

Abstract
Background
Acute myeloid leukemia with complex karyotype (CK-AML) is a distinct biological entity associated with a very poor outcome. Since complex karyotypes frequently contain deletions of the chromosomal region 12p13 encompassing the tumor suppressor genes ETV6 and CDKN1B, we aimed to unravel their modes of inactivation in CK-AML.
Results
To decipher deletions, mutations and methylation of ETV6 and CDKN1B, arrayCGH, SNP arrays, direct sequencing of all coding exons and pyrosequencing of the 5′UTR CpG islands of ETV6 and CDKN1B were performed. In total, 39 of 79 patients (49%) showed monoallelic deletions of 12p13 according to karyotypic data and 20 of 43 patients (47%) according to genomic profiling. Genomic profiling led to the minimal deleted region covering the 3′-UTR of ETV6 and CDKN1B. Direct sequencing revealed one novel monoallelic frameshift mutation in ETV6 while no mutations in CDKN1B were identified. Furthermore, methylation levels of ETV6 and CDKN1B did not indicate transcriptional silencing of any of these genes. ETV6 and CDKN1B had reduced expression levels in CK-AML patients with deletion in 12p13 as compared to CK-AML without deletion in 12p13, while the other genes (BCL2L14, LRP6, DUSP16 and GPRC5D) located within the minimal deleted region in 12p13 had very low or missing expression in CK-AML irrespective of their copy number status.
Conclusions
ETV6 and CDKN1B are mainly affected by small monoallelic deletions, whereas mutations and hypermethylation play a minor role in CK-AML. Reduced gene dosage led to reduced gene expression levels, pointing to haploinsufficiency as the relevant mechanism of inactivation of ETV6 and CDKN1B in CK-AML.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2164-15-784) contains supplementary material, which is available to authorized users.
Keywords: Acute myeloid leukemia (AML), Complex karyotype, Haploinsufficiency, ETV6, CDKN1B, ArrayCGH, Methylation, Gene expression
Background
Acute myeloid leukemia (AML) is a hematopoietic malignancy of clonal myeloid progenitor cells arrested at an immature differentiation stage. There is substantial phenotypic and genetic heterogeneity due to the acquisition of different genetic and/or epigenetic alterations in leukemia-initiating cells [1]. Acute myeloid leukemia with complex karyotype (CK-AML) is a distinct biological entity, traditionally defined by the presence of at least three independent chromosome aberrations, excluding t(8;21), inv(16)/t(16;16), and t(15;17) and is associated with a very poor outcome [2, 3]. More than 150 genes have been shown to be differentially expressed in CK-AML compared to AML with normal karyotype, including several genes located on 5q and 7q as well as genes involved in DNA repair, chromosome segregation, and within the actin cytoskeleton [4]. CK-AML often contains deletions of 5q, 7q, and 17p and shows high incidence of somatic alterations of TP53 [5–7].
Besides these characteristic deletions, complex karyotypes frequently contain deletions of the chromosomal region 12p13 [8–10]. The reported minimal deleted region of 12p13 spans two putative tumor suppressor genes, ETV6 and CDKN1B [11–13]. For both genes, haploinsufficiency as the underlying mechanism was previously suggested, as they are located within the minimal deleted region and inactivation of the second allele is rarely seen [12, 14, 15]. Furthermore, haploinsufficiency of CDKN1B is strongly implicated in numerous cancer types, the vast majority of lymphatic origin [16, 17].
ETV6 (ets translocation variant gene 6), a member of the ETS transcription factor family, shows several properties of a putative tumor suppressor gene like induction of G1 arrest and blocking of Ras-induced transformation [18], induction of apoptosis [19], and activation of TP53 dependent pathways [20]. CDKN1B encodes the p27CDKN1B protein which belongs to the CIP/KIP class of cyclin dependent kinase inhibitors and inactivates the cyclin E/CDK2 complex via binding of CDKN1B to cyclin E/CDK2 [21]. CDKN1B is hence an important negative regulator of the cell cycle.
In this study, we aimed to investigate whether and how ETV6 and CDKN1B are inactivated by (small) deletions, mutations or DNA methylation in the specific subgroup of CK-AML.
Results
Standard karyotyping and genomic profiling of 12p13
We extensively characterized a cohort of 79 patients with CK-AML. Thirty-nine of them (49%) showed a loss of 12p13 according to karyotyping (Additional file 1: Table S1). This included deletions of 12p13 due to interstitial deletions, unbalanced translocations, and monosomy 12. A monosomy 12 was observed in 13 patients, a deletion of 12p13 either by interstitial or terminal deletion or by additive chromosomal material in 21 patients and a dicentric or derivative chromosome with loss of 12p in five patients. The frequency of cytogenetically detectable -5/5q-, -7/7q- or -17/17p- did not differ in the groups of CK-AML with and without 12p13 deletion (analyzed with Fisher’s exact test, p-values: -5/5q- 0.81, -7/7q- 0.17, -17/17p- 0.36).
In 43 patients, DNA was available for genomic profiling [array comparative genomic hybridization (arrayCGH) or single-nucleotide polymorphism (SNP) arrays] to determine the allelic status of 12p13 and delineate the breakpoints of 12p more closely. In 26 of these patients we used arrayCGH methods (244 k array, 2.8 k array and 8.0 k array), five patients were analyzed by using the SNP 250 k array and 12 patients were analyzed by using the SNP 6.0 array. In summary, in 20 of 43 patients (47%), losses of 12p13 could be identified by genomic profiling encompassing the ETV6 and CDKN1B genes. With this approach, three patients were newly detected to carry a deletion in 12p13 (#61, #64, #77), however in six patients (#13, #22, #36, #41, #69, #70) a loss of 12p13 according to karyotyping was not confirmed (Additional file 1: Table S1). Small interstitial deletions within the chromosomal region 12p13 were identified in 13 of 43 patients with CK-AML analyzed by arrayCGH and SNP arrays. The distal breakpoints mapped within a region of 30 kb directly 5′ to ETV6, except in one patient (#48), where the first deleted probe was localized in the 3′UTR of ETV6. The proximal breakpoints were all downstream of GPRC5D (G-protein-coupled receptor, family C, group 5). The minimal deleted region in our cohort spanned 1.43 Mb and included CDKN1B and the 3′UTR of ETV6 (Figure 1).
Figure 1.
