

Abstract
Innate signaling–induced antimicrobial response represents a key protective host feature against infectious microorganisms such as Campylobacter species. In this study, we investigated the role of nucleotide-binding oligomerization domain-containing protein 2 (NOD2) in Campylobacter jejuni–induced intestinal inflammation. Specific-pathogen-free Il10−/−, Nod2−/−, and Il10−/−; Nod2−/− mice were infected with C. jejuni (109 colony-forming units/mouse) 24 hours after a 7-day course of antibiotic treatment. Three weeks later, host responses were determined. The nitric oxide (NO) donor sodium nitroprusside was injected intraperitoneally (2 mg/kg daily) to supplement NO. Although healthy in specific-pathogen-free conditions, Il10−/−; Nod2−/− mice developed severe intestinal inflammation following C. jejuni infection, compared with Nod2−/− and Il10−/− mice. The onset of colitis was associated with elevated neutrophil accumulation, crypt abscesses, and expression of the endogenous proinflammatory mediators Il-1β, Tnfα, and Cxcl1. Fluorescence in situ hybridization and culture assay showed enhanced C. jejuni invasion into the colon and mesenteric lymph nodes in Il10−/−; Nod2−/− mice, compared with Il10−/− mice. C. jejuni–induced bactericidal NO production was reduced in peritoneal macrophages from Il10−/−; Nod2−/− mice, compared with Il10−/− mice. Importantly, sodium nitroprusside attenuated C. jejuni–induced colitis in Il10−/−; Nod2−/− mice. Our findings suggest that NOD2 signaling is critical to control campylobacteriosis in Il10−/− mice, a process involving NOD2-mediated bactericidal responses.
Keywords: innate immunity, intestinal inflammation, campylobacteriosis, bactericide, Il10−/−mice
Inflammatory bowel diseases (IBDs), including ulcerative colitis and Crohn disease, afflict 1.4 million people in the United States alone. The exact etiology of IBD remains to be defined, but the host genetic susceptibility, microbiota, and environmental factors play critical roles in the development of the diseases [1]. Nucleotide-binding oligomerization domain (NOD) proteins are members of a large family of proteins named “NOD-like receptors” whose functions are essential in innate/adaptive host responses to various commensal and pathogenic bacteria [2–5]. NOD2 senses the bacterial cell wall component peptidoglycan and its derivative byproduct, muramyl dipeptide [4, 5]. In addition, NOD2 senses live intracellular pathogenic microorganisms such as Salmonella enterica, Listeria monocytogenes, Mycobacterium tuberculosis, and Streptococcus pneumoniae [6–9]. This host response is central to the elimination of the damaging agents and to the reestablishment of homeostasis. For example, defective NOD2 signaling impaired intestinal epithelial cells in their clearance of Salmonella Typhimurium in vitro and decreased host responses to L. monocytogenes infection in vivo [7, 8]. Similarly, NOD2-deficient mice are more susceptible than wild-type mice to S. Typhimurium infection [10]. The molecular mechanism by which NOD2 controls host response to bacteria is unclear, but its role in autophagy and microbial killing has recently been documented in vitro [11, 12]. For example, NOD2-mediated autophagy is required for bacterial handling and antigen presentation in dendritic cells [11]. Importantly, patients with IBD are often subject to a relapsing episode following infection with enteric bacterial pathogens such as Salmonella or Campylobacter species [13].
Campylobacter jejuni is one of the most prevalent food-borne bacterial pathogens in the world. Millions of people are infected with C. jejuni every year in the United States [14]. Clinical features of acute campylobacteriosis include bloody diarrhea, abdominal cramps, and severe intestinal inflammation. Despite the prevalence of campylobacteriosis, its pathogenesis remains largely unknown because of the limited availability of experimental animal models mimicking human disease.
Interleukin 10 (IL-10) is secreted by a variety of immune cells, including monocytes/macrophages and T cells, upon stimulation with various agents, such as lipopolysaccharide and bacteria [15]. IL-10 plays an essential role in maintaining intestinal homeostasis by regulating expression of proinflammatory cytokines, chemokines, and costimulatory molecules [16]. Indeed, 129SvEv Il10−/− mice born and raised in specific-pathogen-free (SPF) conditions developed intestinal inflammation after a couple of months but remained healthy when raised in germ-free conditions, showing the key role of bacteria in intestinal inflammation. Interestingly, C57BL/6 Il10−/− mice raised in SPF conditions and infected with C. jejuni strain 11168 displayed mild intestinal inflammation at day 35 after infection [17]. More recently, we reported that germ-free C57BL6/129SvEv and SPF 129 Il10−/− mice infected with C. jejuni 84–176 showed acute campylobacteriosis [18–20]. In addition, Haag et al showed that C. jejuni induces acute intestinal inflammation in C57BL/10 mice pretreated with antibiotics [21]. Using germ-free 129SvEv Il10−/−; Rag2−/− mice, we recently showed that innate immune cells are essential in mediating the early phase of campylobacteriosis [20], suggesting an important role for innate immunity in controlling C. jejuni infection. These findings illustrate the benefit of using Il10−/− mice for studying mechanisms implicated in campylobacteriosis.
Reactive nitrogen species (RNS) such as nitric oxide (·NO) and superoxide (O2·−) are a group of antimicrobial intermediate molecules produced by macrophages and are involved in eliminating intercellular and intracellular pathogens [22, 23]. The formation of RNS molecules is mediated by inducible nitric oxide synthase 2 (iNOS) and nicotinamide adenine dinucleotide phosphate oxidase. Interestingly, C. jejuni survival in vitro can be reduced by chemically generated RNS, using an acidified nitrite solution [24]. However, the role of host-generated NO in defense against C. jejuni infection remains unknown. Furthermore, no studies have directly assessed the relationship between NOD2 and RNS in the host response to bacterial infection in vivo.
