

Abstract
AIM: To explore the role and mechanisms of extracellular signal-regulated protein kinase-mitogen-activated protein kinase (ERK-MAPK) signaling in pentagastrin-regulated growth of large intestinal carcinoma.
METHODS: HT-29 cells were incubated in different media and divided into the control group, pentagastrin group, proglumide group, and pentagastrin + proglumide group. No reagent was added to the control group, and other groups were incubated with reagent at different concentrations. Changes in proliferation of HT-29 cells were detected by MTT assay, and the optimal concentrations of pentagastrin and proglumide were determined. The changes in proliferation index (PI) and apoptosis rate (AR) of HT-29 cells were detected by Annexin V-fluorescein isothiocyanate flow cytometry. mRNA expression of pentagastrin receptor/cholecystokinin-B receptor (CCK-BR), ERK1/2 and K-ras were detected by reverse transcriptase polymerase chain reaction. The protein and phosphorylation level of ERK1/2 and K-ras were detected by western blotting. All data were analyzed by analysis of variance and SNK-q test.
RESULTS: The proliferation of HT-29 cells was stimulated by pentagastrin at a concentration of 6.25-100 mg/L, and the optimal concentration of pentagastrin was 25.0 mg/L (F = 31.36, P < 0.05). Proglumide had no obvious effect on the proliferation of HT-29 cells, while it significantly inhibited the proliferation of HT-29 cells stimulated by pentagastrin when the concentration of proglumide was 8.0-128.0 mg/L, and the optimal concentration was 32.0 mg/L (F = 24.31, P < 0.05). The PI of the pentagastrin (25.0 mg/L) group was 37.5% ± 5.2%, which was significantly higher than 27.7% ± 5.0% of the control group and 27.3% ± 5.8% of the pentagastrin (25.0 mg/L) + proglumide (32.0 mg/L) group (Q = 4.56-4.75, P < 0.05). The AR of the pentagastrin (25.0 mg/L) group was 1.9% ± 0.4%, which was significantly lower than 2.5% ± 0.4% of the control group and 2.4% ± 0.3% of the pentagastrin (25.0 mg/L) + proglumide (32.0 mg/L) group (Q = 4.23-4.06, P < 0.05). mRNA expression of CCK-BR was detected in HT-29 cells. The phosphorylation levels of ERK1/2 protein and phosphorylated K-ras protein of the pentagastrin group were 0.43% ± 0.04% and 0.45% ± 0.06%, which were significantly higher than 0.32% ± 0.02% and 0.31% ± 0.05% of the control group (Q = 7.78-4.95, P < 0.05), and 0.36% ± 0.01% and 0.35% ± 0.04% of the pentagastrin + proglumide group (Q = 5.72-4.08, P < 0.05). There were no significant differences in the mRNA and protein expression of ERK1/2 and K-ras among the control, pentagastrin, proglumide and pentagastrin + proglumide groups (F = 0.52, 0.72, 0.78, 0.28; P > 0.05).
CONCLUSION: Gastrin stimulates proliferation of HT-29 cells and inhibits apoptosis by upregulating phosphorylation of ERK and K-ras through the Ras-Raf-MEK1/2-ERK1/2 pathway, and this is restrained by proglumide.
Keywords: Gastrin, Mitogen-activated protein kinase, Extracellular signal-regulated protein kinase 1/2, K-ras, Large intestinal carcinoma
Core tip: We have demonstrated that large intestinal carcinoma is related to abnormal expression of gastrin. Gastrin promoted growth of large intestinal cancer cell via gastrin receptor, but the detailed molecular mechanism is not fully known. We elucidated the molecular mechanisms of gastrin-induced growth of large intestinal cancer in relation to the extracellular signal-regulated protein kinase-mitogen-activated protein kinase (ERK-MAPK) signaling pathway. We attempted to establish a new gastrin-ERK-MAPK pathway in gastrin-dependent large intestinal cancer; provide a new way to interfere with the signaling pathway of gastrin-induced growth of large intestinal cancer; and to offer a new target for gene therapy of large intestinal carcinoma.
INTRODUCTION
Large intestinal carcinoma including colon and rectal cancer, is one of the most common malignant tumors of the human digestive tract. In developed countries such as North America, Australia, New Zealand and parts of Europe, large intestinal carcinoma is considered to be a disease of Western lifestyle, and has an incidence of 35-50/10 million people. Large intestinal carcinoma is the second to third leading cause of gastrointestinal cancer-related mortality worldwide[1-5]. In China, it now ranks fourth to fifth[6-8]. In recent years, there has been a trend towards an increase in the incidence of large intestinal cancer in China.
Although major progress has been made in understanding the molecular mechanisms of large intestinal cancer and several therapeutic agents have been developed, it is still difficult to be cured and poses a serious threat to human health, and remains a major killer in China. The general survival rate of patients with large intestinal cancer is no more than 40%. Therefore, it is crucial to know which cytokines regulate the growth of large intestinal cancer, which will help to elucidate the etiology of the tumor. In addition, to explore the mechanisms of uncontrollable tumor cell growth in light of signal transduction pathway could enable us to find a new way to treat these malignant tumors.
Previous studies have demonstrated that the occurrence of large intestinal carcinoma is directly related to the abnormal expression of gastrin[9-12]. At the same time, some studies have found that gastrin promotes growth and inhibits apoptosis of large intestinal cancer cells. However, the detailed molecular mechanism of gastrin-regulated growth of large intestinal carcinoma cells is still not clear.
We used the MTT assay to detect changes in proliferation of HT-29 cells; annexin V-fluorescein isothiocyanate (FITC) flow cytometry to detect the proliferation index (PI) and apoptosis rate (AR) of HT-29 cells; reverse transcriptase polymerase chain reaction (RT-PCR) to detect expression of pentagastrin receptor/cholecystokinin-B receptor (CCK-BR), extracellular signal-regulated kinase (ERK)1/2 and K-ras; and Western blotting to detect the protein and phosphorylation level of ERK1/2 and K-ras. The purpose of the present study was to explore the effects and mechanisms of the ERK-mitogen-activated protein kinase (MAPK) signal transduction pathway in pentagastrin-induced cell proliferation and apoptosis of large intestinal carcinoma.
