

Abstract
Multiple sclerosis (MS) is a neurodegenerative disease with a major inflammatory component that constitutes the most common progressive and disabling neurological condition in young adults. Injectable immunomodulatory medicines such as interferon drugs and glatiramer acetate have dominated the MS market for over the past two decades but this situation is set to change. This is because of: (i) patent expirations, (ii) the introduction of natalizumab, which targets the interaction between leukocytes and the blood–CNS barrier, (iii) the launch of three oral immunomodulatory drugs (fingolimod, dimethyl fumarate and teriflunomide), with another (laquinimod) under regulatory review and (iv) a number of immunomodulatory monoclonal antibodies (alemtuzumab, daclizumab and ocrelizumab) about to enter the market. Current and emerging medicines are reviewed and their impact on people with MS considered.
Keywords: clinical trials, disease-modifying drugs, drug development, immunomodulatory drugs, monoclonal antibodies, neuroinflammation
Introduction
Multiple sclerosis (MS) is an acquired autoimmune disease that affects the central nervous system and optic nerves. It leads to neuroinflammation, neuronal dysfunction (particularly loss of myelin, the lipophilic insulating sheath around axons) and neuronal loss. These changes manifest as a variety of symptoms that include deterioration in motor function, sense perception, mental function, visual function, pain and fatigue [1–3]. MS presents in different forms that follow distinct patterns of evolution and rates of disability progression [4]. The most common form is relapsing–remitting MS (RRMS), which affects about 85% of people with MS. It is more common in females than males, by a ratio of 2:1, and has an average age at diagnosis of 29 years [5]. RRMS is characterized by acute attacks (relapses) followed by partial or full recovery (remission). It contrasts with primary progressive MS (PPMS), which affects about 10–15% of people with MS, has no gender bias, is diagnosed (on average) at age 40 years, and is characterized by a steady and irreversible progression of functional impairments [6–8]. PPMS begins insidiously whereas the harbinger of RRMS is clinically isolated syndrome (CIS), a transient impairment in motor or sensory function accompanied by white matter abnormalities shown by magnetic resonance imaging (MRI) [9,10]. Most cases of CIS (80% after 20 years) convert to RRMS [11] and, after two or three decades, RRMS converts to secondary progressive MS (SPMS), which has a remission-free progression [12–14]. A final subset is progressive relapsing MS (PRMS), which affects less than 5% of people with MS and is characterized by a steady decline in neurologic function and clear superimposed exacerbations [4]. This sub-categorization of MS is made on the basis of clinical and MRI data and probably reflects different expressions of a single disease. Indeed, a large longitudinal study of people with MS has compared disability progression in those with relapsing and progressive disease. Both groups reached Kurtzke disability status scales scores of 3 and 6 at the same age, but the time taken to reach each of these points was lower in the progressive onset phenotype compared with the relapsing onset phenotype (Figure 1).
Figure 1.
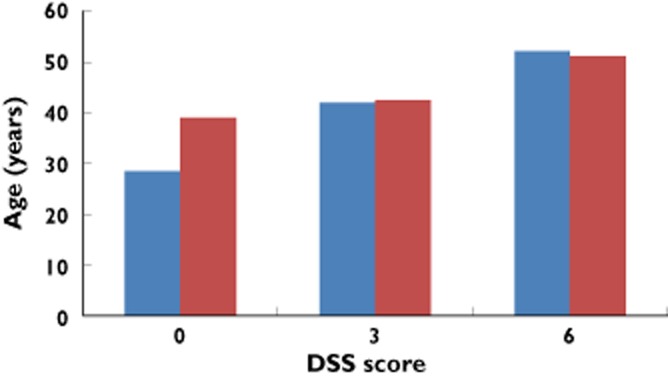
Kaplan–Meier estimated median age at multiple sclerosis clinical onset and at Disability Status Score (DSS) score of both 3 (n = 2054) and 6 (n = 1415) in people with MS with relapsing (blue columns) and progressive (red columns) onset. Data derived from Figure 2 of Leray et al. [89]
MS is a complex disease that is caused by a combination of genetic, environmental and lifestyle factors, knowledge of which is still emerging. What is known is that there is a complex interaction between dozens of genetic variations, particularly in the major histocompatibility complex (MHC) [15–17], and environmental factors, such as Epstein-Barr virus seropositivity, cigarette smoking and low plasma concentrations of vitamin D3 [18–22]. The disease is characterized by multiple plaques throughout the CNS that range in size from a few mm to more than 1 cm and reflect regions of neuroinflammation, demyelination and neuronal loss [23–25]. They are formed by the paracellular movement of activated immune cells from the bloodstream into the CNS principally across the blood–brain barrier (BBB), but also the blood–spinal cord barrier (BSCB) and blood–CSF barriers. These three permeability barriers are collectively referred to as the blood–CNS barrier (BCNSB) [26–28]. Plaques can be assessed in the intact brain and upper spinal cord by magnetic resonance imaging (MRI). This technique is used in (i) patient selection and stratification, (ii) as the primary endpoint in proof-of-concept clinical trials and (iii) as the secondary endpoint in definitive phase III trials [29]. MRI images (either T1-or T2-weighted) allow areas of demyelination to be visualized and measured and sensitivity can be improved by the use of contrast agents such as gadolinium diethylenetriaminepentaacetic acid. It also allows breaches in the BBB and BSCB to be assessed and the emergence of new plaques to be monitored [28,30].