In this study, we investigated the role of NOD2 in C. jejuni–induced intestinal inflammation. Using genetic manipulation, we showed that NOD2 enhances myeloid cell–induced bactericidal capacity and attenuates C. jejuni–induced intestinal inflammation through RNS generation. These findings provide a basis for developing novel approaches with the potential to control campylobacteriosis by targeting NOD2-mediated RNS.
METHODS
Mice and Tissue Processing
All animal protocols were approved by the institutional animal care and use committees of the University of North Carolina at Chapel Hill and the University of Florida. SPF 8–12-week-old C57BL/6 Nod2−/−, Il10−/−; Nod2−/−, and Il10−/− mice received a single dose of 109 colony-forming units of C. jejuni (strain 81–176) by gavage [25] 24 hours after conclusion of a 7-day course of treatment with an antibiotic cocktail (streptomycin 2 g/L, bacitracin 1 g/L, gentamicin 0.5 g/L, and ciprofloxacin 0.125 g/L) [19]. All mice were euthanized 21 days after infection. To supplement NO in vivo, mice were injected intraperitoneally daily from days 0 to 21 after infection with sodium nitroprusside (2 mg/kg). Tissue samples from the colon, spleen, and mesenteric lymph nodes (MLNs) were collected for protein, RNA analysis, and histological analysis as described previously [19]. C. jejuni culture assays were performed as described previously [19]. Histological images were acquired, and intestinal inflammation was scored on a scale of 0 to 4 as described before [18, 19]. Crypt abscesses in colonic tissues were identified on the basis of morphological features, using hematoxylin-eosin–stained sections, and were counted as the number of abscesses per 100 crypts, using a microscope.
Peritoneal Macrophage Isolation and Nitrite Assay
Il10−/− and Il10−/−; Nod2−/− mice were injected intraperitoneally with 2 mL of 3% fluid thioglycollate medium (Difco Laboratories, Detroit, MI) previously autoclaved for 15 minutes under 104 kPa. After 4 days, mice were euthanized by CO2 intoxication, and macrophages in the peritoneal cavity were retrieved by lavaging 3 times with 3 mL of ice-cold HBSS supplemented with 0.15 mM ethylenediaminetetraacetic acid. The macrophages were resuspended in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 2% fetal bovine serum (FBS), and cell viability (>95%) was determined by trypan blue exclusion. Cells were infected with C. jejuni (multiplicity of infection [MOI], 50) for 4 hours, collected by centrifugation, and lysed in TRIzol (Invitrogen) for RNA extraction.
To measure NO production, macrophages were plated in triplicate in 24-well plates and then infected with C. jejuni (MOI, 50) for 12 hours. Cells were washed 3 times and incubated with 100 µg/mL gentamicin for 1 hour, and then medium was replaced with RPMI 1640 medium supplemented with 2% FBS, and 10 µg/mL gentamicin was added. After 12 hours, NO production in the supernatant was determined by the Griess assay [23].
Gentamicin Protection Assay to Determine C. jejuni Invasion of Peritoneal Macrophages
The C. jejuni gentamicin protection assay was performed by infecting 106 macrophages at an MOI of 50 in 12-well plates as described previously [19]. Briefly, macrophages were plated in triplicate in 12-well plates and infected with C. jejuni for 4 hours. After incubation for 4 hours, the macrophages were washed 3 times with phosphate-buffered saline (PBS) and incubated with fresh RPMI 1640 medium containing gentamicin (100 µg/mL) for an additional 1 hour. For sample collection at 0 hours, cells were washed 3 times before lysis in 0.1% Triton X-100. For sample collection at 1 and 2 hours, cells were incubated in fresh RPMI 1640 medium with gentamicin (10 µg/mL) for an additional 1 and 2 hours, respectively. Samples were then washed 3 times and lysed as described above. The lysates were plated on Remel plates to recover live C. jejuni from infected macrophages.
Confocal Microscopy to Determine C. jejuni Invasion
Macrophages (104) were plated in triplicate in 8-well chamber plates and infected for 4 hours with C. jejuni preincubated with 5 mM 5-Cyano-2,3-di-(p-tolyl)tetrazolium chloride (CTC). A gentamicin protection assay was then performed as described above, and the cells were fixed with 4% paraformaldehyde. The fixed cells were stained with DAPI medium (Vector Laboratory) and were visualized using confocal microscopy (Zeiss LSM710). Acquired images were analyzed using BioimageXD [26].
Fluorescence In Situ Hybridization (FISH)
Cy3-tagged 5′-AGCTAACCACACCTTATACCG-3′ was used to probe the presence of C. jejuni in the intestinal tissue sections as previously described [19]. Briefly, tissues were deparaffinized, hybridized with the probe, washed, stained with DAPI, and imaged using a Zeiss LSM710 Spectral Confocal Laser Scanning Microscope system with ZEN 2008 software. Acquired images were analyzed using BioimageXD.
Immunohistochemical (IHC) Analysis
Neutrophils in intestinal tissues were detected using anti-myeloperoxidase (MPO) IHC analysis as described previously [19]. Briefly, intestinal tissue sections were deparaffinized, blocked, and incubated with an anti-MPO antibody (1:400; Thermo Scientific) overnight. After incubation with anti-rabbit biotinylated antibody, avidin/biotin complex (Vectastain ABC Elite Kit, Vector Laboratories), diaminobenzidine (Dako), and hematoxylin-eosin (Fisher Scientific), the sections were imaged.
C. jejuni Quantification in Tissues
Colon, MLNs, and spleen were aseptically resected. Colon tissue was opened and washed 3 times in sterile PBS. The tissues were weighed, homogenized in PBS, serially diluted, and plated on Campylobacter-selective blood plates (Remel) for 48 hours at 37°C, using the GasPak system (BD). C. jejuni colonies were counted, and data are presented as CFU per gram of tissue.
