatin-1 effects in different liver disease models
| Disease model | Probable necroptosis inducer | Downstream signaling | Nec-1 effects | Ref. |
| Ethanol induced liver injury | TNF-α | JNK acts downstream of RIP3 | No effect | [16] |
| Ethanol induced liver injury | TNF-α | No information | No information | [31] |
| APAP-induced liver toxicity | Unknown | JNK acts downstream of RIP3 | Protective | [17] |
| APAP-induced liver toxicity | Unknown | No information | Protective | [32] |
| APAP-induced liver toxicity | Unknown | JNK acts downstream of RIP1 | Protective | [15] |
| Con-A induced hepatitis model | TRAIL | PARP-1 acts downstream of RIP3 | Protective | [33] |
| Con-A-induced hepatitis | Unknown | No information | Protective | [34] |
| Liver sepsis model | FasL | No information | Hepatotoxic | [25] |
APAP: Acetaminophen; Con-A: Concanavalin; TNF-α: Tumor necrosis factor-α; JNK: C-Jun N-terminal kinase; RIP3: Receptor-interacting serine-threonine kinase 3; TRAIL: TNF-related apoptosis-inducing ligand; Nec: Necrostatin.
Although several other forms of regulated necrosis such as PARP-1 mediated regulated necrosis, mitochondrial permeability transition-induced necrosis, pyroptosis, ferroptosis have also been identified (Figure 2), the current knowledge about how these different regulated necrosis pathways interact with each other and what clinical significance can be achieved by combined inhibition of these pathways is still limited[38]. The pyroptosis denotes a caspase-1-dependent cell death exhibiting necrotic and/or apoptotic morphologies and is associated with the release of IL-1β and IL-18[36], while the ferroptosis is an intracellular iron-dependent cell death with distinct morphology[42].
Figure 2.
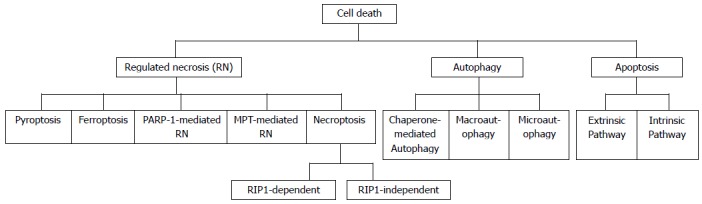
Types of cell deaths. The classification shown in not generalized and is presented only for descriptional purpose. RIP: Receptor interacting proteins.
OTHER TYPES OF PROGRAMMED NECROSIS; PARALLEL OR ALTERNATIVE PATHWAYS OF NECROSIS
Whenever the term programmed necrosis is used usually it refers to necroptosis. However, interestingly, in addition to necroptosis other pathways of programmed necrosis can also concurrently co-exist. Moreover, similar to apoptosis, necroptosis is also thought to be executed from both extrinsic as well as intrinsic mechanisms[43]. It is reported that TNF-α mediated necroptosis and PARP-1 mediated necrosis represent two distinct pathways of programmed necrosis that are separate from each other[37]. Linkermann et al[38] also pointed that in mouse model of kidney ischemic reperfusion injury, cypD-mediated and RIP3-mediated necrosis represent two distinct pathways of regulated necrosis and blockage of both pathways had additional protective effect than the blockage of an individual pathway. Interestingly, it is also reported that mitochondrial PGAM5 could be the converging point of multiple necrosis pathways[44]. Alternatively, the presence of mitochondria is not necessary for necroptosis execution[45]. Similarly, in hepatic APAP toxicity model despite RIP3 elimination the protection against necrosis was transient and effect was short lived and later thought to be dominated by some other alternate pathway[17] probably due to parallel existing alternative pathway of necrosis. Moreover, Sharma et al[32] also reported that as RIP1 induces oxidative stress through JNK activation and MLK3 controls the initial stage of JNK activation. The blockage of RIP1 or MLK3 activity did not had any effect on each other; however, the combined blockage was more protective against APAP toxicity probably due to independent mechanism or pathway.
The interaction between MAPK and necroptosis pathways as well as nature and signaling pathway of MLKL, a RIP3 downstream signaling molecule, are yet to be clarified. For instance, it is reported that MLKL is required for ROS generation and for late phase of JNK activation[46]. Alternatively, it is also reported that MLKL is a kinase dead protein[47]. Moreover, TNF-α and LPS induced JNK and p38 activation were unaffected with MLKL deletion suggesting that MLKL has no role in MAPK signaling activation[48]. However, to further clarify the pathway, there is still a need to find other molecular targets upstream and downstream of MLKL[46-48].