The cascade of inflammatory change is probably mediated by a coordinated attack by T cells, monocytes and B cells against CNS tissue. Although the site and the antigen that triggers the inflammatory sequelae has not been clearly established, the most prominent hypothesis is that antigens (probably proteins associated with myelin or microbial antigens cross-reactive with myelin-associated proteins) are presented to naïve T cells by MHC molecules on specialized antigen-presenting cells (Figure 2) [31–33]. Once exposed to such antigens, these T cells differentiate into T helper cells 1, 2 and 17 expressing the glycoprotein called cluster of differentiation 4 (CD4+) and cytotoxic T cells expressing the CD8+ glycoprotein. These autoreactive T cells then enter the CNS through a sequential and coordinated process involving the binding of adhesion molecules with their respective ligands that tether, roll and anchor the activated immune cells to the inside surface of blood vessels [34]. Anchoring is mediated by the interaction between α4β1-integrin on the leukocytes and vascular cell adhesion molecule-1 on the endothelial cell layer. The expansion of lymphocytes in the CNS is amplified by pro-inflammatory cytokines through the recruitment of naive microglia [35,36]. The resultant perivascular plaques throughout CNS white matter cause transient breakdown of the BCNSB, partly through the action of interleukins 17 and 22 [28]. BCNSB disruption permits the movement of more leukocytes into the CNS where they contribute to the loss of both myelin and the glial cells that form myelin in the CNS (oligodendrocytes) and culminate in neuronal loss by a mechanism that is not yet fully understood [3,34,37–40] [41,42].
Figure 2.
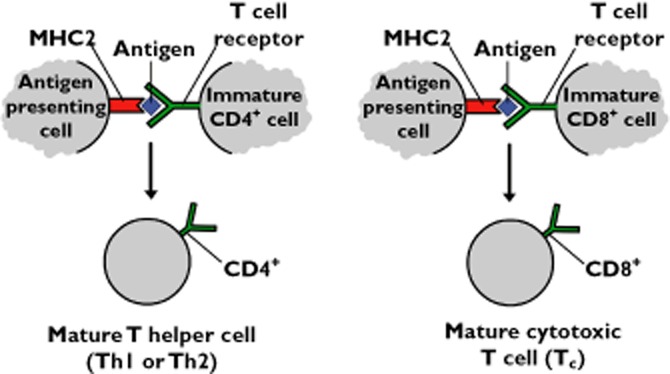
Antigen presentation stimulates naïve T cells to become either cytotoxic CD8+ cells or helper CD4+ cells. CD4+ and CD8+ are receptors that assist the binding of T cell receptors with an antigen presenting cell. The inflammatory sequelae associated with MS may be triggered by an antigen with some epitope homology with one or more myelin-associated protein
Pharmacotherapy for MS has focussed, almost exclusively, on RRMS. The first generation of MS medicines were β-interferon (IFNβ) drugs and glatiramer acetate [2,3]. The clinical impact of these biologic immunomodulatory drugs will be considered, along with a new generation of immunomodulatory MS medicines.
First generation immune-targeted MS drugs (Table 1)
Table 1.
Drugs approved for the treatment of multiple sclerosis [3]
| Generic name (brand name) | Mechanism of action | Route of administration (dose) | Location of molecular target | Therapeutic efficacy |
|---|---|---|---|---|
| IFNβ-1a (Avonex and Rebif)) | Suppression of Th1 and enhancement of Th2 immune response | Avonex: Once a week, i.m. (30 μg). | Circulating compartment* | Reduced relapse rate (32%) and MRI lesions (circa 80%) |
| Rebif: Three times a week; subcutanous (44 μg) | ||||
| IFNβ-1b (Betaseron and Extavia) | Suppression of Th1 and enhancement of Th2 immune response | Betaseron: Every other day, subcutanous (250 μg). | Circulating compartment | Reduced relapse rate (32%) and MRI lesions (circa 80%) |
| Extavia: Three times a week, subcutanous (250 μg) | ||||
| Glatiramer acetate (Copaxone) | Tolerization with myelin-like antigens and modulation of autoreactive T cells by inducing a shift from Th1 toTh2 cells. | Every day, subcutanous (20 mg) | Circulating compartment | Reduced relapse rate (29%) and MRI lesions (35%) |
| Mitoxantrone (Novantrone) | Inhibition of the proliferation of T cells, B cells and macrophages | Four times a year; intravenous. The lifetime cumulative dose is limited to 8–12 doses over 2–3 years (140 mg). | Circulating compartment | Reduced relapse (67%), and MRI lesions (85%) and disease progression |
| Natalizumab (Tysabri) | A humanized monoclonal antibody to α4β1 integrin that prevents the movement of leukocytes from the bloodstream into the CNS | Every 4 weeks by intravenous infusion (300 mg) | Circulating compartment | Reduced relapse (66%), and MRI lesions (90%) and disease progression |
| Fingolimod (Gilenya/Gilenia) | Reduction in the number of lymphocytes in the blood by preventing their egress from lymph nodes through modulation of the sphingosine-1-phosphate receptor 1. | Every day; oral (0.5 mg) | Circulating compartment | Reduced relapse rate (54%) and MRI lesions (67%) |
| Teriflunomide (Aubagio) | An immunomodulator with anti-inflammatory properties, probably through inhibition of dihydro-orotate dehydrogenase. | Every day; oral (7 or 14 mg) | Circulating compartment | Reduced relapse rate (31%) and MRI lesions |
| Dimethyl fumarate (Tecfidera) | TCA intermediate with immune modulation. Activator of the Nrf2 pathway (the innate cellular phase 2 detoxifying pathway). Nrf2 is: (i) a transcription factor that binds to anti-oxidant response elements and elicits changes in anti-oxidant gene transcription pathway and (ii) Nrf2 has major role in cellular neuroprotective and anti-inflammatory effects | Twice a day, oral (240 mg) | Circulating compartment | Reduced relapse, rate (51%), and new MRI lesions (69%) and disease progression |
Blood plasma and lymph fluid. BCNSB, blood–central nervous system barrier; CNS, central nervous system; PNS, peripheral nervous system; Th, T helper cell.