Real-Time Reverse-Transcription Polymerase Chain Reaction (PCR)
Total RNA from intestinal tissues or peritoneal macrophages was extracted using TRIzol (Invitrogen) following the manufacture's protocol, and RNA was reverse transcribed using M-MLV (Invitrogen). Messenger RNA (mRNA) expression of the proinflammatory mediators Il1β, Cxcl1, iNos and Tnfα was measured using SYBR Green PCR Master mix (Applied Biosystems) on an ABI 7900HT Fast Real-Time PCR System and normalized to Gapdh. The PCR primer sequences were previously described [19]; iNos_forward is GTGGTGACAAGCACATTTGG, and iNos_reverse is GGCTGGACTTTTCACTCTGC. The PCR reactions were performed for 40 cycles according to the manufacturer's recommendation, and relative RNA fold-changes were calculated using the ΔΔCt method.
Statistical Analysis
Values are shown as mean ± standard error of the mean as indicated. Differences between groups were analyzed using the nonparametric Mann–Whitney U test or the t test. Experiments were considered statistically significant if P values were <.05. All calculations were performed using Prism 5.0 software.
RESULTS
NOD2 Deficiency Exacerbates C. jejuni–Induced Intestinal Inflammation
We previously showed that C. jejuni–infected Il10−/− mice but not wild-type mice developed colitis [18]. In addition, in our housing conditions and unlike 129SvEv Il10−/− mice, C57BL/6 Il10−/− mice did not develop inflammation, making them a valuable tool to investigate the contribution of susceptibility alleles such as NOD2 to C. jejuni–induced intestinal inflammation. To examine the role of NOD2 in controlling the bacteria/host interaction, we generated Il10−/−; Nod2−/− mice. Interestingly, over a period of 12 months in our housing facility and husbandry conditions, none of the Il10−/− and Il10−/−; Nod2−/− mice showed evidence of spontaneous intestinal inflammation, as evaluated by histological analysis (Supplementary Figure 1). This suggests that absence of the Il10 gene or Nod2 gene on a C57BL/6 background does not disrupt the host's innate responses to the commensal microbiota. To determine the function of NOD2 following infection with the human relevant clinical isolate, Il10−/−, Nod2−/−, and Il10−/−; Nod2−/− mice were treated for 7 days with an antibiotic cocktail regimen. After a 24-hour washout period, mice received a single dose of C. jejuni (109 CFU/mouse) by gavage and were housed for an additional 21 days. The mice were euthanized, and intestinal samples were obtained for histological analysis. Interestingly, C. jejuni–infected Nod2−/− mice failed to develop intestinal inflammation (Figure 1A and 1B). This result suggests that NOD2 deficiency alone is not sufficient to promote C. jejuni–induced colitis. However, as previously reported [18], Il10−/− mice showed evidence of inflammation, compared with uninfected mice, as seen by immune cell infiltration, goblet cell depletion, and epithelial cell hyperplasia. Most importantly, C. jejuni–induced intestinal inflammation was exacerbated in Il10−/−; Nod2−/− mice, compared with Il10−/− mice, as measured by histological scoring (2.51 vs 1.18 [P = .03]; Figure 1A and 1B). Therefore, we focused our attention on the differential response of Il10−/− mice and Il10−/−; Nod2−/− mice to C. jejuni infection. At the molecular level, mRNA expression of the C. jejuni–induced proinflammatory mediators Il1β, Tnfα, and Cxcl1 increased by 47%, 66%, and 39%, respectively, in C. jejuni–infected Il10−/−; Nod2−/− mice, compared with levels in Il10−/− mice (Figure 1C).
Figure 1.
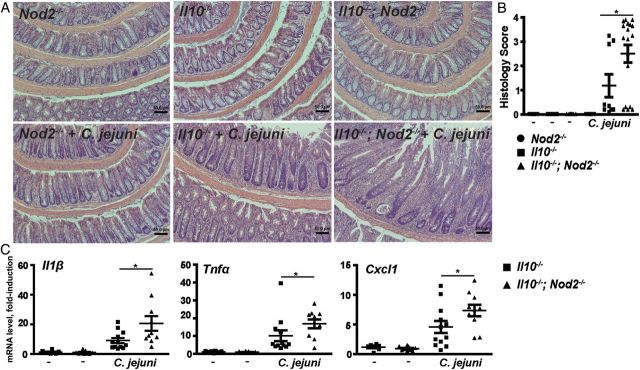
Nucleotide-binding oligomerization domain 2 (NOD2) attenuates Campylobacter jejuni–induced intestinal inflammation. Cohorts of 6–18 Nod2−/−, Il10−/−, and Il10−/−; Nod2−/− mice raised in specific-pathogen-free conditions received a single dose of 109 C. jejuni/mouse by gavage. After 21 days of infection, colons were resected for hematoxylin-eosin staining or for RNA extraction for gene expression analysis. A, Representative intestinal histological images of C. jejuni–induced inflammation in Nod2−/−, Il10−/−, and Il10−/−; Nod2−/− mice. B, Quantification of histological intestinal damage score mediated by C. jejuni infection. C, Il1β, Tnfα, and Cxcl1 messenger RNA accumulation was quantified using an ABI 7900HT Fast Real-Time PCR System and specific primers, and data were normalized to Gapdh. All graphs depict means ± standard errors of the mean. *P < .05. Scale bar is 200 μm. Results are representative of 3 independent experiments.
C. jejuni–induced crypt abscesses are an important clinical feature of the Il10−/− murine model [19]. Remarkably, crypt abscesses were elevated 9-fold in C. jejuni–infected Il10−/−; Nod2−/− mice, compared with Il10−/− mice (Figure 2A). In accordance with this finding, MPO staining revealed that C. jejuni–induced neutrophil infiltration into colonic tissues was strongly enhanced in Il10−/−; Nod2−/− mice, compared with Il10−/− mice (Figure 2B).