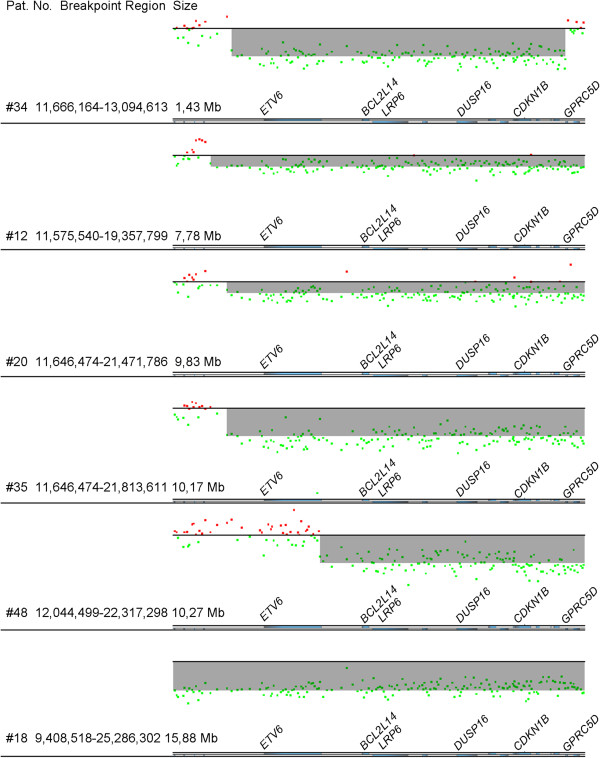
Interstitial 12p13 deletions. ArrayCGH results (244 k, Agilent) showing interstitial 12p13 deletions in six patients. The genomic profiles are zoomed in to the minimal deleted region covering ETV6 at the telomeric site and CDKN1B at the centromeric site. Deleted regions are highlighted in gray. Next to the breakpoint region given in Mb, the size of the deletion is shown. The ETV6 gene is orientated in 5′-3′ direction. Mean log ratios of the deletions correlate with the clone size according to karyotyping.
12p13 deletion breakpoints
A graphic overview of the 12p13 deletion breakpoints including or excluding ETV6 and CDKN1B from different studies also comprising our data is shown in Figure 2. Most of these studies refer to (CK-) AML or contain a large number of patients with AML. Some studies reported minimal deleted regions containing either ETV6 or CDKN1B. However, in the majority of studies, the minimal deleted region covered both ETV6 and CDKN1B.
Figure 2.
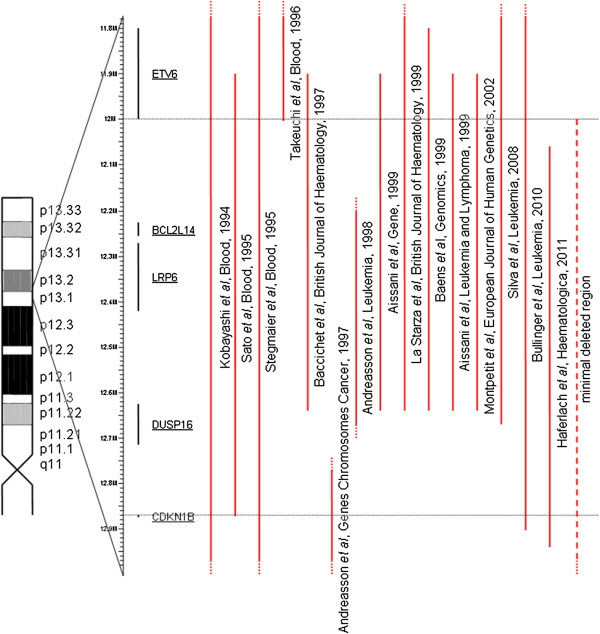
12p minimal deleted regions from different studies. The ideogram of the chromosome arm 12p is shown on the left. The region from 11.8 Mb to 12.9 Mb is zoomed in to the genes ETV6, BCL2L14, LRP6, DUSP16, and CDKN1B given as black bars. The red bars to the right of the genes indicate the minimal deleted regions as described in the different studies shown below. Most of these studies refer to (CK-) AML or contain a large number of patients with AML. The red bar on the right displays the minimal deleted region identified in this study. A dashed line indicates that the border of the deletion is not exactly determined or exceeds the selected chromosomal region.
Mutation analyses of ETV6, CDKN1Band TP53
To further determine the modes of inactivation of ETV6 [NM_001987] and CDKN1B [NM_004064], we performed mutation analyses of all coding exons of ETV6 in 56 patients and of CDKN1B in 67 patients with and without 12p13 deletions for which DNA was available (Additional file 1: Table S1). In our cohort of CK-AML, no CDKN1B mutations were identified. However, in ETV6 we found a distinct and novel heterozygous frameshift mutation of exon 4, c.391dupT, p.(Ser131PhefsTer23) in one patient (#21) (Figure 3). This frameshift mutation lies within the N-terminal homodimerization domain and theoretically would disturb homodimerization, thus leading to a complete loss of the DNA-binding ETS domain. A deleterious effect was confirmed by using the platform PROVEAN for non-synonymous or indel variants and prediction of functional effects (http://provean.jcvi.org/index.php) [22]. The mutation was detected at the time point of relapse. Importantly, in the first diagnostic sample of this patient still showing a normal karyotype, the ETV6 mutation was present, indicating that this mutation emerged early in the course of the disease. In the following samples during the course of the disease, a complex karyotype with several balanced translocations and a terminal deletion of 14q was detected (Additional file 1: Table S1). By means of arrayCGH, no gains or losses in the region of the ETV6 gene were evident. Notably, no TP53, FLT3 nor CEBPA mutations (data not shown) and no typical cytogenetic aberrations of CK-AML like del(5q), -7/del(7q), or del(17p) were present in this patient. However an NPM1 mutation (c.860_863dup, p.(Trp288CysfsTer12)) was identified (data not shown).
Figure 3.
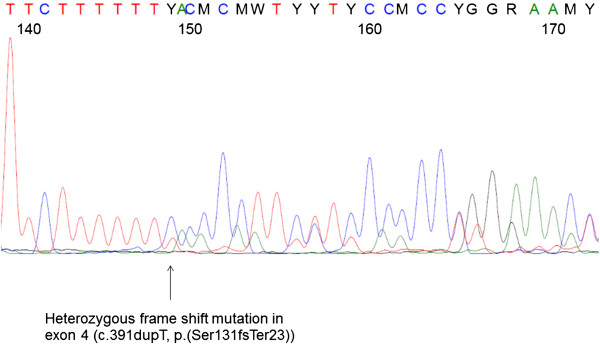
ETV6 mutation. Heterozygous frame shift mutation in exon 4, c.391dupT, p.(Ser131PhefsTer23), leading to truncated protein due to a newly generated stop codon (patient #21).
To determine the frequency of TP53 mutations in the subgroup with and without 12p13 deletions, we performed mutation analysis of all coding exons of TP53 in all 79 patients. In 44 of 79 patients (56%), TP53 mutations were identified (see Additional file 2: Table S2). The frequency of 12p13 alterations was similar in patients with and without a monoallelic or biallelic inactivation of TP53 (45.5% versus 45.7%, p = 0.99).