IFNβs drugs and glatiramer acetate
IFNβ drugs have been available as first line therapy for RRMS for nearly two decades. The first IFNβ drug to gain Food and Drug Administration (FDA) approval was Betaseron (1993), followed by Avonex (1996), Rebif (2002) and Extavia (2009). These recombinant proteins are produced by expression in either Chinese hamster ovary cells (IFNβ-1a; Avonex and Rebif) or in Escherichia coli (IFNβ-1b; Betaseron and Extavia). They are all administered by injection, reduce relapse rates by about a third and make relapses milder [43]. Glatiramer acetate, a random polymer of four amino acids, gained FDA approval in 1996. Injection of all these biologic drugs is often associated with side effects, principally injection site reactions, moderate to severe flu-like symptoms and the potential for liver damage. Such side effects can be burdensome and lead to poor adherence [44,45]. In addition, they all produce neutralizing antibodies in a substantial minority of those treated [46] and there is a high proportion of non-responders [47]. Neutralizing antibodies are less of an issue with glatiramer acetate than the IFNβ drugs and so this drug provides a useful alternative for people with MS expressing neutralizing antibodies to IFNβ drugs [48]. On the basis of both systematic analysis and long term studies, it appears that although IFNβ drugs and glatiramer acetate reduce relapse rates in RRMS, they have little impact on disease progression [49] [50]. These drugs also have no impact on the course of either SPMS or PPMS [50–52], but they do delay the conversion from CIS to RRMS [53–55].
Second generation immune-targeted MS drugs (Table 1)
Mitoxantrone
Mitoxantrone is an immunosuppressant drug (unlike IFNβ drug and glatiramer acetate) as it inhibits the proliferation of T cells, B cells and macrophages. It gained FDA approval for use in the reduction of neurological disability progression and/or frequency of clinical relapses in SPMS, PRMS or worsening RRMS [56]. Mitoxantrone treatment over a 6 month period was found to reduce both the risk of disability and relapse rate compared with IFNβ-1b in people with aggressive RRMS in a 3 year randomized clinical trial [57]. This, together with data showing that mitoxantrone (administered every 3 months for 24 months) slowed the progression of disability in people with worsening RRMS and SPMS [58], supports the use of mitoxantrone in the treatment of MS, particularly in rapidly worsening phenotypes. The dose of mitoxantrone that is used is strictly limited to 140 mg m−2 because of the long term risk of infertility, leukaemia and cardiotoxicity [56,59].
Natalizumab
Natalizumab, a humanized monoclonal antibody (mAb) to α4β1-integrin, blocks the interaction between α4β1-integrin on leukocytes and cell vascular cell adhesion molecule-1 on endothelial cells. Thus, it inhibits the transmigration of immune cells into the CNS. A systematic review and meta-analysis of all double-blind, randomized, controlled trials of studies of natalizumab in people with RRMS, that included one placebo-controlled trial and two add-on placebo-controlled trials (one plus glatiramer acetate and the other plus IFNβ-1a), indicated that the drug displayed robust evidence in favour of a reduction in both relapses and disability at 2 years, along with diminished MRI disease [60]. The reduction in annualized relapse rate resulting from treatment with natalizumab is about twice that seen with IFNβ drugs and glatiramer acetate (Table 2), which is supported by a head-to-head study with IFNβ-1a (s.c. 44 μg; Rebif) [61].
Table 2.
Drug candidates in late-stage development for the treatment of multiple sclerosis [3]
| Generic name (brand name) | Mechanism of action | Route of administration (dose) | Therapeutic efficacy |
|---|---|---|---|
| Alemtuzumab (Lemdra) | mAb for CD52 antigen expressed on T and B cell lymphocytes, neutrophils and natural killer cells that reduces the immune response. | Treatment is by intravenous infusion administered over 3 to 5 consecutive days once a year. | Reduced relapse rates by 55% compared with IFNβ-1a (Rebif), but there was no difference in accumulated disability between the two groups. |
| Daclizumab | mAb for CD25 that increases a subset of dendritic cells which have a regulatory activity and limits T cell expansion by blocking IL-2 signalling | 1 or 2 mg kg−1 by intravenous infusion every 4 weeks. | Reduced relapse rate and MRI lesions |
| 150 mg or 300 mg administered subcutaneously every 4 weeks. | |||
| Ocrelizumab | mAb for CD20 | Every day, subcutanous (20 mg). | Reduced relapse rate and MRI lesions |
| Laquinimod | Shifts immune response from Th1 to Th2, | Once a day (0.3 mg) | Reduced relapse, and MRI lesions and disease progression |
| Firategrast | α4β1-integrin antagonist | Every day; oral (0.5 mg) | Reduced relapse rate and MRI lesions |
| NU100 | recombinant human IFNβ-1b | Every day; oral (7 or 14 mg) | Reduced relapse rate and MRI lesions |
| BII-B017 | PEGylated IFNβ-1a | Twice a day (10 mg) | Reduced relapse rate and MRI lesions |
All compounds act on targets in the circulating compartment (blood plasma and lymph fluid). mAb, monoclonal antibody. Th, T helper cell.
Natalizumab renders the CNS immune-compromised by blocking immune-surveillance. This permits reactivation of dormant viruses, such as JC-1, which causes progressive multifocal leukoencephalopathy (PML), a rare and potentially fatal disorder characterized by progressive damage to white matter. Reports of PML in people with MS administered natalizumab led to its withdrawal from the market in 2005. It was re-introduced after no additional cases were identified in previously treated patients. The risk of people with MS treated with natalizumab developing PML has been quantified [62]. The major risk factors are the presence of anti-JC virus antibodies in blood, prior use of immunosuppressants, and increasing duration of natalizumab treatment. Regular monitoring of these factors, coupled with appropriate action, substantially reduces the risk of people with MS administered natalizumab developing PML [63].