Figure 2.
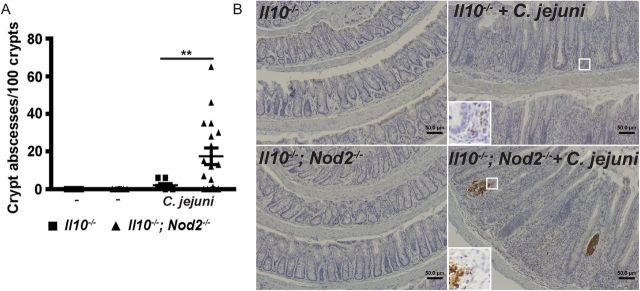
Nucleotide-binding oligomerization domain 2 (NOD2) deficiency exacerbates neutrophil infiltration–induced crypt abscesses in Campylobacter jejuni–infected mice. Cohorts of 9–18 Il10−/− and Il10−/−; Nod2−/− mice raised in specific-pathogen-free conditions were infected as indicated in Figure 1. A, Number of crypt abscesses in C. jejuni–infected mice. B, Representative images of immunohistochemical analysis of myeloperoxidase (MPO) expression (brown dots), showing neutrophil infiltration. All graphs depict means ± standard errors of the mean. **P < .01. Scale bar is 50 μm. Results are representative of 3 independent experiments.
NOD2 Alleviates C. jejuni Colonic Invasion
Since C. jejuni is an invasive intestinal pathogenic bacterium, we next investigated the impact of NOD2 signaling on C. jejuni invasion into intraintestinal and extraintestinal tissues. Following infection, C. jejuni DNA was visualized in the colon of Il10−/− and Il10−/−; Nod2−/− mice, using FISH and confocal microscopy. The presence of C. jejuni was enhanced in inflamed crypts and lamina propria of Il10−/−; Nod2−/− mice, compared with infected Il10−/− mice (Figure 3A). To quantify viable C. jejuni in intestinal and extraintestinal tissues, we aseptically collected samples from the colon and MLNs and enumerated the bacteria on Remel Campylobacter-selective plates. Consistent with the FISH results, counts of viable C. jejuni increased by 132% in colon samples and by 200% in MLNs, respectively, from of Il10−/−; Nod2−/− mice, compared with Il10−/− mice (Figure 3B).
Figure 3.
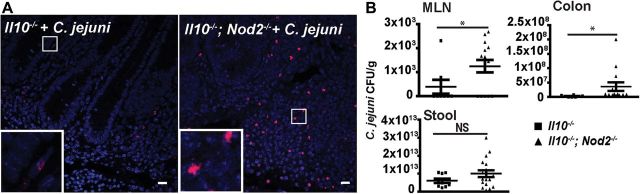
Nucleotide-binding oligomerization domain 2 (NOD2) prevents Campylobacter jejuni invasion into the colon and mesenteric lymph nodes. Cohorts of 9–18 Il10−/− and Il10−/−; Nod2−/− mice raised in specific-pathogen-free conditions were infected as indicated in Figure 1. A, C. jejuni (red dots) in colonic sections of infected mice was detected using fluorescence in situ hybridization. Scale bar represents 10 μm. B, C. jejuni bacterial count in the colon, mesenteric lymph nodes (MLNs), and stool of mice. Data represent means ± standard errors of the mean. *P < .05. Results are representative of 3 independent experiments. Abbreviations: CFU, colony-forming units; NS, not significant.
NOD2 Mediates Bactericidal Capacity and Antiinflammatory Responses
Since NOD2 deficiency elevated C. jejuni invasion into gastrointestinal tissues and is an indispensable cellular component of host recognition on S. pneumoniae [6] and S. enterica [11], we next investigated the role of this innate sensor on C. jejuni clearance. NOD2-derived macrophage and dendritic cell signaling is a critical event in bacterial clearance [11, 27]. We then investigated the contribution of NOD2 in C. jejuni clearance, using primary thioglycollate-elicited peritoneal macrophages. As shown in Figure 4A, C. jejuni invasion into macrophages isolated from Il10−/−; Nod2−/− mice was comparable to findings observed for macrophages from Il10−/− mice at 0 hours. After 2 hours of incubation, C. jejuni survival was 69% higher in Il10−/−; Nod2−/− macrophages, compared with Il10−/− cells (Figure 4A). Fluorescence microscopy showed the persistence of C. jejuni in Il10−/−; Nod2−/− macrophages, compared with infected Il10−/− macrophages (Figure 4B). These results suggest that NOD2 deficiency impaired bacterial killing capacity in macrophages.
Figure 4.

Nucleotide-binding oligomerization domain 2 (NOD2) enhances Campylobacter jejuni clearance in macrophages. Elicited peritoneal macrophages from Il10−/− and Il10−/−; Nod2−/− mice were infected with C. jejuni, and bacterial survival was determined using a gentamicin assay. A, C. jejuni invasion in macrophages at 0 and 2 hours was enumerated by plating. B, Representative images of C. jejuni (red dots) invasion into macrophages, using 5-Cyano-2,3-di-(p-tolyl) tetrazolium chloride-labelled C. jejuni. Scale bar is 10 μm. Data represent means ± standard errors of the mean. Results are representative of 3 independent experiments. *P < .05. Abbreviations: CFU, colony-forming units; NS, not significant.