It is thus fascinating that in addition to RIP1/RIP3 mediated programmed necrosis several other parallel pathways of programmed necrosis could also co-exist. Moreover, in addition to blocking necroptosis by necrostatin-1 or by blocking RIP3, an additional blockage of other programmed necrosis pathway could significantly enhance the protection against organ damages[37,38]. However, the current knowledge about parallel existence of other programmed necrosis pathways in various liver diseases is limited and demands future evaluation. There is a need to understand and explore the potential role of other regulated necrosis pathways in various liver disease models.
CONCLUSION
There is an increasing need to comprehend the exact mechanism of necroptosis as well as functional tasks of RIP1, RIP3, and MLKL in liver related disease conditions. The protective effects of nec-1 have been reported in a number of liver disease conditions, and nec-1 might be helpful in understanding necroptosis as well as contribution of other potential death pathways in liver related diseases. However, it is still not clear whether the necroptosis in liver related disease conditions is RIP3-dependent alone or both RIP1 and RIP3 together are involved in these conditions. As several other kinases might be involved in necroptosis signaling[8], and some studies only report RIP3-dependent necroptosis[16,49,50], and nec-1 has RIP1-independent effects as well[22], it is thus not clear whether to modulate the function of RIP1 and/or RIP3 would be beneficial in liver related disease conditions. Although as suggested by findings mentioned by Cho et al[22] and Takahashi et al[51] results obtained with nec-1 should be carefully interpreted and should be confirmed with other approaches, nec-1 still provides an excellent opportunity not only for understanding the mechanism of necroptosis in liver related clinical conditions but also for exploring new potential cell death pathways with therapeutic significance.
Footnotes
Supported by A grant of the Korea Healthcare technology R and D Project, Ministry of Health and Welfare, South Korea, NO. A121185
P- Reviewer: Chuang WL, Doganay L, Videla LA, Yang SC S- Editor: Gou SX L- Editor: A E- Editor: Ma S
References
- 1.Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Degterev A, Yuan J. Expansion and evolution of cell death programmes. Nat Rev Mol Cell Biol. 2008;9:378–390. doi: 10.1038/nrm2393. [DOI] [PubMed] [Google Scholar]
- 3.Majno G, Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol. 1995;146:3–15. [PMC free article] [PubMed] [Google Scholar]
- 4.Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361:1570–1583. doi: 10.1056/NEJMra0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 6.Meylan E, Tschopp J. The RIP kinases: crucial integrators of cellular stress. Trends Biochem Sci. 2005;30:151–159. doi: 10.1016/j.tibs.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 7.Chan FK, Shisler J, Bixby JG, Felices M, Zheng L, Appel M, Orenstein J, Moss B, Lenardo MJ. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem. 2003;278:51613–51621. doi: 10.1074/jbc.M305633200. [DOI] [PubMed] [Google Scholar]
- 8.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fulda S. The mechanism of necroptosis in normal and cancer cells. Cancer Biol Ther. 2013;14:999–1004. doi: 10.4161/cbt.26428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu W, Liu P, Li J. Necroptosis: an emerging form of programmed cell death. Crit Rev Oncol Hematol. 2012;82:249–258. doi: 10.1016/j.critrevonc.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 11.Sun X, Yin J, Starovasnik MA, Fairbrother WJ, Dixit VM. Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J Biol Chem. 2002;277:9505–9511. doi: 10.1074/jbc.M109488200. [DOI] [PubMed] [Google Scholar]
- 12.Festjens N, Vanden Berghe T, Cornelis S, Vandenabeele P. RIP1, a kinase on the crossroads of a cell’s decision to live or die. Cell Death Differ. 2007;14:400–410. doi: 10.1038/sj.cdd.4402085. [DOI] [PubMed] [Google Scholar]