Methylation analyses of ETV6and CDKN1B
To determine the methylation status of ETV6 and CDKN1B, quantitative methylation analysis of the 5′UTRs of both genes was performed by pyrosequencing. We examined 23 single CpG sites within the ETV6 5′UTR CpG island and 22 single CpG sites within the CDKN1B 5′UTR CpG island in 55 patients (27 with 12p13 deletions, 28 without 12p13 deletions). The mean methylation levels of ETV6 and CDKN1B ranged from 0% to 1.43% and 0.09% to 1.77%, respectively (Figure 4, Additional file 1: Table S1). These results did not exceed the cut-off levels of 5% indicative for increased methylation. Neither did the methylation degree of single CpG sites in the patients exceed the cut-off level. In summary, no evidence of increased methylation of the 5′UTR of either gene was found.
Figure 4.
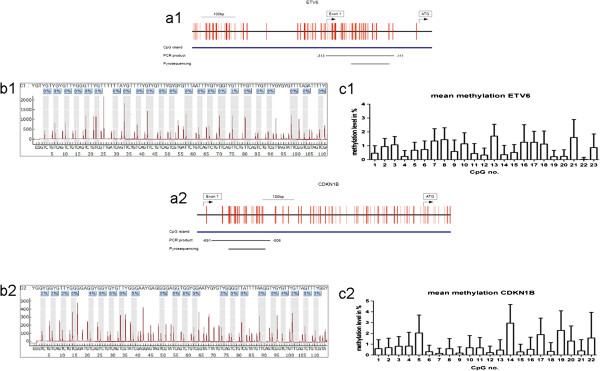
Methylation analyses of ETV6 and CDKN1B . a1/a2: Schematic view of the ETV6/CDKN1B-CpG islands: each vertical bar represents a single CpG site. Arrows indicate the start of the non-coding exon 1 and the start codon. The positions of the PCR product and the product used for pyrosequencing are indicated below as horizontal bars. b1/b2: A representative pyrogram determining the nucleotide sequence within the CpG island of ETV6/CDKN1B. The sequence in the upper part of the pyrogram contains a Y at positions under investigation, the sequence below indicates the injected bases during pyrosequencing. Highlighted areas in the pyrogram indicate variable CpG positions (light gray). The methylation level of each CpG site is indicated in blue boxes on top of the pyrogram. c1/c2: Mean methylation levels of the 23 CpG sites in the 5′UTR of ETV6 and the 22 CpG sites in the 5′UTR of CDKN1B of all patients analyzed are shown as a percentage. The standard deviation was determined using the Kruskal-Wallis test.
Expression profiling of the genes within the minimal deleted region
As we showed that gene dosages of ETV6 and CDKN1B were reduced to half the normal level in CK-AML samples with 12p13 deletion, we were interested to determine whether this is reflected on the transcriptional level. Therefore, we first evaluated the gene dosage effect based on our previously published gene expression profiling [23, 24]. Expression and genomic data were available for four of our own patients with CK-AML and deletion in 12p13 and for 28 cases without deletion in 12p13. The four CK-AML patients with deletion in 12p13 showed a significantly lower expression level for the deleted genes than CK-AML without deletion in 12p13 (p < 0.001, unpaired t-test, Figure 5A). Within the four cases with 12p13 deletion we compared the expression level of the genes located in the critical region with those on chromosome 12 outside the deleted region and found a lower expression level for the deleted genes (Figure 5B).
Figure 5.
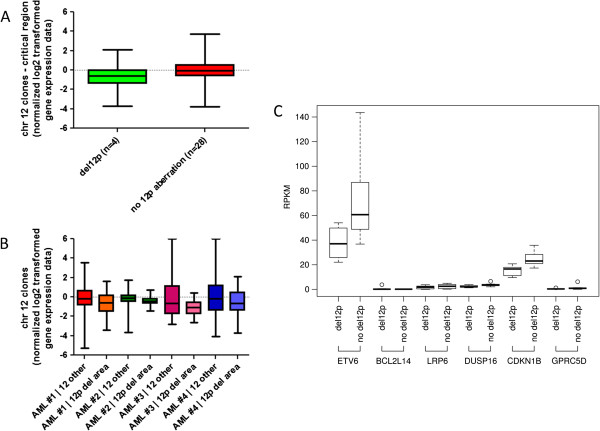
12p13 deletion-associated gene dosage effect. A: Comparison of CK-AML with (n = 4) and without 12p13 deletion (n = 28). The box plot shows the expression values for the genes located in the critical 12p region comprising the following genes: ETV6, BCL2L14, LRP6, DUSP16, CDKN1B and GPRC5D (normalized log2 transformed gene expression values). Cases with a 12p13 deletion show a significantly lower expression level for the deleted genes (p < 0.001, unpaired t-test). B: Comparison of the clones located within the critical region and clones located in unaltered chromosome 12 regions shows a significantly lower expression for the 12p critical region clones in AML cases with a 12p13 deletion (p = 0.041, ANOVA one-way analysis of variance). C: Comparison of CK-AML cases with (n = 8) and without 12p13 deletion (n = 8). The box plot shows the RPKM values of RNAseq data representing the expression level of the genes ETV6, BCL2L14, LRP6, DUSP16, CDKN1B and GPRC5D. Cases with a 12p13 deletion show a significantly lower expression level for ETV6 (p < 0.03, unpaired t-test) and CDKN1B (p < 0.003, unpaired t-test). BCL2L14, LRP6, DUSP16 and GPRC5D show no or very low expression levels. All data represented here were obtained from the TCGA network (http://cancergenome.nih.gov).
In addition, we screened the publicly available data of The Cancer Genome Atlas (TCGA, http://cancergenome.nih.gov/) for CK-AML patients with and without deletion in 12p13. We compared eight cases of CK-AML with deletion in 12p13 and eight cases of CK-AML without deletion in 12p13. ETV6 and CDKN1B are expressed in CK-AML with and without deletion in 12p13 (ETV6/del12p: 37.68 ± 12.95 RPKM, ETV6/no del12p: 71.63 ± 34.43 RPKM; CDKN1B/del12p: 15.19 ± 4.06 RPKM, CDKN1B/no del12p: 24.73 ± 5.86 RPKM). Notably, the expression level of ETV6 and CDKN1B decreased significantly by 0.53-fold (p < 0.03) and 0.62-fold (p < 0.003), respectively, in cases with 12p13 deletions compared to those without 12p13 deletions. The other candidate genes located within the minimal deleted region (BCL2L14, LRP6, DUSP16 and GPRC5D) showed no or very low expression levels irrespective of their copy number status (0.13 – 3.72 RPKM) (see Figure 5C).
Discussion
12p13 deletions are common in a broad spectrum of hematological malignancies, notably in myelodysplastic syndrome (MDS) with monosomy 7 [25] and CK-AML [8–10]. ETV6 and CDKN1B are the candidate tumor suppressor genes within the region 12p13 [12, 13]. Other candidate genes within the minimal deleted region like BCL2L14, LRP6, DUSP16 and GPRC5D may also play a role in tumorigenesis and leukemogenesis.