NOD2-Mediated NO Expression Correlates With Bactericidal Capacity
Since NO production plays an essential role in macrophage-mediated bacterial killing [28, 29], we examine expression of inducible NO. Primary peritoneal macrophages were isolated from Il10−/− and Il10−/−; Nod2−/− mice and infected with C. jejuni. Interestingly, C. jejuni–induced iNos mRNA expression was reduced by 67% in Il10−/−; Nod2−/− macrophages, compared with Il10−/− cells, whereas Tnfα expression was not affected (Figure 5A). These findings indicate that NOD2 controls expression of specific proinflammatory genes, including the bactericidal mediator iNos.
Figure 5.

Nucleotide-binding oligomerization domain 2 (NOD2) promotes Campylobacter jejuni–induced iNos but attenuates Cxcl1 and Il1β messenger RNA (mRNA) expression in macrophages. Elicited peritoneal macrophages from Il10−/− and Il10−/−; Nod2−/− mice were infected with C. jejuni, and mRNA accumulation was determined using real-time polymerase chain reaction. Data represent means ± standard errors of the mean. *P < .05. Results are representative of 3 independent experiments.
Since NOD2 deficiency impaired C. jejuni–induced iNos mRNA expression, we next investigated the level of NO production, using the Griess assay. Notably, C. jejuni infection strongly induced nitrite production in Il10−/− macrophages, an effect attenuated by 35% in Il10−/−; Nod2−/− cells (Figure 6A). To evaluate the impact of NO on C. jejuni killing, we infected peritoneal macrophages with C. jejuni in the presence of sodium nitroprusside and assessed bacterial survival. C. jejuni survival was 33% higher in macrophages isolated from Il10−/−; Nod2−/− mice, compared with Il10−/− cells, at 2 hours (Figure 6B). Interestingly, supplementation with sodium nitroprusside significantly reduced C. jejuni survival in macrophages isolated from Il10−/−; Nod2−/− mice.
Figure 6.
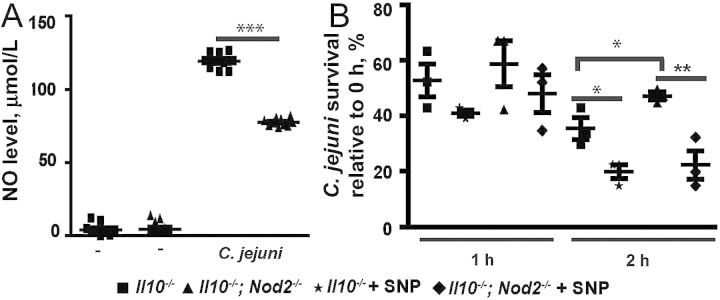
Nucleotide-binding oligomerization domain 2 (NOD2) enhances nitric oxide (NO) production and promotes Campylobacter jejuni clearance in macrophages. Elicited peritoneal macrophages from Il10−/− and Il10−/−; Nod2−/− mice were infected with C. jejuni. A, Production of NO as measured by the Griess assay. B, C. jejuni survival in the presence of NO donor sodium nitroprusside as determined by culture assay. Data represent means ± standard errors of the mean. ***P < .001, **P < .01, *P < .05. Results are representative of 3 independent experiments.
NO-Enhanced Bactericidal Capacity Attenuates C. jejuni–Induced Colitis in Il10−/−; Nod2−/− Mice
To further evaluate the role of NO in C. jejuni–induced intestinal inflammation, Il10−/− and Il10−/−; Nod2−/− mice were infected as described above and injected intraperitoneally with PBS or sodium nitroprusside (2 mg/kg in PBS, daily) for 21 days. As shown above, C. jejuni induced stronger intestinal inflammation in Il10−/−; Nod2−/− mice, as demonstrated by increased immune cell infiltration, goblet cell depletion, and crypt hyperplasia/abscesses, compared with Il10−/− mice (2.89 vs 1.91 [P = .047]; Figure 7A and 7B). Notably, sodium nitroprusside treatment attenuated C. jejuni–induced intestinal inflammation (by approximately 60%) in Il10−/−; Nod2−/− mice, compared with untreated mice (1.15 vs 2.89; P = .026). Interestingly, sodium nitroprusside treatment did not significantly attenuate C. jejuni–induced colitis in Il10−/− mice.
Figure 7.

Supplementation with nitric oxide (NO) attenuates Campylobacter jejuni–induced intestinal inflammation. Cohorts of 5–8 Il10−/− and Il10−/−; Nod2−/− mice raised in specific-pathogen-free conditions received a single dose of 109 C. jejuni/mouse by gavage and daily intraperitoneal injection of phosphate-buffered saline or sodium nitroprusside (SNP). After 21 days, colons were resected for hematoxylin-eosin staining or fluorescence in situ hybridization (FISH). A, Representative intestinal histological images of C. jejuni–induced inflammation in Il10−/− and Il10−/−; Nod2−/− mice in the presence of SNP. B, Quantification of histological intestinal damage score mediated by C. jejuni infection. C, C. jejuni (red dots) in colonic sections of infected mice was detected using FISH. Scale bars are 50 μm (A) and 20 μm (C). Data represent means ± standard errors of the mean. Results are representative of 3 independent experiments. ***P < .001, **P < .01, *P < .05.
To assess the contribution of NO in bacterial clearance in intestinal tissue, we evaluated C. jejuni DNA in colonic tissues, using FISH. Sodium nitroprusside strongly diminished C. jejuni translocation into colonic tissues of both Il10−/−; Nod2−/− and Il10−/− mice (Figure 7C). Overall, these findings suggest that NOD2 deficiency impairs C. jejuni clearance and exacerbates colitis, an effect rescued by restoring bactericidal capacity through NO supplementation.