- 13.Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–495. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 14.Moriwaki K, Chan FK. RIP3: a molecular switch for necrosis and inflammation. Genes Dev. 2013;27:1640–1649. doi: 10.1101/gad.223321.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.An J, Mehrhof F, Harms C, Lättig-Tünnemann G, Lee SL, Endres M, Li M, Sellge G, Mandić AD, Trautwein C, et al. ARC is a novel therapeutic approach against acetaminophen-induced hepatocellular necrosis. J Hepatol. 2013;58:297–305. doi: 10.1016/j.jhep.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 16.Roychowdhury S, McMullen MR, Pisano SG, Liu X, Nagy LE. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology. 2013;57:1773–1783. doi: 10.1002/hep.26200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramachandran A, McGill MR, Xie Y, Ni HM, Ding WX, Jaeschke H. Receptor interacting protein kinase 3 is a critical early mediator of acetaminophen-induced hepatocyte necrosis in mice. Hepatology. 2013;58:2099–2108. doi: 10.1002/hep.26547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 19.Mannon RB. Necroptosis in solid organ transplantation: a missing link to immune activation? Am J Transplant. 2013;13:2785–2786. doi: 10.1111/ajt.12450. [DOI] [PubMed] [Google Scholar]
- 20.Lau A, Wang S, Jiang J, Haig A, Pavlosky A, Linkermann A, Zhang ZX, Jevnikar AM. RIPK3-mediated necroptosis promotes donor kidney inflammatory injury and reduces allograft survival. Am J Transplant. 2013;13:2805–2818. doi: 10.1111/ajt.12447. [DOI] [PubMed] [Google Scholar]
- 21.Vandenabeele P, Vanden Berghe T, Festjens N. Caspase inhibitors promote alternative cell death pathways. Sci STKE. 2006;2006:pe44. doi: 10.1126/stke.3582006pe44. [DOI] [PubMed] [Google Scholar]
- 22.Cho Y, McQuade T, Zhang H, Zhang J, Chan FK. RIP1-dependent and independent effects of necrostatin-1 in necrosis and T cell activation. PLoS One. 2011;6:e23209. doi: 10.1371/journal.pone.0023209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 25.McNeal SI, LeGolvan MP, Chung CS, Ayala A. The dual functions of receptor interacting protein 1 in fas-induced hepatocyte death during sepsis. Shock. 2011;35:499–505. doi: 10.1097/SHK.0b013e31820b2db1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Los M, Mozoluk M, Ferrari D, Stepczynska A, Stroh C, Renz A, Herceg Z, Wang ZQ, Schulze-Osthoff K. Activation and caspase-mediated inhibition of PARP: a molecular switch between fibroblast necrosis and apoptosis in death receptor signaling. Mol Biol Cell. 2002;13:978–988. doi: 10.1091/mbc.01-05-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meurette O, Huc L, Rebillard A, Le Moigne G, Lagadic-Gossmann D, Dimanche-Boitrel MT. TRAIL (TNF-related apoptosis-inducing ligand) induces necrosis-like cell death in tumor cells at acidic extracellular pH. Ann N Y Acad Sci. 2005;1056:379–387. doi: 10.1196/annals.1352.018. [DOI] [PubMed] [Google Scholar]
- 28.Linkermann A, Bräsen JH, Himmerkus N, Liu S, Huber TB, Kunzendorf U, Krautwald S. Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int. 2012;81:751–761. doi: 10.1038/ki.2011.450. [DOI] [PubMed] [Google Scholar]
- 29.Rosenbaum DM, Degterev A, David J, Rosenbaum PS, Roth S, Grotta JC, Cuny GD, Yuan J, Savitz SI. Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J Neurosci Res. 2010;88:1569–1576. doi: 10.1002/jnr.22314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang YQ, Wang L, Zhang MY, Wang T, Bao HJ, Liu WL, Dai DK, Zhang L, Chang P, Dong WW, et al. Necrostatin-1 suppresses autophagy and apoptosis in mice traumatic brain injury model. Neurochem Res. 2012;37:1849–1858. doi: 10.1007/s11064-012-0791-4. [DOI] [PubMed] [Google Scholar]