MATERIALS AND METHODS
Materials and reagents
Ht-29 human large intestinal carcinoma cells were purchased from Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences. Pentagastrin was purchased from Shanghai Tian Yuan Biotechnology Co. Ltd. The proglumide and propidium iodide staining kit were purchased from Sigma (St. Louis, MO, United States). K-ras goat anti-human polyclonal antibody, ERK1/2 rabbit polyclonal anti-human antibody, phosphorylated K-ras (p-K-ras) goat anti-human polyclonal antibody, phosphorylated ERK1/2 (p-ERK1/2) rabbit anti-human polyclonal antibody, β-actin antibody, and ECL chromogenic detection kit were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, United States). The Annexin V-EGFP/PI apoptosis detection kit was purchased from Mbchem (Shanghai, China). Reverse transcription kit, SK312 Trizol reagent, Taq DNA polymerase, Marker, and dNTP were purchased from Promega (Madison, WI, United States). The gastrin receptor/CCK-BR, ERK1/2, K-ras and muscle β-actin mRNA amplification primers were synthesized by Beijing Sunbiotech Co. Ltd. (Beijing, China).
Cell culture and grouping
HT-29 cells strain recovery and cell culture after resuscitation: Cells were subcultured when the cells reached 80% confluence on United States corning 50 cm cell culture flasks containing 10% fetal calf serum RPMI-1640. Resuscitated cells with a higher cell density were subjected to timely passage, generally 2-3 d for a new generation. Cells in the logarithmic growth phase after digestion and centrifugation with removal of digestive fluid and culture medium were placed for cryopreservation in a -70 °C liquid nitrogen tank for later use. The cells were divided into four groups: control group, pentagastrin group, proglumide group, and pentagastrin + proglumide group.
Detection of cell proliferation and viability
In the logarithmic growth phase, HT-29 cells were digested into a single cell suspension in 10% fetal bovine serum (FBS) medium, and the cell concentration was adjusted to 5 × 104 cells/mL. The cells were seeded in 96-well culture plates (each well, 200 μL) and cultured for 24 h. The culture medium was discarded after the cells became adherent. After washing with D-Hank’s solution, serum-free medium was added and the cells were cultured for 24 h. We added 100 μL l% FBS medium to each well, along with 100 μL of each drug. In the pentagastrin group, pentagastrin concentration was 6.25, 12.5, 25.0, 50.0 or 100.0 mg/L. In the proglumide group, proglumide concentration was 8.0, 16.0, 32.0, 64.0 or 128.0 mg/L. In the pentagastrin + proglumide group, pentagastrin concentration was 25.0 mg/L and proglumide concentration was 8.0, 16.0, 32.0, 64.0 or 128.0 mg/L. The control group had no drugs added. Each group of six wells was cultured for 48 h. For the MTT method, MTT (5 mg/mL) was added to each well of the 96-well plate, and incubated at 37 °C for 4 h. The culture medium was then discarded, and 150 μL DMSO was added to each well, and the plate was shaken for 10 min at room temperature. OD492 was measured by ELISA, and the results of each six wells were presented as mean ± SD and the experiments were repeated three times.
Detection of cell proliferation and apoptosis
Cells in the logarithmic growth phase were digested with 0.25% trypsin and adjusted to a concentration of 1.7 × 105 cells/mL in 10% FBS medium in six-well plates (2 mL/well). After 24 h, FBS medium was replaced with serum-free medium, and cells were cultured for a further 24 h to absorb the supernatant. Then, we added 1 mL 1% FBS medium to each well, along with 1 mL of the following: pentagastrin group (25.0 mg/L pentagastrin); proglumide group (32.0 mg/L proglumide); pentagastrin + proglumide group (25.0 mg/L pentagastrin + 32.0 mg/L proglumide) group; control group (no drugs). Each group had five wells, and the cells were cultured for 48 h. After digestion with 0.25% trypsin, the cells were centrifuged at 12000 × g at 4 °C for 10 min, and the supernatant was discarded. One milliliter of cold PBS was added, and the cell suspension was gently shaken. The supernatant was discarded after centrifugation at 12000 × g at 4 °C for 10 min. After washing twice with cold PBS, the cells were resuspended in 200 μL binding buffer, fixed with 1 mL 70% ethanol, DNA and protein stained, and determined by flow cytometry for proportion and the PI of cell cycle.
After 48 h incubation with or without drugs, cells were collected from the control, pentagastrin (25.0 mg/L), proglumide (32.0 mg/L), and proglumide (25.0 mg/L) + pentagastrin (32.0 mg/L) groups, and adjusted to a concentration of 2 × 106/mL. One milliliter of cells was taken from each group, and centrifuged at 12000 × g at 4 °C for 10 min, and the supernatant was discarded. One milliliter of cold PBS was added to resuspend the cells, with gentle shaking. The cell suspension was centrifuged again at 12000 × g at 4 °C for 10 min. The supernatant was removed; the cells were washed twice with cold PBS, and resuspended in 200 μL binding buffer. We added 10 μL Annexin V-FITC and 5 μL propidium iodide, and mixed gently in the dark at room temperature for 15 min. Then, 300 μL binding buffer was added, and detected the cell AR in one hour on the machine.
Detection of CCK-BR, ERK1/2 and K-ras mRNA
Total RNA extraction and cDNA synthesis were performed as described previously[13]. mRNA expression of CCK-BR, ERK1/2 and K-ras was detected by RT-PCR. The primer sequences are listed in Table 1, and the specific steps were carried out according to the kit instructions (Promega, Madison, United States). The mean optical density (OD) values of the amplified product bands were determined by Tanon gel imaging and Image System version 4.0. Results were expressed as the semi-quantitative density ratio of the samples such as CCK-BR, ERK1/2, K-ras and β-actin.
Table 1.