Fingolimod
Fingolimod is the first orally available immunomodulatory medicine for the treatment of RRMS. It was approved by the FDA in 2010 and the European Medicines Agency (EMA) in 2011. It is indicated in the US as first line treatment of RRMS, at a recommended dose of 0.5 mg once daily, to reduce the frequency of clinical exacerbations and delay the accumulation of physical disability. In the EU, fingolimod is indicated for the treatment of people with highly active RRMS despite treatment with IFNβ drugs, or people with MS with rapidly-evolving severe RRMS. Fingolimod is a structural analogue of intracellular sphingosine that is phosphorylated by the enzyme sphingosine kinase-2 and exerts its immunomodulatory effect by mimicking sphingosine 1-phosphate (S1P) and binding to four of the five S1P receptors on lymphocytes. This binding leads to internalization of activated S1P receptors, and their down regulation. In the absence of S1P receptor signalling, CD4+ and CD8+ T cells and B cells are unable to egress from secondary lymphoid tissue. This reduces the number of lymphocytes in the blood by about 70%, which attenuates their movement into the CNS [64]. Fingolimod showed superior efficacy over IFNβ-1a (Avonex) on the basis of head-to-head studies comparing relapse rates in people with RRMS [65]. A systematic review and meta-analyses of treatments in RRMS concluded that fingolimod significantly reduces relapse frequency in people with RRMS compared with IFNβ drugs and glatiramer acetate [66]. Because of effects on heart rate and atrioventricular conduction that may cause bradycardia and heart block, fingolimod requires enhanced cardiovascular monitoring on first dose or when recommencing dosing following treatment interruption. In addition, dimethyl fumarate has been linked to four cases of PML [67]. The FDA is currently investigating a case of PML in a patient with MS taking fingolimod.
Teriflunomide
Teriflunomide, another oral drug, gained FDA in 2012 and EMA approval the following year, for the treatment of RRMS. It is an active metabolite of leflunomide, the first disease modifying anti-rheumatic drug, and inhibits the mitochondrial enzyme dihydro-orotate dehydrogenase. It thus reduces pyrimidine synthesis and, because the production of activated T cells largely depends on de novo pyrimidine synthesis, inhibits immune cell proliferation [68]. On the basis of five phase III studies, teriflunomide reduced MRI lesions and annual relapse rates, to an extent similar to that of IFNβ drugs and glatiramer acetate. There were no serious adverse events and the most common ones were liver dysfunction, alopecia, diarrhoea, influenza, nausea and paraesthesia [69–71].
Dmethyl fumarate
Dimethyl fumarate (BG-12) is the methyl ester of fumaric acid, an intermediate in the tricarboxylic acid cycle, that is formed by the oxidation of succinate by the enzyme succinate dehydrogenase. Although the biological basis of its immunomodulatory action is not fully understood, fumarate does induce IL-4-producing Th2 cells. These cells generate type II dendritic cells that produce IL-10 instead of IL-12 and IL-23 [72].
Results from two phase III trials were recently published. DEFINE compared 240 mg dimethyl fumarate, administered either twice or three times daily with placebo in people with RRMS [73]. CONFIRM compared dimethyl fumarate treatment with both placebo and glatiramer acetate in 1200 people with RRMS [74]. Both studies showed that dimethyl fumarate reduced relapse rate by 44–53%, with some evidence of a reduced risk of disability progression. Dimethyl fumarate also reduced new gadolinium lesion development across a range of subgroups (defined on the basis of baseline disease characteristics or demographics) by 49–89% [75]. In 2013, dimethyl fumarate gained FDA approval and the Committee for Medicinal Products for Human Use of the EMA recommended the granting of marketing authorization. Adverse events associated with dimethyl fumarate included flushing and gastrointestinal (such as diarrhoea, nausea, and upper abdominal pain), along with reduced numbers of lymphocytes, elevated liver aminotransferase activity and evidence of acute kidney injury [73].
Emerging immune-targeted MS drugs (Table 2)
Alemtuzumab
Alemtuzumab is a humanized monoclonal antibody for CD52, a surface glycoprotein of unknown function that is expressed throughout the immune system on T and B lymphocytes, natural killer cells, dendritic cells and most monocytes. Alemtuzumab causes complement and antibody-dependent cytoxicity of lymphocytes that leads to a profound depletion of circulating T and B cells. While B cells rapidly recover, CD4+ T helper cells take years to recover to pre-treatment numbers. Other factors are probably also involved, including increased numbers of regulatory T cells and fewer memory T cells [76].
The results from two phase III trials were recently published: (i) CARE-MS1 compared alemtuzumab and IFNβ-1a (Rebif) in people with MS in the first few years after diagnosis and were immunomodulatory drug free [77] and (ii) CARE-MS2 examined people with MS who had continued to have relapses despite treatment with IFNβ drugs or glatiramer acetate [78]. The results indicated that, compared with comparator drugs, alemtuzumab reduced the relapse rate by 49–55%, improved radiological markers of MS pathology, reduced the risk of sustaining disability and partially reversed disability. Alemtuzumab was also found to increase the risk of developing other autoimmune diseases, particularly autoimmune thyroid-related problems and idiopathic thrombocytopenic purpura [79]. Marketing authorization applications have been submitted for the use of alemtuzumab in MS to the FDA and EMA.