- 31.Roychowdhury S, Chiang DJ, Mandal P, McMullen MR, Liu X, Cohen JI, Pollard J, Feldstein AE, Nagy LE. Inhibition of Apoptosis Protects Mice from Ethanol-Mediated Acceleration of Early Markers of CCl4-Induced Fibrosis but not Steatosis or Inflammation. Alcohol Clin Exp Res. 2012;36:1139–1147. doi: 10.1111/j.1530-0277.2011.01720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharma M, Gadang V, Jaeschke A. Critical role for mixed-lineage kinase 3 in acetaminophen-induced hepatotoxicity. Mol Pharmacol. 2012;82:1001–1007. doi: 10.1124/mol.112.079863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jouan-Lanhouet S, Arshad MI, Piquet-Pellorce C, Martin-Chouly C, Le Moigne-Muller G, Van Herreweghe F, Takahashi N, Sergent O, Lagadic-Gossmann D, Vandenabeele P, et al. TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ. 2012;19:2003–2014. doi: 10.1038/cdd.2012.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou Y, Dai W, Lin C, Wang F, He L, Shen M, Chen P, Wang C, Lu J, Xu L, et al. Protective effects of necrostatin-1 against concanavalin A-induced acute hepatic injury in mice. Mediators Inflamm. 2013;2013:706156. doi: 10.1155/2013/706156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vanlangenakker N, Bertrand MJ, Bogaert P, Vandenabeele P, Vanden Berghe T. TNF-induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis. 2011;2:e230. doi: 10.1038/cddis.2011.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sosna J, Voigt S, Mathieu S, Lange A, Thon L, Davarnia P, Herdegen T, Linkermann A, Rittger A, Chan FK, et al. TNF-induced necroptosis and PARP-1-mediated necrosis represent distinct routes to programmed necrotic cell death. Cell Mol Life Sci. 2014;71:331–348. doi: 10.1007/s00018-013-1381-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Linkermann A, Bräsen JH, Darding M, Jin MK, Sanz AB, Heller JO, De Zen F, Weinlich R, Ortiz A, Walczak H, et al. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci USA. 2013;110:12024–12029. doi: 10.1073/pnas.1305538110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang P, Dong W, Zhang M, Wang Z, Wang Y, Wang T, Gao Y, Meng H, Luo B, Luo C, et al. Anti-necroptosis chemical necrostatin-1 can also suppress apoptotic and autophagic pathway to exert neuroprotective effect in mice intracerebral hemorrhage model. J Mol Neurosci. 2014;52:242–249. doi: 10.1007/s12031-013-0132-3. [DOI] [PubMed] [Google Scholar]
- 40.Tamura Y, Chiba Y, Tanioka T, Shimizu N, Shinozaki S, Yamada M, Kaneki K, Mori S, Araki A, Ito H, et al. NO donor induces Nec-1-inhibitable, but RIP1-independent, necrotic cell death in pancreatic β-cells. FEBS Lett. 2011;585:3058–3064. doi: 10.1016/j.febslet.2011.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Biton S, Ashkenazi A. NEMO and RIP1 control cell fate in response to extensive DNA damage via TNF-α feedforward signaling. Cell. 2011;145:92–103. doi: 10.1016/j.cell.2011.02.023. [DOI] [PubMed] [Google Scholar]
- 42.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou Z, Han V, Han J. New components of the necroptotic pathway. Protein Cell. 2012;3:811–817. doi: 10.1007/s13238-012-2083-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012;148:228–243. doi: 10.1016/j.cell.2011.11.030. [DOI] [PubMed] [Google Scholar]
- 45.Tait SW, Oberst A, Quarato G, Milasta S, Haller M, Wang R, Karvela M, Ichim G, Yatim N, Albert ML, et al. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep. 2013;5:878–885. doi: 10.1016/j.celrep.2013.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, Liu ZG. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci USA. 2012;109:5322–5327. doi: 10.1073/pnas.1200012109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 48.Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, Ma J, Chen W, Zhang Y, Zhou X, et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 2013;23:994–1006. doi: 10.1038/cr.2013.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Upton JW, Kaiser WJ, Mocarski ES. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe. 2010;7:302–313. doi: 10.1016/j.chom.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Linkermann A, Bräsen JH, De Zen F, Weinlich R, Schwendener RA, Green DR, Kunzendorf U, Krautwald S. Dichotomy between RIP1- and RIP3-mediated necroptosis in tumor necrosis factor-α-induced shock. Mol Med. 2012;18:577–586. doi: 10.2119/molmed.2011.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takahashi N, Duprez L, Grootjans S, Cauwels A, Nerinckx W, DuHadaway JB, Goossens V, Roelandt R, Van Hauwermeiren F, Libert C, et al. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis. 2012;3:e437. doi: 10.1038/cddis.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]