Primers used for nested reverse transcriptase polymerase chain reaction amplification of cholecystokinin-B receptor, extracellular signal-regulated protein kinase 1/2 and K-ras
| Name | Primer sequence | PCR conditions | Size (bp) |
| CCK-BR | 1: 5’TCTCGCGAGCTCTACTTAGGG3’ | 94 °C, 30 s | 185 |
| 2: 5’ACCGACGATGCACGTTGAAG3’ | 62 °C, 30 s | ||
| 72 °C, 30 s | |||
| ERK1/2 | 1: 5’TATTCCCGGGCAAGCACTATTT3’ | 94 °C, 30 s | 243 |
| 2: 5’CGGGCTCATCATTCGGGTCGTA3’ | 54 °C, 30 s | ||
| 72 °C, 30 s | |||
| K-ras | 1: 5’ACAGTGCAATGAGGGACCAGTA3’ | 94 °C, 30 s | 275 |
| 2: 5’GTATAGAAGGCATCATCAACACC3’ | 50 °C, 30 s | ||
| 72 °C, 30 s | |||
| Actin | 1: 5’ATGATATCGCCGCGCTCGTCGTC3’ | 94 °C, 30 s | 342 |
| 2: 5’CGCGGTTGGCCTTGGGGTTCAG3’ | 60 °C, 30 s | ||
| 72 °C, 30 s |
PCR: Polymerase chain reaction; CCK-BR: Cholecystokinin-B receptor; ERK1/2: Extracellular signal-regulated protein kinase 1/2.
Detection of protein expression and phosphorylation of ERK1/2 and K-ras
The protein expression and phosphorylation level of ERK1/2 and K-ras were detected by western blotting. The specific steps were carried out according to the kit instructions. The primary antibody was diluted with Tris-buffered saline with Tween to a concentration of 1:500, and treated with electrophoresis, transfer film and ECL development, and Gel Image System software was used for the average OD analysis. The results were expressed as the relative ratios of the target gene expression respectively and the β-actin expression.
Statistical analysis
Statistical evaluation was performed using the Student-Newman-Keul Q test, and the data are shown as mean ± SD. All data were analyzed with SPSS version 10.0. P < 0.05 was considered statistically significant.
RESULTS
HT-29 cell proliferation
The MTT assay showed that pentagastrin stimulated the proliferation of HT-29 cells and inhibited apoptosis in a dose-dependent manner (6.25-100 mg/L), and optimal dose was 25 mg/L (F = 31.36, P < 0.05). As the pentagastrin concentration continued to increase, OD did not. Proglumide had no obvious effect on HT-29 cell proliferation at a dose of 8.0-128.0 mg/L (F = 1.38, P > 0.05). However, proglumide markedly inhibited gastrin-induced proliferation of large intestinal cancer cells when pentagastrin was at the optimal dose of 25 mg/L. However, when the proglumide concentration was 32.00 mg/L or more, the active cell number in the pentagastrin + proglumide group tended to be constant and the optimal dose of proglumide was 32 mg/L ( F = 24.31, P < 0.05) (Tables 2 and 3).
Table 2.
Effect of different pentagastrin and proglumide concentrations on the proliferation of HT-29 cells (mean ± SD)
| Groups | n |
Pentagastrin |
Proglumide |
||
| Concentration (mg/L) | OD value | Concentration (mg/L) | OD value | ||
| Control group | 6 | 0 | 0.44 ± 0.00 | 0 | 0.44 ± 0.02 |
| Experimental group | 6 | 6.25 | 0.48 ± 0.03a | 8 | 0.43 ± 0.03 |
| 6 | 12.5 | 0.52 ± 0.03a | 16 | 0.44 ± 0.04 | |
| 6 | 25 | 0.57 ± 0.04a | 32 | 0.43 ± 0.03 | |
| 6 | 50 | 0.58 ± 0.04a | 64 | 0.45 ± 0.02 | |
| 6 | 100 | 0.57 ± 0.04a | 128 | 0.45 ± 0.04 | |
P < 0.05 vs control group. OD: Optical density.
Table 3.
Effect of combined pentagastrin and proglumide on the proliferation of HT-29 cells (mean ± SD)
| Groups | n |
Pentagastrin + proglumide |
||
| Pentagastrin (mg/L) | Proglumide (mg/L) | OD value | ||
| Control group | 6 | 25 | 0 | 0.58 ± 0.02 |
| Experimental group | 6 | 25 | 8 | 0.54 ± 0.03a |
| 6 | 25 | 16 | 0.51 ± 0.04a | |
| 6 | 25 | 32 | 0.47 ± 0.04a | |
| 6 | 25 | 64 | 0.47 ± 0.03a | |
| 6 | 25 | 128 | 0.46 ± 0.03a | |
P < 0.05 vs control group. OD: Optical density.
PI and AR
In the pentagastrin group (25.0 mg/L), the PI was 37.5% ± 5.2%, which was significantly higher than that in the control (27.7% ± 5.0%) and pentagastrin (25.0 mg/L) + proglumide (32.0 mg/L) (27.3% ± 5.8%) groups (Q = 4.56, 4.75, P < 0.05). In the proglumide (32.0 mg/L) group, the PI was 27.4% ± 2.7%, which did not differ significantly from the control group (Q = 0.14, P > 0.05) (Table 4, Figure 1). In the pentagastrin group (25.0 mg/L), the AR was 1.9% ± 0.4%, which was significantly lower than in the control (2.5% ± 0.4%) (Q = 4.23, P < 0.05) and pentagastrin (25.0 mg/L) + proglumide (32.0 mg/L) (2.4% ± 0.3%) (Q = 4.06, P < 0.05) groups. In the proglumide (32.0 mg/L) group, the AR was 2.8% ± 0.5%, which did not differ significantly from the control group (Q = 1.74, P > 0.05) (Table 4, Figure 2).
Table 4.
Comparison of proliferation index and apoptotic rates between the experimental groups and control group (mean ± SD)
| Group | n |
PI |
AR |
||||
| PI (%) | F | P value | AR (%) | F | P value | ||
| Control group | 5 | 27.72 ± 5.00 | 2.53 ± 0.36 | ||||
| Pentagastrin group | 5 | 37.54 ± 5.17a | 7.59 | 0.01 | 1.91 ± 0.42a | 6.50 | 0.01 |
| Proglumide group | 5 | 27.36 ± 2.68c | 2.79 ± 0.45c | ||||
| Pentagastrin + proglumide group | 5 | 27.31 ± 5.76c | 2.41 ± 0.30c | ||||
P < 0.05 vs control group;
P < 0.05 vs pentagastrin group. AR: Apoptosis rate; PI: Proliferation index.