Daclizumab
Daclizumab is a monoclonal antibody that binds to CD25, a receptor for IL-2 on the surface of lymphocytes and thus prevents T cell activation [80]. A recent systematic analysis of the use of daclizumab (both alone and combined with other immunomodulatory drugs) in the treatment of RRMS concluded that high dose daclizumab (300 mg month−1) reduces the number of new or enlarged gadolinium contrast-enhancing lesions, with the caveat that more studies are required to evaluate properly its efficacy and safety [81].
Ocrelizumab
Ocrelizumab is a mAb that targets CD20 on the surface of B cells and thus attenuates the abnormal immune response associated with MS. It is under development for the treatment of both RRMS and PPMS. Ocrelizumab reduced the total number of gadolinium-enhancing and T1-weighted MRI lesions in a phase II study [82] and phase III trials for both RRMS and PPMS are on-going.
Laquinimod
Laquinimod is an oral immunomodulatory agent for RRMS that is thought to act by shifting the immune response from Th1 to Th2, thus decreasing the concentration of pro-inflammatory cytokines (particularly IFNγ and tumour necrosis factor) and increasing production of IL-4, an anti-inflammatory cytokine [83]. The results from two phase III trials were recently published. ALLEGRO compared 0.6 mg laquinimod, administered once daily, with placebo in people with RRMS. BRAVO compared laquinimod 0.6 mg with IFNβ-1a (Avonex) with placebo. In the ALLEGRO study, laquinimod produced a modest (23%) reduction in the relapse rate compared with placebo and reduced the risk of disability progression, although laquinimod and placebo groups showed no difference in their Multiple Sclerosis Functional Composite scores [84]. Results from the BRAVO study have not yet been published fully, but initial reports indicate a similar modest effect. Laquinimod has been well-tolerated in clinical studies, with the most frequent side effects being back pain, increased liver enzyme activity and headache [84–86] A study is about to commence to compare two doses of laquinimod (0.6 mg and 1.2 mg) in approximately 1800 people for up to 24 months. The main outcome measure will be progression of disability measured by the Expanded Disability Status Scale (EDSS).
Firategrast
Firategrast is an orally active, small molecule α4β1-integrin antagonist with a shorter half-life than natalizumab. It showed efficacy, on the basis of imaging endpoints, in a phase II study of individuals with RRMS. One of four twice daily treatment regimens was applied: (i) 150 mg, (ii) 600 mg, (iii) 900 mg (women) or 1200 mg (men) and (iv) placebo. A 49% reduction was observed in the cumulative number of new gadolinium-enhancing lesions at the highest dose tested, with no cases of PML or evidence of JC-1 reactivation [87]. No planned phase III study is listed on the U.S. National Institutes of Health website http://www.clinicaltrials.gov.
NU100
NU100 is a proprietary recombinant human IFNβ-1b being developed for the treatment of RRMS. A phase III study is underway in people with RRMS randomized to receive NU100, a marketed IFNβ-1b or placebo over a 12 month period [3].
BII-B017
BII-B017 is polyethylene glycol covalently bonded to IFNβ-1a and so represents an extended-release version of Avonex and Rebif. Thus, it only needs to be administered every 2–4 weeks, rather than one to three times weekly. The top-line results from a phase III trial comparing BII-B017 with placebo indicate that it reduces relapse rate at 1 year and has favourable safety and tolerability profiles (Biogen Idec press release, January 24, 2013).
Conclusions
The first IFNβ drug gained regulatory approval in 1993 and glatiramer acetate in 1997 and have dominated the MS market ever since [88]. However, a number of factors are now changing this situation. Patent expirations are imminent and a number of oral immunomodulatory drugs (fingolimod, dimethyl fumarate and teriflunomide) have recently been launched. More orally available MS drugs are in the pipeline, with laquinimod submitted for approval and firategrast is in late stage development. The emergence of a number of immunomodulatory mAb drugs (alemtuzumab, daclizumab and ocrelizumab) and medicines to manage symptoms, including motor dysfunction (dalfampride and botulinum toxin) and spasticity (nabiximols), are also changing the market landscape.
A two stage disease process has been proposed on the basis of a large longitudinal study of people with MS [89]. It suggests that immunomodulatory drugs that reduce demyelination will slow the course of MS by reducing the extent of subsequent neurodegeneration [90]. However, although IFNβ drugs and glatiramer acetate are described as ‘disease modifying’, they have little impact on the course of RRMS, unless given during the prodromal stage of MS (CIS) [3]. In addition, no immune-targeted therapy has shown efficacy in the treatment of progressive forms of MS [49–55]. There are three possible explanations for this: (i) non-immune based mechanisms are critically involved in the progressive development of the symptoms, (ii) IFNβ drugs and glatiramer acetate have insufficient impact on the process of demyelination to meaningfully alter disease progression, or (iii) a combination of both. It remains to be established whether the new and emerging immunomodulatory MS drugs (Tables 1 and 2) have efficacy in slowing the course of MS over the long term. The magnitude of the reduction in relapse rate with natalizumab is twice that seen with IFNβ drugs and glatiramer acetate and appears to slow disease progression (Table 1) [60].
Grey matter damage has attracted much less interest than white matter lesions, mainly because it has been difficult to see cortical grey matter lesions using conventional histochemical staining procedures. However, greater MRI resolution now permits the detection of grey matter changes in the intact CNS [91]. The importance of such change is demonstrated by loss of grey matter (but not white matter) correlating with long term disability [92]. The loss of corticospinal axons is the major contributor to the disability associated with both PPMS and SPMS [93,94]. On the basis of recent MRI studies, cortical lesions and cortical atrophy have been observed in patients with early multiple sclerosis [95–99]. This has been confirmed on the basis of immune-histochemical studies of cortical biopsy tissue from people with early stage MS that showed that cortical demyelinating lesions were frequent, inflammatory and strongly associated with meningeal inflammation [100].