However, we now demonstrate according to the expression data provided by TCGA in confirmation of data published by Haferlach et al, that these genes do not show any expression or very low expression in blood or bone marrow cells of AML patients [26]. Likewise, using the platform HemaExplorer (http://servers.binf.ku.dk/hemaexplorer/), they show a low expression in normal hematopoietic stem and progenitor cells, compared to the expression level of ETV6 (5- to 40-fold higher) and CDKN1B (3- to 20-fold higher) in these cells [26]. It seems unlikely that they play a major role in leukemogenesis (see Figure 5C). Recent data suggest that the function of some tumor suppressor genes can be disrupted solely by haploinsufficiency leading to reduced gene dosage which might be sufficient to contribute to tumorigenesis [27–29]. Popular examples are TP53, PTEN, NPM1, NF1 and RPS14 [27, 29]. Likewise, for the genes APC, ATM, BRCA1/2 and RB haploinsufficiency contributes to tumorigenesis [28]. Haploinsufficiency of CDKN1B is strongly implicated in numerous cancer types, the vast majority of lymphatic origin [16, 17]. CDKN1B heterozygous mice are predisposed to tumors in multiple tissues [30]. ETV6 and CDKN1B are known to be inactivated mostly by monoallelic deletions. Yet, the mode of inactivation has not been investigated in detail in CK-AML with a high frequency of 12p13 deletions.
In our study, 12p13 deletions, mostly small interstitial deletions, were present in nearly half (49%) of the analyzed CK-AML patients according to karyotype. In 20 of 43 patients (47%), losses of 12p13 could be identified by genomic profiling, of those 13 small interstitial deletions (see Additional file 1: Table S1). The high frequency may be explained by high-resolution arrays used in this study. In previous studies based on karyotyping or SNP arrays of lower resolution, the frequency of 12p13 deletions in CK-AML was 27% [31] and 18% [10], respectively. However, it cannot be excluded that the rather high frequency in our study is due to the relatively small size of our cohort. The minimal deleted region in our cohort spanned 1.43 Mb and included CDKN1B and the 3′UTR of ETV6 (see Figure 1). In one patient (#48) the distal breakpoint is located within a region between exon 8 and the 3′UTR of the ETV6 gene. As the end of the ETS DNA-binding domain and the highly conserved polyadenylation signal lay within the deleted region and differential polyadenylation of the 3′UTR of ETV6 plays a major role in posttranscriptional modification [32], we assume that ETV6 is contained in the minimal deleted region.
ETV6 and CDKN1B mutations have never been investigated specifically in CK-AML. Somatic ETV6 mutations are rare events in newly diagnosed AML [33], AML-M0 [34] and MDS [35]. We detected one heterozygous frameshift mutation among 56 patients (see Figure 3, Additional file 1: Table S1). Our results thereby confirm the low rate of ETV6 mutations in the distinct subgroup of CK-AML. The detected frameshift mutation lies within the N-terminal homodimerization domain leading to a complete loss of the DNA-binding ETS domain. The vast majority of all reported ETV6 mutations results in inactivation of one ETV6 allele which is consistent with haploinsufficiency as the underlying mechanism. There was no typical driver alteration like TP53 or FLT3 mutations and no typical cytogenetic aberrations of CK-AML like del(5q), -7/del(7q), or del(17p) present in this patient, which could provide a proliferative advantage or even initiate leukemogenesis. This might strengthen the role of this ETV6 mutation as a driver rather than a passenger mutation. Unfortunately, no fibroblast DNA was available to prove whether the ETV6 mutation was of germline origin. However, this seems to be unlikely as ETV6 is a critical regulator in the survival of multiple cell types during early embryonic development and ETV6 knockout mice are embryonically lethal [36].
According to the literature, CDKN1B mutations have been previously reported in childhood leukemia [37] and rarely in T-cell prolymphocytic leukemia [16] as well as in adult T-cell leukemia [38]. We did not identify a mutation in the CDKN1B gene in our analyzed patient cohort (see Additional file 1: Table S1).
As expected, in more than half of the analyzed patients, TP53 mutations were detected (see Additional file 2: Table S2). However, the frequency of 12p13 deletions did not differ between patients with and without a monoallelic or biallelic alteration of TP53 (45.5% versus 45.7%, p = 0.99).
One mechanism of haploinsufficiency is increased methylation leading to reduced gene dosage due to transcriptional silencing. For ETV6, increased methylation was suggested as a possible mechanism since decreased ETV6 protein expression was reported in AML patients [33]. Hypermethylation of CDKN1B was excluded in not further defined AML and MDS, but found in the lymphoblast-like cell line Raji [39]. We demonstrate here that the CpG islands within the ETV6 and CDKN1B 5′UTRs are not hypermethylated in CK-AML (see Figure 4). To our knowledge, this is the first report that excludes 5′UTR methylation leading to ETV6 and CDKN1B inactivation in CK-AML and supports haploinsufficiency by heterozygous deletions as mode of inactivation.
We show by reanalysis of our previously published gene expression profiling [23, 24] of patients with CK-AML and deletion in 12p13 a significantly lower expression level for the deleted genes (p < 0.001) compared to a group of CK-AML patients without 12p13 alteration (see Figure 5A + B). Furthermore, we screened the publicly available data of TCGA and demonstrate that the other candidate genes located within the minimal deleted region (BCL2L14, LRP6, DUSP16 and GPRC5D) show no or very low expression levels irrespective of their copy number. In contrast, ETV6 and CDKN1B are expressed in CK-AML and their expression levels significantly decreased in cases with 12p13 deletions (see Figure 5C). Thus, ETV6 and CDKN1B are the sole genes within the minimal deleted region with expression levels reduced to approximately half. These data strongly support our hypothesis that haploinsufficiency is the underlying mechanism of inactivation of ETV6 and CDKN1B.
Conclusion
In summary, we demonstrate that the putative tumor suppressor genes ETV6 and CDKN1B are frequently inactivated by loss of one copy, most frequently by small deletions of 12p13, in CK-AML. The expression level of ETV6 and CDKN1B is significantly decreased in cases with 12p13 deletions whereas the other potential candidate genes within the minimal deleted region do not show any or very low expression irrespective of their copy number status. Mutations and hypermethylation as mode of inactivation were largely excluded. It is possible that the genomic complexity leads to acquisition of the deletion in 12p13. Future studies investigating clonal evolution should clarify, whether haploinsufficiency of both genes may cooperate early in the process of leukemic transformation by disordering key processes of differentiation and proliferation and whether they may also play a critical role in the induction of chromosomal instability finally resulting in the development of clones with complex karyotypes.
Methods
Patients
79 patients with CK-AML were analyzed (see Additional file 1: Table S1). Complex karyotype was defined by the presence of at least three chromosomal abnormalities in the absence of the prognostically favorable t(8;21)(q22;q22), inv(16)(p13q22) or t(16;16)(p13;q22) and t(15;17)(q22;q12). The diagnosis of AML was made according to the French-American-British Cooperative Group criteria. The karyotypes were described according to the International System for Human Cytogenetic Nomenclature (ISCN) (2013) [40]. DNA for analysis was extracted from bone marrow or peripheral blood-derived cell pellets stored at -196°C in liquid nitrogen and from methanol/acetic acid fixed cells stored at -80°C, using the Qiagen QIAamp® DNA Micro/Midi Kit (Qiagen, Hilden, Germany).