Figure 1.
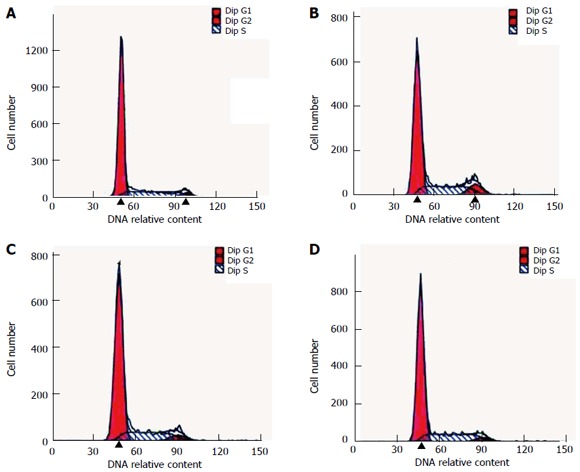
Analysis of cell cycle diagram by flow cytometry. A: Control group, B: Pentagastrin group; C: Proglumide group; D: Pentagastrin + proglumide group.
Figure 2.
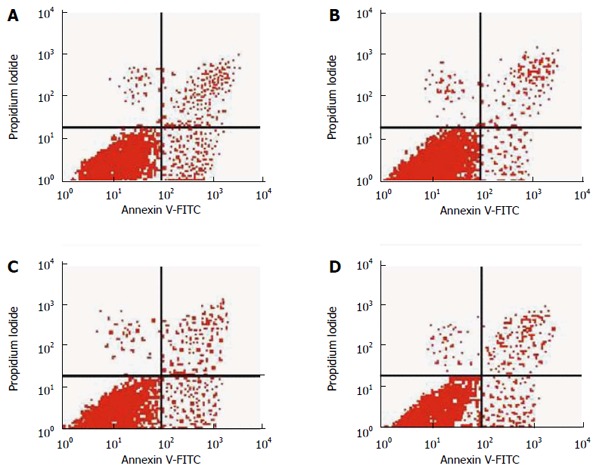
Analysis of apoptosis by flow cytometry. A: Control group; B: Pentagastrin group; C: Proglumide group; D: Pentagastrin + proglumide group. FITC: Fluorescein isothiocyanate.
Expression of CCK-BR, ERK1/2 and K-ras mRNA
HT-29 cells showed obvious CCK-BR mRNA expression. The amplification products of CCK-BR, ERK1/2, K-ras and β-actin mRNA were 185, 243, 275 and 342 bp, respectively. There was no obvious change in expression of ERK1/2 and K-ras mRNA in the control, pentagastrin, proglumide and pentagastrin + proglumide groups (F = 0.52, 0.72, 0.78 and 0.28, P > 0.05) (Table 5, Figures 3, 4 and 5).
Table 5.
Comparison of protein, mRNA and phosphorylation levels of extracellular signal-regulated protein kinase 1/2 and K-ras between the experimental groups (mean ± SD) and control group
| Group | n |
ERK1/2 |
K-ras |
||||
| mRNA | Protein | p-ERK1/2 | mRNA | Protein | p-K-ras | ||
| Control group | 5 | 0.76 ± 0.04 | 0.56 ± 0.05 | 0.32 ± 0.02 | 0.54 ± 0.08 | 0.56 ± 0.04 | 0.31 ± 0.05 |
| Pentagastrin group | 5 | 0.79 ± 0.05 | 0.60 ± 0.04 | 0.43 ± 0.04a | 0.59 ± 0.06 | 0.57 ± 0.04 | 0.45 ± 0.06a |
| Proglumide group | 5 | 0.74 ± 0.06 | 0.55 ± 0.07 | 0.31 ± 0.02c | 0.53 ± 0.05 | 0.55 ± 0.04 | 0.27 ± 0.06c |
| Pentagastrin + proglumide group | 5 | 0.77 ± 0.05 | 0.58 ± 0.05 | 0.36 ± 0.01c | 0.55 ± 0.06 | 0.57 ± 0.04 | 0.35 ± 0.04c |
P < 0.05 vs control group;
P < 0.05 vs pentagastrin group. ERK1/2: Extracellular signal-regulated protein kinase 1/2.
Figure 3.
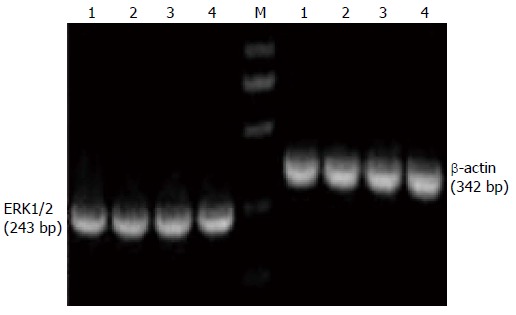
Analysis of extracellular signal-regulated protein kinase1/2 and β-actin mRNA expression by nested reverse transcriptase polymerase chain reaction in HT-29 cells. Lane M: DNA marker; Lane 1: Control group; Lane 2: Pentagastrin group; Lane 3: Proglumide group; Lane 4: Pentagastrin + proglumide group.
Protein expression and phosphorylation levels of ERK1/2 and K-ras
After incubation of HT-29 cells with drugs for 72 h, there were no significant differences in ERK1/2 and K-ras protein levels among the control, pentagastrin (25.0 mg/L), proglumide (32.0 mg/L) and pentagastrin (25.0 mg/L) + proglumide (32.0 mg/L) groups (F = 0.72, 0.28, P > 0.05). The protein phosphorylation levels of ERK1/2 and K-ras in the pentagastrin (25.0 mg/L) group were higher than in the control (Q = 7.78, 4.95, P < 0.05) and pentagastrin (25.0 mg/L) + proglumide (32.0 mg/L) (Q = 5.72, 4.08, P < 0.05) groups. There were no significant differences in protein phosphorylation levels of ERK1/2 and K-ras between the proglumide (32 mg/L) and control groups (Q = 0.71, 1.63, P > 0.05) (Table 5, Figure 5).