DISCUSSION
The importance of the innate sensor NOD2 in regulating innate response and intestinal homeostasis has long been recognized, which is evident by increased susceptibility to IBD (eg, Crohn disease) in individuals carrying NOD2 polymorphisms (eg, R702W, G908R, and 1007fs) [30]. The monocyte/macrophage lineage in patients with IBD displayed an increased inflammatory response following stimulation with commensal bacteria [31] and inferior Escherichia coli clearance in the gastrointestinal tract [32]. The impact of NOD2 on bacterial host responses could involve the regulation of proinflammatory molecule expression, control of Toll-like receptor signaling intensity, regulation of antimicrobial peptide production, and control of microbial composition [8, 33–37]. Whether host-derived NOD2 signaling utilizes some or all of these mechanisms to respond to enteric pathogen, especially C. jejuni, is unknown. Our study showed that NOD2 deficiency exacerbates C. jejuni–induced colitis in Il10−/− mice, as demonstrated by increased hyperplasia, immune cell infiltration and crypt abscesses. Interestingly, C. jejuni-infected Nod2−/− mice failed to develop colitis. Therefore, this defective innate response to C. jejuni infection is insufficient to promote colitis, and a more global immune alteration, such as the one afforded by absence of IL-10 signaling in Il10−/− mice is necessary to reveal NOD2 contribution. This two hit model is compatible with the limited risk factor confer by NOD2 polymorphisms in humans, which highlights the polygenic nature of IBD [38, 39].
The increased colitis among Il10−/−; Nod2−/− mice was coupled with elevated expression of the inflammatory mediators Il-1β and Cxcl1, as well as with increased C. jejuni invasion into the colon and MLN, compared with Il10−/− mice. Interestingly, the inability to eliminate C. jejuni from Il10−/−; Nod2−/− macrophages was associated with reduced C. jejuni–induced bactericidal NO production. Supplementation with the NO-donor sodium nitroprusside promoted C. jejuni eradication and attenuated C. jejuni–induced colitis in Il10−/−; Nod2−/− mice. Therefore, we have found a unique role for NOD2 in protecting Il10−/− mice from enteric bacterial pathogen C. jejuni–induced colitis, likely through NOD2-induced bactericidal responses.
Interestingly, Jamontt et al recently showed spontaneous colitis is attenuated in Il10−/−; Nod2−/− mice, compared with Il10−/− mice, at 9 weeks of age [40], suggesting a deleterious role for NOD2 signaling in the intestine. In contrast, both Il10−/− and Il10−/−; Nod2−/− mice raised in our SPF facility remained healthy for >56 weeks, suggesting a different environmental trigger (eg, microbial composition) between our animal facility and that of Jamontt et al. Further investigation would be necessary to resolve this phenotypic discrepancy.
Using FISH and culture assays, we found that NOD2 deficiency impairs C. jejuni clearance in the colon and in peritoneal macrophages in vitro, leading to increased bacterial invasion/survival. Mammalian cells (eg, epithelial cells and macrophages) possess a variety of tools to eliminate invading bacterial pathogens, including bactericidal and autophagy responses [6, 7, 11]. We previously found that rapamycin-induced autophagy enhances C. jejuni clearance in Il10−/− splenocytes [19]. Interestingly, generation of the C. jejuni–induced autophagy protein LC3 II is reduced in Il10−/−; Nod2−/− splenocytes, compared with Il10−/− cells (data not shown). The diminished bacterial clearance observed in C. jejuni–infected Il10−/−; Nod2−/− mice may involve an impaired autophagic response. Further investigation would be required to demonstrate the role of NOD2/autophagy in C. jejuni clearance.
In addition to autophagy, host-derived RNS such as the bactericidal agent NO actively participate in the elimination of intercellular and intracellular bacteria and suppresses the growth of E. coli and Salmonella in vitro [22, 41]. C. jejuni survival in vitro is reduced in the presence of chemically generated NO [24]. Here, we found that C. jejuni–induced iNos expression and NO generation is attenuated in Il10−/−; Nod2−/− macrophages, compared with Il10−/− cells. Impaired NO generation by Il10−/−; Nod2−/− macrophages is associated with increased C. jejuni survival in these cells, whereas the NO-donor sodium nitroprusside attenuated bacterial survival. Furthermore, sodium nitroprusside attenuated C. jejuni invasion and intestinal inflammation in Il10−/−; Nod2−/− mice. Interestingly, sodium nitroprusside did not significantly diminish intestinal inflammation in Il10−/− mice, which correlated with lower levels of bacterial invasion, compared with Il10−/−; Nod2−/− mice. These findings indicate that iNOS expression and NO production are dependent on functional NOD2 signaling and likely represent an important mechanism for controlling campylobacteriosis.
NOD2 deficiency is associated with defective intestinal antimicrobial peptide expression and microbial dysbiosis [8, 42, 43]. Interestingly, microbial composition is an important environmental factor influencing the susceptibility to C. jejuni infection in experimental models [44]. However, recent findings by Shanahan et al showed that Nod2−/− mice and cohoused wild-type littermates showed comparable antimicrobial peptide expression [45]. Moreover, microbial composition was not influenced by NOD2 status [45, 46]. These findings suggest that increased C. jejuni translocation and intestinal inflammation in Il10−/−; Nod2−/− mice is mostly driven by defective bactericidal activity rather than by impaired antimicrobial peptide production and microbial dysbiosis. Further experiments would be necessary to fully assess the contribution of microbial composition to C. jejuni infection.
Taken together, our findings highlight an essential role for NOD2 in defending the host against enteropathogenic C. jejuni. The protective function of NOD2 seems to depend on enhancing bactericidal capacity through NO production. Targeting NOD2-mediated NO production may represent an alternative therapeutic approach to treating campylobacteriosis.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org/). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank Brigitte Allard, for technical assistance throughout this project; Mr Jens Durruthy-Durruthy, for conducting preliminary experiments on bacterial invasion and host proinflammatory response; members of our laboratory, for reviewing this manuscript; Ms Carolyn Batchelor Suitt, for her assistance with processing histology samples; and Dr Robert Bagnell and Mr Steven Ray (Microscopy Services Laboratory, University of North Carolina–Chapel Hill) and Mr Robert Currin (manager, Cell and Molecular Imaging Facility and UNC–Olympus Center, University of North Carolina–Chapel Hill), for their assistance with fluorescent microscopy.