All patients signed informed consent forms, and the project was approved by the Ethics Committee of Hannover Medical School (approval no. 2899 of 20.06.2011). The research has been conducted in compliance with the Helsinki declaration.
Array-based genomic profiling
ArrayCGH using the 2.8 k and/or the 8.0 k platform and SNP analyses using Affymetrix GeneChip Human Mapping 250 K Array (Affymetrix, Santa Clara, California) and/or Genome-Wide Human SNP 6.0 Array (Affymetrix, Santa Clara, California) were performed as previously described by Rücker et al [41]. Results obtained from BAC/PAC arrayCGH are given in Additional file 3: Table S3. In brief, cut-off levels for gains and losses were determined for each individual experiment. After computing the ratios from dye-swap hybridization and subsequent normalization, an individual set of balanced clones for each experiment was used to calculate the mean and standard deviations. The cut-off levels were defined as mean plus/minus three times the standard deviation. Frequently affected regions recently detected as copy number polymorphisms (5q11, 7q22, 7q35, 14q32, and 15q11) were excluded from data analysis.
For arrayCGH using the Agilent Human Genome Microarray Kit 244A (Agilent Technologies, Waldbronn, Germany), a high resolution 60-mer oligonucleotide-based microarray, the procedures for DNA labeling, hybridization and washing were performed according to the manufacturer’s instructions (protocol version 6.1.) with some modifications as previously described by Praulich et al [42]. The slides were scanned on an Agilent Microarray Scanner and captured images were analyzed with Feature Extraction Software (v.10.7) (Agilent Technologies, Waldbronn, Germany). Data imaging and analysis were performed by the Agilent CGH Analytics software (v.5.0.14) with hg18 and Aberration Detection Method 2 (ADM-2) analysis algorithms set to specified thresholds and filter settings. All raw data from Agilent and Affymetrix are available under GEO (Gene expression omnibus, http://www.ncbi.nlm.nih.gov/geo/). Aberration summaries are archived under GSE55535. GEO accession numbers are given in Additional file 4: Table S4.
Direct sequencing
Exons 1-8 of ETV6 (ENSG00000139083), exons 1-2 of CDKN1B (ENSG00000111276), and exons 2-11 of TP53 (ENSG00000141510) were PCR-amplified from genomic DNA using FastStart Taq DNA Polymerase (Roche, Mannheim, Germany). After purification with the magnetic bead-based CleanSEQ® system (Beckman Coulter, Krefeld, Germany), PCR fragments were sequenced in both directions using the GenomeLab™ DTCS Quick Start Kit and CEQTM 8000 Genetic Analysis System (Beckman Coulter, Krefeld, Germany). Cloning of PCR products was performed in patients with complex mutations to describe the mutations properly using the TOPO TA Cloning® Kit (Invitrogen, Karlsruhe, Germany). All mutations were described according to the nomenclature for the description of sequence variations of the Human Genome Variation Society (HGVS, http://www.hgvs.org/).
Pyrosequencing
CpG islands were identified using CpG island searcher (http://cpgislands.usc.edu). We used the following settings: a G + C content of at least 55% GC, an observed CpG/expected CpG in excess of 0.65, and a minimum length of 500 bp. The pyrosequenced regions were also selected in the context of primer design, optimal PCR conditions and stringency.
Sodium bisulfite treatment of genomic DNA was performed using the EZ DNA Methylation Direct Kit™ (Zymo Research, Freiburg, Germany). Fragments for pyrosequencing were generated by PCR using the FastStart Taq™ DNA Polymerase Kit (Roche, Basel, Switzerland) with the following protocol: 10pmol of forward and reverse PCR primers (Metabion, Martinsried, Germany), and 0.78 units of FastStart Taq™ DNA Polymerase as well as different amounts of dNTPs and MgCl2. Cycle conditions were as follows: for fragment ETV6 denaturation at 97°C for 7 min, touchdown for 10 cycles including denaturation at 96°C for 30 s, annealing at 58°C for 30 seconds (which was decreased by 0.5°C in each cycle) and extension at 72°C for 1 min, followed by 25 cycles at 96°C for 30 s, annealing at 53°C for 30 s and 72°C for 1 min, finished with 72°C for 7 min; for fragment CDKN1B denaturation at 97°C for 7 min, touchdown for 10 cycles including denaturation at 96°C for 30 s, annealing at 53°C for 30 seconds (which was decreased by 0.5°C in each cycle) and extension at 72°C for 1 min, followed by 25 cycles at 96°C for 30 s, annealing at 48°C for 30 s and 72°C for 1 min, finished with 72°C for 7 min. Primer sequences were as follows: fragETV6-fw: 5′-GYGGGTGGGAGGAGAG-3′; fragETV6-rev-biot: 5′-biotin-TTCTTCCAACATCTCTCCC-3′; fragCDKN1B-fw: 5′-GTAGGTTTGTTGGTAGTAG-3′; fragCDKN1B-rev-biot: 5′-biotin-AAAAAAAATCCATTAATTAC-3′.
For purification of biotinylated fragments, 5 to 13 μL of PCR products were added to a mixture consisting of 3 μL Streptavidin Sepharose HP™ Beads (Amersham Biosciences, Freiburg, Germany) and 47 μL binding buffer (Qiagen, Hilden, Germany). Single-stranded fragments were purified using the Vacuum Prep Tool™ (Qiagen, Hilden, Germany). Sepharose beads with the single-stranded templates attached were added to a PSQ 96 Plate Low™ (Qiagen, Hilden, Germany) containing a mix of 11 mL annealing buffer (Qiagen, Hilden, Germany) and 800 nmmol/L of the corresponding sequencing primers (Metabion, Martinsried, Germany) with following sequences: fragETV6-seq: 5′-GATTTG TAGATTT-3′; fragCDKN1B-seq: 5′-GATTAGTTAATTTTT-3′.
Pyrosequencing was performed in a PyroMark MD™ System (Qiagen, Hilden, Germany) with the PyroGold SQA™ Reagent Kit (Qiagen, Hilden, Germany) containing nucleotides and prepared mixtures with enzymes or substrates. For pyrogram exposure including CpG-site methylation calculation, the Pyro Q-CpG™ Software (Biotage, Uppsala, Sweden; Version 1.0.9.) was applied. Only pyrograms including sharp peaks with satisfactory heights for each injected nucleotide of interest and without peaks for unsuccessful bisulfite treatment or background controls were considered.
Gene expression profiling analysis
Based on our previously published gene expression profiling [23, 24], we evaluated a 12p deletion associated gene dosage effect of our own patients by comparing complex karyotype cases with 12p13 deletion (n = 4) and cases without (n = 28). In brief, normalized log2 transformed gene expression levels of the minimally deleted region were averaged and compared to the average unaltered chromosome 12 regions.