Figure 5.
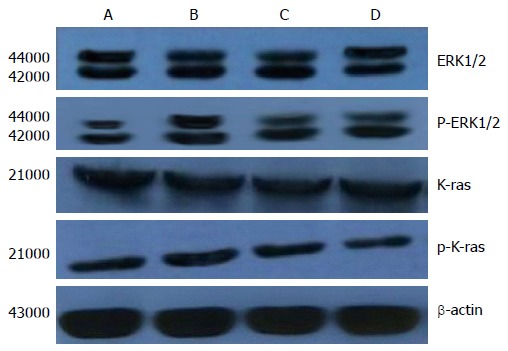
Analysis of extracellular signal-regulated protein kinase 1/2, p- extracellular signal-regulated protein kinase1/2, K-Ras and p-K-Ras protein expression of each experimental group by Western blotting.
DISCUSSION
Gastrin was discovered as early as 1905 in the extracts of gastric antral mucosa and is associated with gastric acid secretion. In 1964, it was isolated and identified by Gregory and Tracy who explained its chemical structure and confirmed it as a gastrointestinal hormone[14,15]. Gastrin is an important gastrointestinal hormone and type of gastrointestinal peptide that is mainly secreted by gastrointestinal G cells. The G cells are typical open-type cells found in the antrum. As a nutritional gastrointestinal hormone, gastrin stimulates the growth of normal gastrointestinal tract mucosa, as well as growth of gallbladder, pancreatic, colorectal and gastric carcinoma and other tumor cells[9,16-18]. Most studies to date have shown that gastrin is an autocrine growth factor that exerts its function through autocrine, paracrine and endocrine ways[19,20]. The combination of gastrin and its receptor can regulate tumor cell growth by intracellular signaling transduction, and exert biological effects[17,21,22]. Recent studies have demonstrated that the incidence of colorectal cancer is related to abnormal expression of gastrin. Some researchers have referred to this type of colorectal cancer as a hormone-dependent tumor, which promotes cell proliferation through generation and secretion of gastrin, which acts on its own receptor on the cell membrane to exert a biological effect, but its effects can be inhibited by gastrin receptor antagonist[23,24]. However, the molecular mechanism involved in the regulation of colon cancer proliferation remains unclear.
In recent years, gene chip technology has been used to detect colorectal-cancer-related gene expression. There were significant differences in gene expression in many internal and external networks, and the cell signal transduction pathway is closely related with the inhibition of regulation, and is directly involved in cell metabolism, proliferation, differentiation and apoptosis[25-29]. MAPK is an intracellular serine/threonine protein kinase that is present widely in the cytoplasm and nuclei of most mammalian cells. MAPK is involved in transduction of extracellular stimulation to the cell and its nucleus, and regulates cell proliferation, differentiation, development and apoptosis[30,31]. MAPK consists of ERK, c-Jun N-terminal kinase (JNK)/stress-activated protein kinase (SAPK), p38 and ERK5/BMK1 subgroup of MAPK. Multiple signal pathways are made up of these subgroups. It has been shown that mammalian cells have three parallel MAPK pathways: ERK, p38 MAPK, and JNK/SAPK signal pathways. Among these, the JNK/SAPK and p38MAPK pathways are related to cellular stress and apoptosis, and the ERK pathway is closely related to cell proliferation and differentiation[32-34]. Many studies have suggested that the MAPK signaling pathway is involved in the regulation of growth of colon cancer cells. The MAPK signal transduction pathway plays an important role in the gastrin-regulated proliferation of colon cancer cells, but the mechanism is still unclear.
The abnormal expression of gastrin can lead to uncontrolled growth of colon cancer cells to form tumors, but this effect can be inhibited by the gastrin receptor antagonist proglumide[11,13,19,35]. In this study, we found that the concentration gradient of gastrin within a certain dose range promoted HT-29 cell proliferation and inhibited apoptosis. As the concentration increased from 25 mg/L, proliferation no longer continued to increase, which was consistent with the receptor saturation theory. It was clear that the number of gastrin receptors and their affinity determined the strength of the effects on colorectal cancer cells. We also found that gastrin promoted the proliferation of HT-29 cells and inhibited apoptosis through accelerating the S and G2 phases of the cell cycle. The gastrin-promoted colon cancer cell proliferation and inhibition of apoptosis were inhibited by the gastrin receptor antagonist proglumide. In addition, our study also showed that there was no significant impact on the colorectal cancer cell cycle if we used proglumide alone. When we combined pentagastrin with proglumide, PI decreased and AR increased, although this did not differ significantly from the control group. Proglumide markedly inhibited gastrin-induced proliferation of large intestinal cancer cells in a dose-dependent manner within a certain range, and the optimal dose was 32 mg/L, which is also in line with the receptor saturation theory.
Gastrin can promote cell proliferation in human large intestinal carcinoma and inhibit apoptosis[36-39], but it is still unclear through which signal transduction pathways. To investigate further the specific molecular mechanism, we studied the relationship between gastrin and the ERK-MAPK signal transduction pathway. ERK is an important signal transduction protein kinase and mediates between extracellular signals and intracellular responses in eukaryotic cells. ERK belongs to the MAPK family, and includes two isomers ERK1 and ERK2. The intracellular ERK signal transduction pathway is deemed to be the classical MAPK signal transduction pathway, and it has been shown to mediate a variety of extracellular signals through different intracellular signaling molecules. ERK activation comprises a three-stage process: (1) Ras activates Raf protein; (2) activation of Raf activates MEK1/2, and MEK1/2 is phosphorylated; and (3) phosphorylation of MEK1/2 activates ERK1/2, which promotes growth of tumor cells[32,33,40,41]. Wang et al[6] have reported that ERK-MAPK signaling pathway activation reduces expression of the related genes p21WAF1 and p16INK4a, increases expression of c-myc, reduces cell cycle G0 phase, accelerates G1/S phase transformation, and promotes proliferation of colon cancer cells.