Focal inflammation that results in loss of myelin is probably the pivotal event in the pathophysiology of MS, in all its phenotypes [89]. Myelin plays a critical role in neuronal conduction since it inhibits charge leakage through the axonal membrane. The sheath of myelin does not provide a continuous insulating cover, as there are numerous gaps (nodes of Ranvier) about 1 mm long along the length of the axon. This permits the action potential to move from one node to another. This saltatory conduction both increases the speed of the nerve impulse and reduces energy expenditure at the area of depolarization. The demyelination that occurs in MS therefore slows the speed of nerve transmission and can lead to conduction block, i.e. a failure of an action potential to propagate along a structurally intact axon. This increases the energy required for neurotransmission in a manner that is directly proportional to the extent of myelin loss. Once the energy expenditure required for the propagation of neuronal impulses in myelin-denuded axons reaches a critical point, excitotoxic processes will begin to occur that will eventually lead to neuronal cell death. In terms of directly targeting the neurodegenerative processes occurring in MS, anti-excitotoxic compounds, including AMPA/kainite receptor antagonists and sodium channel blockers, have shown some evidence of neuroprotective efficacy in experimental models of MS. There is also evidence for one sodium channel blocker (lamotrigine) showing neuroprotective efficacy in people with SPMS [101–103]. Another approach showing promise is blockade of acid-sensing ion channel 1 using the drug amiloride, which is licensed for the treatment of hypertension and congestive cardiac failure. This compound has shown neuroprotective efficacy in experimental models of MS [104] and displayed evidence consistent with neuroprotection in a pilot study of people with PPMS [105].
Competing Interests
The author is a Director of MS Therapeutics Ltd.
References
- 1.Bol Y, Smolders J, Duits A, Lange IM, Romberg-Camps M, Hupperts R. Fatigue and heat sensitivity in patients with multiple sclerosis. Acta Neurol Scand. 2012;126:384–389. doi: 10.1111/j.1600-0404.2012.01660.x. [DOI] [PubMed] [Google Scholar]
- 2.Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 3.Palmer A. Pharmacotherapeuetic options for the treatment of multiple sclerosis. Clin Med Insights. 2012;4:1–24. [Google Scholar]
- 4.Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology. 1996;46:907–911. doi: 10.1212/wnl.46.4.907. [DOI] [PubMed] [Google Scholar]
- 5.WHO. Atlas Multiple Sclerosis. Geneva: WHO Press; 2008. [Google Scholar]
- 6.Antel J, Antel S, Caramanos Z, Arnold DL, Kuhlmann T. Primary progressive multiple sclerosis: part of the MS disease spectrum or separate disease entity? Acta Neuropathol. 2012;123:627–638. doi: 10.1007/s00401-012-0953-0. [DOI] [PubMed] [Google Scholar]
- 7.Koch M, Kingwell E, Rieckmann P, Tremlett H. The natural history of primary progressive multiple sclerosis. Neurology. 2009;73:1996–2002. doi: 10.1212/WNL.0b013e3181c5b47f. [DOI] [PubMed] [Google Scholar]
- 8.Miller DH, Leary SM. Primary-progressive multiple sclerosis. Lancet Neurol. 2007;6:903–912. doi: 10.1016/S1474-4422(07)70243-0. [DOI] [PubMed] [Google Scholar]
- 9.Pandey K, Lublin FD. Clinically isolated syndrome and multiple sclerosis: rethinking the arsenal. Curr Treat Options Neurol. 2009;11:193–202. doi: 10.1007/s11940-009-0023-7. [DOI] [PubMed] [Google Scholar]
- 10.Forn C, Rocca MA, Valsasina P, Bosca I, Casanova B, Sanjuan A, Avila C, Filippi M. Functional magnetic resonance imaging correlates of cognitive performance in patients with a clinically isolated syndrome suggestive of multiple sclerosis at presentation: an activation and connectivity study. Mult Scler. 2012;18:153–163. doi: 10.1177/1352458511417744. [DOI] [PubMed] [Google Scholar]
- 11.Fisniku LK, Brex PA, Altmann DR, Miszkiel KA, Benton CE, Lanyon R, Thompson AJ, Miller DH. Disability and T2 MRI lesions: a 20-year follow-up of patients with relapse onset of multiple sclerosis. Brain. 2008;131:808–817. doi: 10.1093/brain/awm329. [DOI] [PubMed] [Google Scholar]
- 12.Koch M, Kingwell E, Rieckmann P, Tremlett H. The natural history of secondary progressive multiple sclerosis. J Neurol Neurosurg Psychiatry. 2010;81:1039–1043. doi: 10.1136/jnnp.2010.208173. [DOI] [PubMed] [Google Scholar]
- 13.Rovaris M, Confavreux C, Furlan R, Kappos L, Comi G, Filippi M. Secondary progressive multiple sclerosis: current knowledge and future challenges. Lancet Neurol. 2006;5:343–354. doi: 10.1016/S1474-4422(06)70410-0. [DOI] [PubMed] [Google Scholar]