SNP level3 datasets (archive: broad.mit.edu_LAML.Genome_Wide_SNP_6.Level_3.25.2004.0) and RNAseq level3 datasets (archive: bcgsc.ca_LAML.IlluminaGA_RNASeq. Level_3.1.7.0) of AML patients were obtained from the TCGA Research Network (http://cancergenome.nih.gov/). Visualization and selection of the SNP data was done using the Integrative Genomics Viewer [43, 44]. The RPKM values (RPKM = (10^9 * C)/(N * L); C = Number of reads mapped to a gene, N = Total mapped reads in the experiment, L = exon length in base-pairs for a gene) of the corresponding gene-quantification files were extracted and analyzed on gene level applying the t-test program of the statistical program R, version 3.1.1 (http://www.R-project.org/) [45]. Accordingly, box plots were made using the boxplot function of R.
Statistical analysis
We used the two-tailed Fisher’s exact test, Kruskal-Wallis one-way analysis of variance, the unpaired t-test and the ANOVA one-way analysis of variance. An effect was considered significant if the p value was <0.05.
Electronic supplementary material
Additional file 1: Table S1: Karyotypes, genomic profiling, sequencing results and methylation analyses of the patient cohort. Karyotypes of the 79 patients with CK-AML investigated in this study (classified according to ISCN recommendations), results obtained by genomic profiling, mutation analyses of ETV6 and CDKN1B (mutations are named according to HGVS (http://www.hgvs.org/)), and mean methylation levels. (XLS 43 KB)
Additional file 2: Table S2: Overview of TP53 mutations. TP53 mutation status of the 79 patients investigated in this study, mutations are named according to HGVS (http://www.hgvs.org/). FISH results with a probe for the locus 17p13 (TP53) are also shown if available. (XLS 26 KB)
Additional file 3: Table S3: Normalized log2 fluorescence ratio of six CK-AML samples obtained from 2.8/8.0 k BAC/PAC arrays. Cut-off levels for gains and losses were determined for each individual experiment. After computing the ratios from dye-swap hybridization and subsequent normalization, an individual set of balanced clones for each experiment was used to calculate the mean and standard deviations. The cutoff levels were defined as mean plus/minus three times the standard deviation. Frequently affected regions recently detected as copy number polymorphisms (5q11, 7q22, 7q35, 14q32, and 15q11) were excluded from data analysis. (XLSX 1 MB)
Additional file 4: Table S4: List of GEO accession numbers. List of GEO accession numbers for all samples run on the Agilent/Affymetrix platform. (DOC 56 KB)
Acknowledgments
Funding was supported by COST Action BM0801 (EuGESMA). We would like to thank Gillian Teicke for editing the manuscript.
Abbreviations
- AML
Acute myeloid leukemia
- CK-AML
Acute myeloid leukemia with complex karyotype
- arrayCGH
Array comparative genomic hybridization
- HGVS
Humane Genome Variation Society
- ISCN
International System for Human Cytogenetic Nomenclature
- MDS
Myelodysplastic syndrome
- SNP array
Single-nucleotide polymorphism array
- TCGA
The cancer genome atlas.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SF, FR, LB, WH, GM, and GG performed experiments. SF, FR, LB, WH, GG, MH, UL, BS and DS analyzed and interpreted data. SF and DS wrote the paper. KD, UL, MH, AG, BS and DS proposed the research goal, supervised the whole studies and provided a critical review of the manuscript. All authors read and approved the final manuscript.
Contributor Information
Simone Feurstein, Email: Feurstein.Simone@mh-hannover.de.
Frank G Rücker, Email: Frank.Ruecker@uniklinik-ulm.de.
Lars Bullinger, Email: Lars.Bullinger@uniklinik-ulm.de.
Winfried Hofmann, Email: Hofmann.Winfried@mh-hannover.de.
Georgi Manukjan, Email: Manukjan.Georgi@mh-hannover.de.
Gudrun Göhring, Email: Goehring.Gudrun@mh-hannover.de.
Ulrich Lehmann, Email: Lehmann.Ulrich@mh-hannover.de.
Michael Heuser, Email: Heuser.Michael@mh-hannover.de.
Arnold Ganser, Email: Ganser.Arnold@mh-hannover.de.
Konstanze Döhner, Email: Konstanze.Doehner@uniklinik-ulm.de.
Brigitte Schlegelberger, Email: Schlegelberger.Brigitte@mh-hannover.de.
Doris Steinemann, Email: Steinemann.Doris@mh-hannover.de.
References
- 1.Schlenk RF, Dohner K, Krauter J, Frohling S, Corbacioglu A, Bullinger L, Habdank M, Spath D, Morgan M, Benner A, Schlegelberger B, Heil G, Ganser A, Dohner H, German-Austrian Acute Myeloid Leukemia Study, Group Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358(18):1909–1918. doi: 10.1056/NEJMoa074306. [DOI] [PubMed] [Google Scholar]
- 2.Haferlach T, Kern W, Schoch C, Schnittger S, Sauerland MC, Heinecke A, Buchner T, Hiddemann W, German AMLCG. A new prognostic score for patients with acute myeloid leukemia based on cytogenetics and early blast clearance in trials of the German AML Cooperative Group. Haematologica. 2004;89(4):408–418. [PubMed] [Google Scholar]
- 3.Schoch C, Haferlach T, Haase D, Fonatsch C, Loffler H, Schlegelberger B, Staib P, Sauerland MC, Heinecke A, Buchner T, Hiddemann W, German A, German A. M. L. Cooperative Study Group Patients with de novo acute myeloid leukaemia and complex karyotype aberrations show a poor prognosis despite intensive treatment: a study of 90 patients. Br J Haematol. 2001;112(1):118–126. doi: 10.1046/j.1365-2141.2001.02511.x. [DOI] [PubMed] [Google Scholar]
- 4.Lindvall C, Furge K, Bjorkholm M, Guo X, Haab B, Blennow E, Nordenskjold M, Teh BT. Combined genetic and transcriptional profiling of acute myeloid leukemia with normal and complex karyotypes. Haematologica. 2004;89(9):1072–1081. [PubMed] [Google Scholar]
- 5.Bowen D, Groves MJ, Burnett AK, Patel Y, Allen C, Green C, Gale RE, Hills R, Linch DC. TP53 gene mutation is frequent in patients with acute myeloid leukemia and complex karyotype, and is associated with very poor prognosis. Leukemia. 2009;23(1):203–206. doi: 10.1038/leu.2008.173. [DOI] [PubMed] [Google Scholar]
- 6.Haferlach C, Dicker F, Herholz H, Schnittger S, Kern W, Haferlach T. Mutations of the TP53 gene in acute myeloid leukemia are strongly associated with a complex aberrant karyotype. Leukemia. 2008;22(8):1539–1541. doi: 10.1038/leu.2008.143. [DOI] [PubMed] [Google Scholar]