In general, phosphorylation is a molecular switch in signal transduction pathways, which controls protein activity in different pathways, such as metabolism, signal transduction, cell division, and other aspects. So, we can establish through phosphorylation if the signaling pathway is activated, and the strength of the activation. In this study, we found that gastrin significantly increased the phosphorylation level of ERK1/2 and K-ras. Proglumide alone had no significant effect on the level of phosphorylation of ERK1/2 and K-ras in HT-29 cells, but combined with gastrin, proglumide significantly antagonized the role of gastrin, and the protein phosphorylation levels of ERK1/2 and K-ras were significantly reduced in HT-29 cells. However, there was no significant difference in the mRNA and protein expression of ERK1/2 and K-ras among the different groups. Our results indicated that gastrin promoted proliferation of human large intestinal cancer cells and inhibited apoptosis, and these effects were inhibited by the gastrin receptor antagonist proglumide. Gastrin perhaps regulates the proliferation and apoptosis of large intestinal cancer cells by the pathway Ras→Raf→MEK1/2→ERK1/2, and upregulated protein phosphorylation levels of ERK1/2 and K-ras.
In conclusion, gastrin promotes proliferation of the human large intestinal cancer HT-29 cells and inhibits apoptosis, but these effects are inhibited by gastrin receptor antagonist proglumide. The ERK-MAPK signaling transduction pathway is involved in gastrin-regulated proliferation and apoptosis of large intestinal cancer cells. However, which target genes are involved in this mechanism under the downstream signaling transduction pathway still needs in-depth study. In addition, we expect to find a new treatment strategy for gastrin-dependent large intestinal carcinoma if we can competitively inhibit the gastrin receptors or reduce the ERK and K-ras phosphorylation levels of the ERK-MAPK signaling pathway.
COMMENTS
Background
Although great progress has been made in understanding the molecular aspects of large intestinal carcinoma and several therapeutic agents have been developed, it is still difficult to cure. The general survival rate of large intestine cancer patients does not exceed 40%. Therefore, it is important to know which cell factors can influence proliferation and apoptosis of cancer cells, to elucidate more clearly the etiology of the tumor. Despite abundant evidence to show that gastrin may play an integral role in promoting tumor growth in large intestinal carcinoma, the detailed molecular mechanisms are not fully known.
Research frontiers
Some recent studies have shown that activation of the extracellular signal-regulated kinase-mitogen-activated protein kinase (ERK-MAPK) signaling pathway is regulated by many kinds of cell factors, including gastrin.
Innovations and breakthroughs
Some studies have found that gastrin promotes proliferation of HT-29 human large intestinal cancer cells and inhibits apoptosis. However, the detailed molecular mechanism by which gastrin mediates cell proliferation and apoptosis of large intestinal carcinoma is not fully known. In this study, the authors analyzed those effects on the expression of ERK1/2 and K-ras protein and phosphorylation level. The authors aimed to elucidate the molecular mechanisms of gastrin-induced growth of large intestinal carcinoma cells from the perspective of the ERK-MAPK signaling pathway. They tried to establish a new gastrin-ERK-MAPK signaling pathway in gastrin-dependent large intestinal carcinoma, to provide a new means of interfering with the signaling pathway of gastrin-induced growth of large intestinal cancer cells. This could provide a novel way for us to treat malignant tumors.
Applications
This study aimed to elucidate the molecular mechanisms of gastrin-induced proliferation of large intestinal carcinoma cells from the perspective of the ERK-MAPK signaling pathway, and to provide a new method for interfering with the signaling pathway of gastrin-induced growth of large intestinal cancer cells, and offer a new target for gene therapy of large intestinal carcinoma.
Terminology
Gastrin is mainly secreted from gastrin-secreting cells (G cells) in the antrum mucosa or upper small intestine and large intestine. Medulla oblongata and dorsal nuclei of the vagus nerve and central nervous system also secrete gastrin. MAPK is an intracellular serine/threonine protein kinase that is widely present in the cytoplasm and nucleus of most mammalian cells. It can mediate extracellular stimulation via signal transduction to the cell and its nucleus, and regulate cell proliferation, differentiation, development and apoptosis. The MAPK family consists of ERK, c-Jun N-terminal kinase/stress-activated protein kinase (SAPK), p38 and the ERK5/BMK1 subgroup. ERK is a recently identified family of protein serine/threonine kinases that occupies a pivotal position in intracellular signaling pathways mediating mitogen/growth factor effects. Ras was originally isolated from rats with sarcoma, and is widely present in the inner plasma membrane of most mammalian cells. It binds GDP and GTP and possesses intrinsic GTPase activity, and is implicated in the regulation of their activity. The human Ras family consists of three proto-oncogenes, c-Harvey (H)-ras, c-Kirsten (K)-ras, and N-ras.
Peer review
Large intestinal cancer is one of the most common malignant tumors of human digestive tract. It is a serious threat to human life. However, the mechanism of development of large intestinal carcinoma cells is still not clear. In this paper, the author aimed to explore the effects and mechanisms of ERK-MAPK signaling transduction pathway in pentagastrin-induced cell proliferation and apoptosis of large intestinal carcinoma. The article is well written on the whole.