- 14.Trojano M, Paolicelli D. The differential diagnosis of multiple sclerosis: classification and clinical features of relapsing and progressive neurological syndromes. Neurol Sci. 2001;22(Suppl 2):S98–102. doi: 10.1007/s100720100044. [DOI] [PubMed] [Google Scholar]
- 15.Gourraud PA, Harbo HF, Hauser SL, Baranzini SE. The genetics of multiple sclerosis: an up-to-date review. Immunol Rev. 2012;248:87–103. doi: 10.1111/j.1600-065X.2012.01134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patsopoulos NA, Bayer Pharma MSGWG. Steering Committees of Studies Evaluating I-b, a CCRA, Consortium AN, GeneMsa, International Multiple Sclerosis Genetics C. Esposito F, Reischl J, Lehr S, Bauer D, Heubach J, Sandbrink R, Pohl C, Edan G, Kappos L, Miller D, Montalban J, Polman CH, Freedman MS, Hartung HP, Arnason BG, Comi G, Cook S, Filippi M, Goodin DS, Jeffery D, O'Connor P, Ebers GC, Langdon D, Reder AT, Traboulsee A, Zipp F, Schimrigk S, Hillert J, Bahlo M, Booth DR, Broadley S, Brown MA, Browning BL, Browning SR, Butzkueven H, Carroll WM, Chapman C, Foote SJ, Griffiths L, Kermode AG, Kilpatrick TJ, Lechner-Scott J, Marriott M, Mason D, Moscato P, Heard RN, Pender MP, Perreau VM, Perera D, Rubio JP, Scott RJ, Slee M, Stankovich J, Stewart GJ, Taylor BV, Tubridy N, Willoughby E, Wiley J, Matthews P, Boneschi FM, Compston A, Haines J, Hauser SL, McCauley J, Ivinson A, Oksenberg JR, Pericak-Vance M, Sawcer SJ, De Jager PL, Hafler DA, de Bakker PI. Genome-wide meta-analysis identifies novel multiple sclerosis susceptibility loci. Ann Neurol. 2011;70:897–912. doi: 10.1002/ana.22609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sawcer S, Hellenthal G, Pirinen M, Spencer CC, Patsopoulos NA, Moutsianas L, Dilthey A, Su Z, Freeman C, Hunt SE, Edkins S, Gray E, Booth DR, Potter SC, Goris A, Band G, Oturai AB, Strange A, Saarela J, Bellenguez C, Fontaine B, Gillman M, Hemmer B, Gwilliam R, Zipp F, Jayakumar A, Martin R, Leslie S, Hawkins S, Giannoulatou E, D'Alfonso S, Blackburn H, Martinelli Boneschi F, Liddle J, Harbo HF, Perez ML, Spurkland A, Waller MJ, Mycko MP, Ricketts M, Comabella M, Hammond N, Kockum I, McCann OT, Ban M, Whittaker P, Kemppinen A, Weston P, Hawkins C, Widaa S, Zajicek J, Dronov S, Robertson N, Bumpstead SJ, Barcellos LF, Ravindrarajah R, Abraham R, Alfredsson L, Ardlie K, Aubin C, Baker A, Baker K, Baranzini SE, Bergamaschi L, Bergamaschi R, Bernstein A, Berthele A, Boggild M, Bradfield JP, Brassat D, Broadley SA, Buck D, Butzkueven H, Capra R, Carroll WM, Cavalla P, Celius EG, Cepok S, Chiavacci R, Clerget-Darpoux F, Clysters K, Comi G, Cossburn M, Cournu-Rebeix I, Cox MB, Cozen W, Cree BA, Cross AH, Cusi D, Daly MJ, Davis E, de Bakker PI, Debouverie M, D'Hooghe MB, Dixon K, Dobosi R, Dubois B, Ellinghaus D, Elovaara I, Esposito F, Fontenille C, Foote S, Franke A, Galimberti D, Ghezzi A, Glessner J, Gomez R, Gout O, Graham C, Grant SF, Guerini FR, Hakonarson H, Hall P, Hamsten A, Hartung HP, Heard RN, Heath S, Hobart J, Hoshi M, Infante-Duarte C, Ingram G, Ingram W, Islam T, Jagodic M, Kabesch M, Kermode AG, Kilpatrick TJ, Kim C, Klopp N, Koivisto K, Larsson M, Lathrop M, Lechner-Scott JS, Leone MA, Leppa V, Liljedahl U, Bomfim IL, Lincoln RR, Link J, Liu J, Lorentzen AR, Lupoli S, Macciardi F, Mack T, Marriott M, Martinelli V, Mason D, McCauley JL, Mentch F, Mero IL, Mihalova T, Montalban X, Mottershead J, Myhr KM, Naldi P, Ollier W, Page A, Palotie A, Pelletier J, Piccio L, Pickersgill T, Piehl F, Pobywajlo S, Quach HL, Ramsay PP, Reunanen M, Reynolds R, Rioux JD, Rodegher M, Roesner S, Rubio JP, Ruckert IM, Salvetti M, Salvi E, Santaniello A, Schaefer CA, Schreiber S, Schulze C, Scott RJ, Sellebjerg F, Selmaj KW, Sexton D, Shen L, Simms-Acuna B, Skidmore S, Sleiman PM, Smestad C, Sorensen PS, Sondergaard HB, Stankovich J, Strange RC, Sulonen AM, Sundqvist E, Syvanen AC, Taddeo F, Taylor B, Blackwell JM, Tienari P, Bramon E, Tourbah A, Brown MA, Tronczynska E, Casas JP, Tubridy N, Corvin A, Vickery J, Jankowski J, Villoslada P, Markus HS, Wang K, Mathew CG, Wason J, Palmer CN, Wichmann HE, Plomin R, Willoughby E, Rautanen A, Winkelmann J, Wittig M, Trembath RC, Yaouanq J, Viswanathan AC, Zhang H, Wood NW, Zuvich R, Deloukas P, Langford C, Duncanson A, Oksenberg JR, Pericak-Vance MA, Haines JL, Olsson T, Hillert J, Ivinson AJ, De Jager PL, Peltonen L, Stewart GJ, Hafler DA, Hauser SL, McVean G, Donnelly P, Compston A. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–219. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Faridar A, Eskandari G, Sahraian MA, Minagar A, Azimi A. Vitamin D and multiple sclerosis: a critical review and recommendations on treatment. Acta Neurol Belg. 2012;112:327–333. doi: 10.1007/s13760-012-0108-z. [DOI] [PubMed] [Google Scholar]