- 7.Schoch C, Kern W, Kohlmann A, Hiddemann W, Schnittger S, Haferlach T. Acute myeloid leukemia with a complex aberrant karyotype is a distinct biological entity characterized by genomic imbalances and a specific gene expression profile. Genes Chromosomes Cancer. 2005;43(3):227–238. doi: 10.1002/gcc.20193. [DOI] [PubMed] [Google Scholar]
- 8.Byrd JC, Mrozek K, Dodge RK, Carroll AJ, Edwards CG, Arthur DC, Pettenati MJ, Patil SR, Rao KW, Watson MS, Koduru PR, Moore JO, Stone RM, Mayer RJ, Feldman EJ, Davey FR, Schiffer CA, Larson RA, Bloomfield CD, Cancer Leukemia Group B Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461) Blood. 2002;100(13):4325–4336. doi: 10.1182/blood-2002-03-0772. [DOI] [PubMed] [Google Scholar]
- 9.Kayser S, Zucknick M, Dohner K, Krauter J, Kohne CH, Horst HA, Held G, von Lilienfeld-Toal M, Wilhelm S, Rummel M, Germing U, Gotze K, Nachbaur D, Schlegelberger B, Gohring G, Spath D, Morlok C, Teleanu V, Ganser A, Dohner H, Schlenk RF, German-Austrian Acute Myeloid Leukemia Study, Group Monosomal karyotype in adult acute myeloid leukemia: prognostic impact and outcome after different treatment strategies. Blood. 2012;119(2):551–558. doi: 10.1182/blood-2011-07-367508. [DOI] [PubMed] [Google Scholar]
- 10.Rucker FG, Bullinger L, Schwaenen C, Lipka DB, Wessendorf S, Frohling S, Bentz M, Miller S, Scholl C, Schlenk RF, Radlwimmer B, Kestler HA, Pollack JR, Lichter P, Dohner K, Dohner H. Disclosure of candidate genes in acute myeloid leukemia with complex karyotypes using microarray-based molecular characterization. J Clin Oncol. 2006;24(24):3887–3894. doi: 10.1200/JCO.2005.04.5450. [DOI] [PubMed] [Google Scholar]
- 11.Montpetit A, Boily G, Sinnett D. A detailed transcriptional map of the chromosome 12p12 tumour suppressor locus. Eur J Hum Genet. 2002;10(1):62–71. doi: 10.1038/sj.ejhg.5200766. [DOI] [PubMed] [Google Scholar]
- 12.Montpetit A, Larose J, Boily G, Langlois S, Trudel N, Sinnett D. Mutational and expression analysis of the chromosome 12p candidate tumor suppressor genes in pre-B acute lymphoblastic leukemia. Leukemia. 2004;18(9):1499–1504. doi: 10.1038/sj.leu.2403441. [DOI] [PubMed] [Google Scholar]
- 13.Sato Y, Suto Y, Pietenpol J, Golub TR, Gilliland DG, Davis EM, Le Beau MM, Roberts JM, Vogelstein B, Rowley JD. TEL and KIP1 define the smallest region of deletions on 12p13 in hematopoietic malignancies. Blood. 1995;86(4):1525–1533. [PubMed] [Google Scholar]
- 14.Andreasson P, Johansson B, Arheden K, Billstrom R, Mitelman F, Hoglund M. Deletions of CDKN1B and ETV6 in acute myeloid leukemia and myelodysplastic syndromes without cytogenetic evidence of 12p abnormalities. Gene Chromosome Canc. 1997;19(2):77–83. doi: 10.1002/(SICI)1098-2264(199706)19:2<77::AID-GCC2>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 15.Baens M, Wlodarska I, Corveleyn A, Hoornaert I, Hagemeijer A, Marynen P. A physical, transcript, and deletion map of chromosome region 12p12.3 flanked by ETV6 and CDKN1B: hypermethylation of the LRP6 CpG island in two leukemia patients with hemizygous del(12p) Genomics. 1999;56(1):40–50. doi: 10.1006/geno.1998.5685. [DOI] [PubMed] [Google Scholar]
- 16.Le Toriellec E, Despouy G, Pierron G, Gaye N, Joiner M, Bellanger D, Vincent-Salomon A, Stern MH. Haploinsufficiency of CDKN1B contributes to leukemogenesis in T-cell prolymphocytic leukemia. Blood. 2008;111(4):2321–2328. doi: 10.1182/blood-2007-06-095570. [DOI] [PubMed] [Google Scholar]
- 17.Paige AJ. Redefining tumour suppressor genes: exceptions to the two-hit hypothesis. Cell Mol Life Sci. 2003;60(10):2147–2163. doi: 10.1007/s00018-003-3027-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fenrick R, Wang L, Nip J, Amann JM, Rooney RJ, Walker-Daniels J, Crawford HC, Hulboy DL, Kinch MS, Matrisian LM, Hiebert SW. TEL, a putative tumor suppressor, modulates cell growth and cell morphology of ras-transformed cells while repressing the transcription of stromelysin-1. Mol Cell Biol. 2000;20(16):5828–5839. doi: 10.1128/MCB.20.16.5828-5839.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Irvin BJ, Wood LD, Wang L, Fenrick R, Sansam CG, Packham G, Kinch M, Yang E, Hiebert SW. TEL, a putative tumor suppressor, induces apoptosis and represses transcription of Bcl-XL. J Biol Chem. 2003;278(47):46378–46386. doi: 10.1074/jbc.M305189200. [DOI] [PubMed] [Google Scholar]
- 20.Yamagata T, Maki K, Waga K, Mitani K. TEL/ETV6 induces apoptosis in 32D cells through p53-dependent pathways. Biochem Biophys Res Commun. 2006;347(2):517–526. doi: 10.1016/j.bbrc.2006.06.127. [DOI] [PubMed] [Google Scholar]
- 21.Toyoshima H, Hunter T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell. 1994;78(1):67–74. doi: 10.1016/0092-8674(94)90573-8. [DOI] [PubMed] [Google Scholar]
- 22.Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7(10):e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bullinger L, Dohner K, Bair E, Frohling S, Schlenk RF, Tibshirani R, Dohner H, Pollack JR. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N Engl J Med. 2004;350(16):1605–1616. doi: 10.1056/NEJMoa031046. [DOI] [PubMed] [Google Scholar]
- 24.Kharas MG, Lengner CJ, Al-Shahrour F, Bullinger L, Ball B, Zaidi S, Morgan K, Tam W, Paktinat M, Okabe R, Gozo M, Einhorn W, Lane SW, Scholl C, Frohling S, Fleming M, Ebert BL, Gilliland DG, Jaenisch R, Daley GQ. Musashi-2 regulates normal hematopoiesis and promotes aggressive myeloid leukemia. Nat Med. 2010;16(8):903–908. doi: 10.1038/nm.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wall M, Rayeroux KC, MacKinnon RN, Zordan A, Campbell LJ. ETV6 deletion is a common additional abnormality in patients with myelodysplastic syndromes or acute myeloid leukemia and monosomy 7. Haematologica. 2012;97(12):1933–1936. doi: 10.3324/haematol.2012.069716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haferlach C, Bacher U, Kohlmann A, Schindela S, Alpermann T, Kern W, Schnitther S, Haferlach T. CDKN1B, encoding the cyclin-dependent kinase inhibitor 1B (p27), is located in the minimally deleted region of 12p abnormalities in myeloid malignancies and its low expression is a favorable prognostic marker in acute myeloid leukemia. Haematologica. 2011;96(6):829–36. doi: 10.3324/haematol.2010.035584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berger AH, Pandolfi PP. Haplo-insufficiency: a driving force in cancer. J Pathol. 2011;223(2):137–146. doi: 10.1002/path.2800. [DOI] [PubMed] [Google Scholar]