Footnotes
Supported by Natural Science Foundation of Anhui Province, No. 1408085MH148; Natural Science Fund of Education Bureau of Anhui Province, No. kj2010b242; Natural Science Fund of Wannan Medical College, No. wk2012zf02; and The key science and technology project of Wuhu City, No. health-2-4
P- Reviewer: Chiang TA, Guo JM S- Editor: Ma YJ L- Editor: O’Neill M E- Editor: Zhang DN
References
- 1.Shin HR, Carlos MC, Varghese C. Cancer control in the Asia Pacific region: current status and concerns. Jpn J Clin Oncol. 2012;42:867–881. doi: 10.1093/jjco/hys077. [DOI] [PubMed] [Google Scholar]
- 2.Mihajlović J, Pechlivanoglou P, Miladinov-Mikov M, Zivković S, Postma MJ. Cancer incidence and mortality in Serbia 1999-2009. BMC Cancer. 2013;13:18. doi: 10.1186/1471-2407-13-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herszényi L, Tulassay Z. Epidemiology of gastrointestinal and liver tumors. Eur Rev Med Pharmacol Sci. 2010;14:249–258. [PubMed] [Google Scholar]
- 4.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 5.Gomez SL, Noone AM, Lichtensztajn DY, Scoppa S, Gibson JT, Liu L, Morris C, Kwong S, Fish K, Wilkens LR, et al. Cancer incidence trends among Asian American populations in the United States, 1990-2008. J Natl Cancer Inst. 2013;105:1096–1110. doi: 10.1093/jnci/djt157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X, Song ZF, Xie RM, Pei J, Xiang MF, Wang H. Analysis of death causes of in-patients with malignant tumors in Sichuan Cancer Hospital of China from 2002 to 2012. Asian Pac J Cancer Prev. 2013;14:4399–4402. doi: 10.7314/apjcp.2013.14.7.4399. [DOI] [PubMed] [Google Scholar]
- 7.Chen W, Zheng R, Zhang S, Zhao P, Li G, Wu L, He J. Report of incidence and mortality in China cancer registries, 2009. Chin J Cancer Res. 2013;25:10–21. doi: 10.3978/j.issn.1000-9604.2012.12.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang G, Wang Y, Zeng Y, Gao GF, Liang X, Zhou M, Wan X, Yu S, Jiang Y, Naghavi M, et al. Rapid health transition in China, 1990-2010: findings from the Global Burden of Disease Study 2010. Lancet. 2013;381:1987–2015. doi: 10.1016/S0140-6736(13)61097-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chueca E, Lanas A, Piazuelo E. Role of gastrin-peptides in Barrett’s and colorectal carcinogenesis. World J Gastroenterol. 2012;18:6560–6570. doi: 10.3748/wjg.v18.i45.6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song LJ, Liu RJ, Zeng Z, Alper SL, Cui HJ, Lu Y, Zheng L, Yan ZW, Fu GH. Gastrin inhibits a novel, pathological colon cancer signaling pathway involving EGR1, AE2, and P-ERK. J Mol Med (Berl) 2012;90:707–718. doi: 10.1007/s00109-011-0851-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kovac S, Anderson GJ, Baldwin GS. Gastrins, iron homeostasis and colorectal cancer. Biochim Biophys Acta. 2011;1813:889–895. doi: 10.1016/j.bbamcr.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chao C, Hellmich MR. Gastrin, inflammation, and carcinogenesis. Curr Opin Endocrinol Diabetes Obes. 2010;17:33–39. doi: 10.1097/MED.0b013e328333faf8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kreeger PK, Mandhana R, Alford SK, Haigis KM, Lauffenburger DA. RAS mutations affect tumor necrosis factor-induced apoptosis in colon carcinoma cells via ERK-modulatory negative and positive feedback circuits along with non-ERK pathway effects. Cancer Res. 2009;69:8191–8199. doi: 10.1158/0008-5472.CAN-09-1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Modlin IM, Kidd M, Marks IN, Tang LH. The pivotal role of John S. Edkins in the discovery of gastrin. World J Surg. 1997;21:226–234. doi: 10.1007/s002689900221. [DOI] [PubMed] [Google Scholar]
- 15.Gregory RA, Tracy HJ. The constitution and properties of two gastrins extracted from hog antral mucosa. Gut. 1964;5:103–114. [PMC free article] [PubMed] [Google Scholar]
- 16.Steigedal TS, Prestvik WS, Selvik LK, Fjeldbo CS, Bruland T, Lægreid A, Thommesen L. Gastrin-induced proliferation involves MEK partner 1 (MP1) In Vitro Cell Dev Biol Anim. 2013;49:162–169. doi: 10.1007/s11626-013-9588-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu W, Chen GS, Shao Y, Li XL, Xu HC, Zhang H, Zhu GQ, Zhou YC, He XP, Sun WH. Gastrin acting on the cholecystokinin2 receptor induces cyclooxygenase-2 expression through JAK2/STAT3/PI3K/Akt pathway in human gastric cancer cells. Cancer Lett. 2013;332:11–18. doi: 10.1016/j.canlet.2012.12.030. [DOI] [PubMed] [Google Scholar]
- 18.Singh P, Sarkar S, Kantara C, Maxwell C. Progastrin Peptides Increase the Risk of Developing Colonic Tumors: Impact on Colonic Stem Cells. Curr Colorectal Cancer Rep. 2012;8:277–289. doi: 10.1007/s11888-012-0144-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kovac S, Xiao L, Shulkes A, Patel O, Baldwin GS. Gastrin increases its own synthesis in gastrointestinal cancer cells via the CCK2 receptor. FEBS Lett. 2010;584:4413–4418. doi: 10.1016/j.febslet.2010.09.046. [DOI] [PubMed] [Google Scholar]
- 20.Patel O, Clyde D, Chang M, Nordlund MS, Steel R, Kemp BE, Pritchard DM, Shulkes A, Baldwin GS. Pro-GRP-derived peptides are expressed in colorectal cancer cells and tumors and are biologically active in vivo. Endocrinology. 2012;153:1082–1092. doi: 10.1210/en.2011-1875. [DOI] [PubMed] [Google Scholar]