C. J. and X. S. conceived and designed the experiments. X. S. performed the experiments. X. S. and C. J. analyzed the data. X. S. and C. J. wrote the manuscript.
Disclaimer. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Financial support. This work was supported by the National Institutes of Health (grants DK047700, DK073338, and AI082319, to C. J.; and grant P30 DK34987, to the CGIBD).
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Abraham C, Medzhitov R. Interactions between the host innate immune system and microbes in inflammatory bowel disease. Gastroenterology. 2011;140:1729–37. doi: 10.1053/j.gastro.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cook DN, Pisetsky DS, Schwartz DA. Toll-like receptors in the pathogenesis of human disease. Nat Immunol. 2004;5:975–9. doi: 10.1038/ni1116. [DOI] [PubMed] [Google Scholar]
- 3.Ulevitch RJ. Therapeutics targeting the innate immune system. Nat Rev Immunol. 2004;4:512–20. doi: 10.1038/nri1396. [DOI] [PubMed] [Google Scholar]
- 4.Franchi L, McDonald C, Kanneganti TD, Amer A, Nunez G. Nucleotide-binding oligomerization domain-like receptors: intracellular pattern recognition molecules for pathogen detection and host defense. J Immunol. 2006;177:3507–13. doi: 10.4049/jimmunol.177.6.3507. [DOI] [PubMed] [Google Scholar]
- 5.Strober W, Murray PJ, Kitani A, Watanabe T. Signalling pathways and molecular interactions of NOD1 and NOD2. Nat Rev Immunol. 2006;6:9–20. doi: 10.1038/nri1747. [DOI] [PubMed] [Google Scholar]
- 6.Opitz B, Puschel A, Schmeck B, et al. Nucleotide-binding oligomerization domain proteins are innate immune receptors for internalized Streptococcus pneumoniae. J Biol Chem. 2004;279:36426–32. doi: 10.1074/jbc.M403861200. [DOI] [PubMed] [Google Scholar]
- 7.Hisamatsu T, Suzuki M, Reinecker HC, Nadeau WJ, McCormick BA, Podolsky DK. CARD15/NOD2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology. 2003;124:993–1000. doi: 10.1053/gast.2003.50153. [DOI] [PubMed] [Google Scholar]
- 8.Kobayashi KS, Chamaillard M, Ogura Y, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–4. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- 9.Yang Y, Yin C, Pandey A, Abbott D, Sassetti C, Kelliher MA. NOD2 pathway activation by MDP or Mycobacterium tuberculosis infection involves the stable polyubiquitination of Rip2. J Biol Chem. 2007;282:36223–9. doi: 10.1074/jbc.M703079200. [DOI] [PubMed] [Google Scholar]
- 10.Meinzer U, Esmiol-Welterlin S, Barreau F, et al. Nod2 mediates susceptibility to Yersinia pseudotuberculosis in mice. PLoS One. 2008;3:e2769. doi: 10.1371/journal.pone.0002769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cooney R, Baker J, Brain O, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16:90–7. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 12.Travassos LH, Carneiro LA, Ramjeet M, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 13.Gradel KO, Nielsen HL, Schønheyder HC, Ejlertsen T, Kristensen B, Nielsen H. Increased short- and long-term risk of inflammatory bowel disease after Salmonella or Campylobacter gastroenteritis. Gastroenterology. 2009;137:495–501. doi: 10.1053/j.gastro.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 14.Centers for Disease Control and Prevention. Campylobacter: general information. 2010 http://www.cdc.gov/nczved/divisions/dfbmd/diseases/campylobacter/ . Accessed 19 October 2010. [Google Scholar]
- 15.Lalani I, Bhol K, Ahmed AR. Interleukin-10: biology, role in inflammation and autoimmunity. Ann Allergy Asthma Immunol. 1997;79:469–83. doi: 10.1016/S1081-1206(10)63052-9. [DOI] [PubMed] [Google Scholar]
- 16.Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol. 2011;29:71–109. doi: 10.1146/annurev-immunol-031210-101312. [DOI] [PubMed] [Google Scholar]
- 17.Mansfield LS, Bell JA, Wilson DL, et al. C57BL/6 and congenic interleukin-10-deficient mice can serve as models of Campylobacter jejuni colonization and enteritis. Infect Immun. 2007;75:1099–115. doi: 10.1128/IAI.00833-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lippert E, Karrasch T, Sun X, et al. Gnotobiotic IL-10; NF-kappaB mice develop rapid and severe colitis following Campylobacter jejuni infection. PLoS One. 2009;4:e7413. doi: 10.1371/journal.pone.0007413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun X, Threadgill D, Jobin C. Campylobacter jejuni induces colitis through activation of mammalian target of rapamycin signaling. Gastroenterology. 2012;142:86–95 e5. doi: 10.1053/j.gastro.2011.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun X, Liu B, Sartor RB, Jobin C. Phosphatidylinositol 3-kinase-gamma signaling promotes Campylobacter jejuni-induced colitis through neutrophil recruitment in mice. J Immunol. 2013;190:357–65. doi: 10.4049/jimmunol.1201825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haag LM, Fischer A, Otto B, et al. Campylobacter jejuni induces acute enterocolitis in gnotobiotic IL-10-/- mice via Toll-like-receptor-2 and -4 signaling. PLoS One. 2012;7:e40761. doi: 10.1371/journal.pone.0040761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Franchini A, Conte A, Ottaviani E. Nitric oxide: an ancestral immunocyte effector molecule. Adv Neuroimmunol. 1995;5:463–78. doi: 10.1016/0960-5428(95)00029-1. [DOI] [PubMed] [Google Scholar]