- 28.Santarosa M, Ashworth A. Haploinsufficiency for tumour suppressor genes: when you don't need to go all the way. Biochim Biophys Acta. 2004;1654(2):105–122. doi: 10.1016/j.bbcan.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 29.Wong JC, Le Beau MM, Shannon K. Tumor suppressor gene inactivation in myeloid malignancies. Best Pract Res Clin Haematol. 2008;21(4):601–614. doi: 10.1016/j.beha.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 30.Fero ML, Randel E, Gurley KE, Roberts JM, Kemp CJ. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature. 1998;396(6707):177–180. doi: 10.1038/24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schoch C, Haferlach T, Bursch S, Gerstner D, Schnittger S, Dugas M, Kern W, Loffler H, Hiddemann W. Loss of genetic material is more common than gain in acute myeloid leukemia with complex aberrant karyotype: a detailed analysis of 125 cases using conventional chromosome analysis and fluorescence in situ hybridization including 24-color FISH. Gene Chromosome Canc. 2002;35(1):20–29. doi: 10.1002/gcc.10088. [DOI] [PubMed] [Google Scholar]
- 32.Baens M, Peeters P, Guo C, Aerssens J, Marynen P. Genomic organization of TEL: the human ETS-variant gene 6. Genome Res. 1996;6(5):404–413. doi: 10.1101/gr.6.5.404. [DOI] [PubMed] [Google Scholar]
- 33.Barjesteh van Waalwijk van Doorn-Khosrovani S, Spensberger D, de Knegt Y, Tang M, Lowenberg B, Delwel R. Somatic heterozygous mutations in ETV6 (TEL) and frequent absence of ETV6 protein in acute myeloid leukemia. Oncogene. 2005;24(25):4129–4137. doi: 10.1038/sj.onc.1208588. [DOI] [PubMed] [Google Scholar]
- 34.Silva FP, Morolli B, Storlazzi CT, Zagaria A, Impera L, Klein B, Vrieling H, Kluin-Nelemans HC, Giphart-Gassler M. ETV6 mutations and loss in AML-M0. Leukemia. 2008;22(8):1639–1643. doi: 10.1038/leu.2008.34. [DOI] [PubMed] [Google Scholar]
- 35.Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, Kantarjian H, Raza A, Levine RL, Neuberg D, Ebert BL. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364(26):2496–2506. doi: 10.1056/NEJMoa1013343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang LC, Kuo F, Fujiwara Y, Gilliland DG, Golub TR, Orkin SH. Yolk sac angiogenic defect and intra-embryonic apoptosis in mice lacking the Ets-related factor TEL. EMBO J. 1997;16(14):4374–4383. doi: 10.1093/emboj/16.14.4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Markaki EA, Stiakaki E, Zafiropoulos A, Arvanitis DA, Katzilakis N, Dimitriou H, Spandidos DA, Kalmanti M. Mutational analysis of the cell cycle inhibitor Kip1/p27 in childhood leukemia. Pediatr Blood Cancer. 2006;47(1):14–21. doi: 10.1002/pbc.20730. [DOI] [PubMed] [Google Scholar]
- 38.Morosetti R, Kawamata N, Gombart AF, Miller CW, Hatta Y, Hirama T, Said JW, Tomonaga M, Koeffler HP. Alterations of the p27KIP1 gene in non-Hodgkin's lymphomas and adult T-cell leukemia/lymphoma. Blood. 1995;86(5):1924–1930. [PubMed] [Google Scholar]
- 39.Chim CS, Wong AS, Kwong YL. Epigenetic inactivation of the CIP/KIP cell-cycle control pathway in acute leukemias. Am J Hematol. 2005;80(4):282–287. doi: 10.1002/ajh.20503. [DOI] [PubMed] [Google Scholar]
- 40.Schaffer L, McGowan-Jordan J, Schmid M. ISCN 2013:An international system for human cytogenetic nomenclature (2013) 2012. [Google Scholar]
- 41.Rucker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H, Habdank M, Kugler CM, Holzmann K, Gaidzik VI, Paschka P, Held G, von Lilienfeld-Toal M, Lubbert M, Frohling S, Zenz T, Krauter J, Schlegelberger B, Ganser A, Lichter P, Dohner K, Dohner H. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2012;119(9):2114–2121. doi: 10.1182/blood-2011-08-375758. [DOI] [PubMed] [Google Scholar]
- 42.Praulich I, Tauscher M, Gohring G, Glaser S, Hofmann W, Feurstein S, Flotho C, Lichter P, Niemeyer CM, Schlegelberger B, Steinemann D. Clonal heterogeneity in childhood myelodysplastic syndromes–challenge for the detection of chromosomal imbalances by array-CGH. Gene Chromosome Canc. 2010;49(10):885–900. doi: 10.1002/gcc.20797. [DOI] [PubMed] [Google Scholar]
- 43.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14(2):178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.R: A Language And Environment For Statistical Computing. I Vienna, Austria: R Foundation for Statistical computing; 2014. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1: Karyotypes, genomic profiling, sequencing results and methylation analyses of the patient cohort. Karyotypes of the 79 patients with CK-AML investigated in this study (classified according to ISCN recommendations), results obtained by genomic profiling, mutation analyses of ETV6 and CDKN1B (mutations are named according to HGVS (http://www.hgvs.org/)), and mean methylation levels. (XLS 43 KB)
Additional file 2: Table S2: Overview of TP53 mutations. TP53 mutation status of the 79 patients investigated in this study, mutations are named according to HGVS (http://www.hgvs.org/). FISH results with a probe for the locus 17p13 (TP53) are also shown if available. (XLS 26 KB)
Additional file 3: Table S3: Normalized log2 fluorescence ratio of six CK-AML samples obtained from 2.8/8.0 k BAC/PAC arrays. Cut-off levels for gains and losses were determined for each individual experiment. After computing the ratios from dye-swap hybridization and subsequent normalization, an individual set of balanced clones for each experiment was used to calculate the mean and standard deviations. The cutoff levels were defined as mean plus/minus three times the standard deviation. Frequently affected regions recently detected as copy number polymorphisms (5q11, 7q22, 7q35, 14q32, and 15q11) were excluded from data analysis. (XLSX 1 MB)
Additional file 4: Table S4: List of GEO accession numbers. List of GEO accession numbers for all samples run on the Agilent/Affymetrix platform. (DOC 56 KB)