- 21.Sanchez C, Escrieut C, Clerc P, Gigoux V, Waser B, Reubi JC, Fourmy D. Characterization of a novel five-transmembrane domain cholecystokinin-2 receptor splice variant identified in human tumors. Mol Cell Endocrinol. 2012;349:170–179. doi: 10.1016/j.mce.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 22.Kato H, Seto K, Kobayashi N, Yoshinaga K, Meyer T, Takei M. CCK-2/gastrin receptor signaling pathway is significant for gemcitabine-induced gene expression of VEGF in pancreatic carcinoma cells. Life Sci. 2011;89:603–608. doi: 10.1016/j.lfs.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 23.Song Y, Xu Y, Wang Z, Chen Y, Yue Z, Gao P, Xing C, Xu H. MicroRNA-148b suppresses cell growth by targeting cholecystokinin-2 receptor in colorectal cancer. Int J Cancer. 2012;131:1042–1051. doi: 10.1002/ijc.26485. [DOI] [PubMed] [Google Scholar]
- 24.Jin G, Westphalen CB, Hayakawa Y, Worthley DL, Asfaha S, Yang X, Chen X, Si Y, Wang H, Tailor Y, et al. Progastrin stimulates colonic cell proliferation via CCK2R- and β-arrestin-dependent suppression of BMP2. Gastroenterology. 2013;145:820–830.e10. doi: 10.1053/j.gastro.2013.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watanabe T, Kobunai T, Tanaka T, Ishihara S, Matsuda K, Nagawa H. Gene expression signature and the prediction of lymph node metastasis in colorectal cancer by DNA microarray. Dis Colon Rectum. 2009;52:1941–1948. doi: 10.1007/DCR.0b013e3181b53684. [DOI] [PubMed] [Google Scholar]
- 26.Lan H, Jin K, Xie B, Han N, Cui B, Cao F, Teng L. Heterogeneity between primary colon carcinoma and paired lymphatic and hepatic metastases. Mol Med Rep. 2012;6:1057–1068. doi: 10.3892/mmr.2012.1051. [DOI] [PubMed] [Google Scholar]
- 27.Hernandez JM, Farma JM, Coppola D, Hakam A, Fulp WJ, Chen DT, Siegel EM, Yeatman TJ, Shibata D. Expression of the antiapoptotic protein survivin in colon cancer. Clin Colorectal Cancer. 2011;10:188–193. doi: 10.1016/j.clcc.2011.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Solier S, Barb J, Zeeberg BR, Varma S, Ryan MC, Kohn KW, Weinstein JN, Munson PJ, Pommier Y. Genome-wide analysis of novel splice variants induced by topoisomerase I poisoning shows preferential occurrence in genes encoding splicing factors. Cancer Res. 2010;70:8055–8065. doi: 10.1158/0008-5472.CAN-10-2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu HH, Wu P, Mao JD, Lu Kun, Lu LM. Profiling analysis of differentially expressed genes in colorectal cancer with or without high expression of gastrin. Zhonghua Shiyan Waike Zazhi. 2012;29:2191–2194. [Google Scholar]
- 30.Mohapatra P, Preet R, Choudhuri M, Choudhuri T, Kundu CN. 5-fluorouracil increases the chemopreventive potentials of resveratrol through DNA damage and MAPK signaling pathway in human colorectal cancer cells. Oncol Res. 2011;19:311–321. doi: 10.3727/096504011x13079697132844. [DOI] [PubMed] [Google Scholar]
- 31.Konicek BW, Stephens JR, McNulty AM, Robichaud N, Peery RB, Dumstorf CA, Dowless MS, Iversen PW, Parsons S, Ellis KE, et al. Therapeutic inhibition of MAP kinase interacting kinase blocks eukaryotic initiation factor 4E phosphorylation and suppresses outgrowth of experimental lung metastases. Cancer Res. 2011;71:1849–1857. doi: 10.1158/0008-5472.CAN-10-3298. [DOI] [PubMed] [Google Scholar]
- 32.Rauch J, Moran-Jones K, Albrecht V, Schwarzl T, Hunter K, Gires O, Kolch W. c-Myc regulates RNA splicing of the A-Raf kinase and its activation of the ERK pathway. Cancer Res. 2011;71:4664–4674. doi: 10.1158/0008-5472.CAN-10-4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duhamel S, Hébert J, Gaboury L, Bouchard A, Simon R, Sauter G, Basik M, Meloche S. Sef downregulation by Ras causes MEK1/2 to become aberrantly nuclear localized leading to polyploidy and neoplastic transformation. Cancer Res. 2012;72:626–635. doi: 10.1158/0008-5472.CAN-11-2126. [DOI] [PubMed] [Google Scholar]
- 34.Ogino S, Shima K, Meyerhardt JA, McCleary NJ, Ng K, Hollis D, Saltz LB, Mayer RJ, Schaefer P, Whittom R, et al. Predictive and prognostic roles of BRAF mutation in stage III colon cancer: results from intergroup trial CALGB 89803. Clin Cancer Res. 2012;18:890–900. doi: 10.1158/1078-0432.CCR-11-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ellrichmann M, Ritter PR, Schrader H, Schmidt WE, Meier JJ, Schmitz F. Gastrin stimulates the VEGF-A promotor in a human colon cancer cell line. Regul Pept. 2010;165:146–150. doi: 10.1016/j.regpep.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 36.Wu P, Mao JD, Yan JY, Rui J, Zhao YC, Li XH, Xu GQ. Correlation between the expressions of gastrin, somatostatin and cyclin and cyclin-depend kinase in colorectal cancer. World J Gastroenterol. 2005;11:7211–7217. doi: 10.3748/wjg.v11.i45.7211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mao JD, Wu P, Yang YL, Wu J, Huang H. Relationship between expression of gastrin, somatostatin, Fas/FasL and caspases in large intestinal carcinoma. World J Gastroenterol. 2008;14:2802–2809. doi: 10.3748/wjg.14.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mao JD, Wu P, Xia XH, Hu JQ, Huang WB, Xu GQ. Correlation between expression of gastrin, somatostatin and cell apoptosis regulation gene bcl-2/bax in large intestine carcinoma. World J Gastroenterol. 2005;11:721–725. doi: 10.3748/wjg.v11.i5.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu P, Mao JD, Zhao YC, Wu J, Huang H. Relationship between the cells proliferation of large intestinal cancer induced by gastrin and signaling transduction pathway of STAT3. Guoji Waikex Xue Zazhi. 2010;37:243–247. [Google Scholar]
- 40.Zhang S, Xu R, Luo X, Jiang Z, Shu H. Genome-wide identification and expression analysis of MAPK and MAPKK gene family in Malus domestica. Gene. 2013;531:377–387. doi: 10.1016/j.gene.2013.07.107. [DOI] [PubMed] [Google Scholar]
- 41.Papageorgis P, Cheng K, Ozturk S, Gong Y, Lambert AW, Abdolmaleky HM, Zhou JR, Thiagalingam S. Smad4 inactivation promotes malignancy and drug resistance of colon cancer. Cancer Res. 2011;71:998–1008. doi: 10.1158/0008-5472.CAN-09-3269. [DOI] [PMC free article] [PubMed] [Google Scholar]