- 23.MacFarlane AS, Schwacha MG, Eisenstein TK. In vivo blockage of nitric oxide with aminoguanidine inhibits immunosuppression induced by an attenuated strain of Salmonella typhimurium, potentiates Salmonella infection, and inhibits macrophage and polymorphonuclear leukocyte influx into the spleen. Infect Immun. 1999;67:891–8. doi: 10.1128/iai.67.2.891-898.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iovine NM, Pursnani S, Voldman A, Wasserman G, Blaser MJ, Weinrauch Y. Reactive nitrogen species contribute to innate host defense against Campylobacter jejuni. Infect Immun. 2008;76:986–93. doi: 10.1128/IAI.01063-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Korlath JA, Osterholm MT, Judy LA, Forfang JC, Robinson RA. A point-source outbreak of campylobacteriosis associated with consumption of raw milk. J Infect Dis. 1985;152:592–6. doi: 10.1093/infdis/152.3.592. [DOI] [PubMed] [Google Scholar]
- 26.Kankaanpää P, Pahajoki K, Marjomäki V, Heino J, White D. BioImageXD-new open source free software for the processing, analysis and visualization of multidimensional microscopic images. Micros Today. 2006;14:12–16. [Google Scholar]
- 27.Juarez E, Carranza C, Hernandez-Sanchez F, et al. NOD2 enhances the innate response of alveolar macrophages to Mycobacterium tuberculosis in humans. Eur J Immunol. 2012;42:880–9. doi: 10.1002/eji.201142105. [DOI] [PubMed] [Google Scholar]
- 28.Gomes RN, Teixeira-Cunha MG, Figueiredo RT, et al. Bacterial clearance in septic mice is modulated by MCP-1/CCL2 and nitric oxide. Shock. 2013;39:63–9. doi: 10.1097/SHK.0b013e31827802b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Y, Sjolinder M, Wang X, et al. Thyroid hormone enhances nitric oxide-mediated bacterial clearance and promotes survival after meningococcal infection. PLoS One. 2012;7:e41445. doi: 10.1371/journal.pone.0041445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lesage S, Zouali H, Cezard JP, et al. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet. 2002;70:845–57. doi: 10.1086/339432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kamada N, Hisamatsu T, Okamoto S, et al. Unique CD14 intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-gamma axis. J Clin Invest. 2008;118:2269–80. doi: 10.1172/JCI34610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Subramanian S, Roberts CL, Hart CA, et al. Replication of colonic Crohn's disease mucosal Escherichia coli isolates within macrophages and their susceptibility to antibiotics. Antimicrob Agents Chemother. 2008;52:427–34. doi: 10.1128/AAC.00375-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watanabe T, Asano N, Murray PJ, et al. Muramyl dipeptide activation of nucleotide-binding oligomerization domain 2 protects mice from experimental colitis. J Clin Invest. 2008;118:545–59. doi: 10.1172/JCI33145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang Z, Fuss IJ, Watanabe T, et al. NOD2 transgenic mice exhibit enhanced MDP-mediated down-regulation of TLR2 responses and resistance to colitis induction. Gastroenterology. 2007;133:1510–21. doi: 10.1053/j.gastro.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watanabe T, Kitani A, Murray PJ, Strober W. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat Immunol. 2004;5:800–8. doi: 10.1038/ni1092. [DOI] [PubMed] [Google Scholar]
- 36.Netea MG, Ferwerda G, de Jong DJ, et al. Nucleotide-binding oligomerization domain-2 modulates specific TLR pathways for the induction of cytokine release. J Immunol. 2005;174:6518–23. doi: 10.4049/jimmunol.174.10.6518. [DOI] [PubMed] [Google Scholar]
- 37.Hedl M, Li J, Cho JH, Abraham C. Chronic stimulation of Nod2 mediates tolerance to bacterial products. Proc Natl Acad Sci U S A. 2007;104:19440–5. doi: 10.1073/pnas.0706097104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kanaan ZM, Eichenberger MR, Ahmad S, et al. Clinical predictors of inflammatory bowel disease in a genetically well-defined Caucasian population. J Negat Results Biomed. 2012;11:7. doi: 10.1186/1477-5751-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474:307–17. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jamontt J, Petit S, Clark N, Parkinson SJ, Smith P. Nucleotide-binding oligomerization domain 2 signaling promotes hyperresponsive macrophages and colitis in IL-10-deficient mice. J Immunol. 2013;190:2948–58. doi: 10.4049/jimmunol.1201332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–5. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 42.Huttner KM, Bevins CL. Antimicrobial peptides as mediators of epithelial host defense. Pediatr Res. 1999;45:785–94. doi: 10.1203/00006450-199906000-00001. [DOI] [PubMed] [Google Scholar]
- 43.Couturier-Maillard A, Secher T, Rehman A, et al. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest. 2013;123:700–11. doi: 10.1172/JCI62236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bereswill S, Fischer A, Plickert R, et al. Novel murine infection models provide deep insights into the “menage a trois” of Campylobacter jejuni, microbiota and host innate immunity. PLoS One. 2011;6:e20953. doi: 10.1371/journal.pone.0020953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shanahan MT, Carroll IM, Grossniklaus E, et al. Mouse Paneth cell antimicrobial function is independent of Nod2. Gut. 2013 doi: 10.1136/gutjnl-2012-304190. doi:10.1136/gutjnl-2012-304190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robertson SJ, Zhou JY, Geddes K, et al. Nod1 and Nod2 signaling does not alter the composition of intestinal bacterial communities at homeostasis. Gut Microbes. 2013;4:222–31. doi: 10.4161/gmic.24373. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.