- 19.Handel AE, Giovannoni G, Ebers GC, Ramagopalan SV. Environmental factors and their timing in adult-onset multiple sclerosis. Nat Rev Neurol. 2010;6:156–166. doi: 10.1038/nrneurol.2010.1. [DOI] [PubMed] [Google Scholar]
- 20.Handel AE, Williamson AJ, Disanto G, Dobson R, Giovannoni G, Ramagopalan SV. Smoking and multiple sclerosis: an updated meta-analysis. PLoS ONE. 2011;6:e16149. doi: 10.1371/journal.pone.0016149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Handel AE, Williamson AJ, Disanto G, Handunnetthi L, Giovannoni G, Ramagopalan SV. An updated meta-analysis of risk of multiple sclerosis following infectious mononucleosis. PLoS ONE. 2010;5:e12496. doi: 10.1371/journal.pone.0012496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wingerchuk DM. Environmental factors in multiple sclerosis: Epstein-Barr virus, vitamin D, and cigarette smoking. Mt Sinai J Med. 2011;78:221–230. doi: 10.1002/msj.20240. [DOI] [PubMed] [Google Scholar]
- 23.Filippi M, Rocca MA, Barkhof F, Bruck W, Chen JT, Comi G, Deluca G, De Stefano N, Erickson BJ, Evangelou N, Fazekas F, Geurts JJ, Lucchinetti C, Miller DH, Pelletier D, Popescu BF, Lassmann H. Association between pathological and MRI findings in multiple sclerosis. Lancet Neurol. 2012;11:349–360. doi: 10.1016/S1474-4422(12)70003-0. [DOI] [PubMed] [Google Scholar]
- 24.Simon JH, Li D, Traboulsee A, Coyle PK, Arnold DL, Barkhof F, Frank JA, Grossman R, Paty DW, Radue EW, Wolinsky JS. Standardized MR imaging protocol for multiple sclerosis: consortium of MS Centers consensus guidelines. AJNR Am J neuroradiol. 2006;27:455–461. [PMC free article] [PubMed] [Google Scholar]
- 25.Polman CH, Reingold SC, Edan G, Filippi M, Hartung HP, Kappos L, Lublin FD, Metz LM, McFarland HF, O'Connor PW, Sandberg-Wollheim M, Thompson AJ, Weinshenker BG, Wolinsky JS. Diagnostic criteria for multiple sclerosis: 2005 revisions to the ‘McDonald Criteria. Ann Neurol. 2005;58:840–846. doi: 10.1002/ana.20703. [DOI] [PubMed] [Google Scholar]
- 26.Palmer AM. The role of the blood-CNS barrier in CNS disorders and their treatment. Neurobiol Dis. 2010;37:3–12. doi: 10.1016/j.nbd.2009.07.029. [DOI] [PubMed] [Google Scholar]
- 27.Holmoy T. The immunology of multiple sclerosis: disease mechanisms and therapeutic targets. Minerva Med. 2008;99:119–140. [PubMed] [Google Scholar]
- 28.Palmer AM. Multiple sclerosis and the blood-central nervous system barrier. Cardiovasc Psychiat Neurol. 2013;2013:530356. doi: 10.1155/2013/530356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ceccarelli A, Bakshi R, Neema M. MRI in multiple sclerosis: a review of the current literature. Curr Opin Neurol. 2012;25:402–409. doi: 10.1097/WCO.0b013e328354f63f. [DOI] [PubMed] [Google Scholar]
- 30.Filippi M, Rocca MA. MR imaging of multiple sclerosis. Radiology. 2011;259:659–681. doi: 10.1148/radiol.11101362. [DOI] [PubMed] [Google Scholar]
- 31.Haegert DG. Multiple sclerosis: a disorder of altered T-cell homeostasis. Mult Scler Int. 2011;2011:461304. doi: 10.1155/2011/461304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ousman SS, Tomooka BH, van Noort JM, Wawrousek EF, O'Connor KC, Hafler DA, Sobel RA, Robinson WH, Steinman L. Protective and therapeutic role for alphaB-crystallin in autoimmune demyelination. Nature. 2007;448:474–479. doi: 10.1038/nature05935. [DOI] [PubMed] [Google Scholar]
- 33.Mathey EK, Derfuss T, Storch MK, Williams KR, Hales K, Woolley DR, Al-Hayani A, Davies SN, Rasband MN, Olsson T, Moldenhauer A, Velhin S, Hohlfeld R, Meinl E, Linington C. Neurofascin as a novel target for autoantibody-mediated axonal injury. J Exp Med. 2007;204:2363–2372. doi: 10.1084/jem.20071053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Engelhardt B, Kempe B, Merfeld-Clauss S, Laschinger M, Furie B, Wild MK, Vestweber D. P-selectin glycoprotein ligand 1 is not required for the development of experimental autoimmune encephalomyelitis in SJL and C57BL/6 mice. J Immunol. 2005;175:1267–1275. doi: 10.4049/jimmunol.175.2.1267. [DOI] [PubMed] [Google Scholar]
- 35.Holmoy T, Vartdal F. The immunological basis for treatment of multiple sclerosis. Scand J Immunol. 2007;66:374–382. doi: 10.1111/j.1365-3083.2007.01982.x. [DOI] [PubMed] [Google Scholar]
- 36.Wekerle H. Immune pathogenesis of multiple sclerosis. Neurol Sci. 2005;26(Suppl 1):S1–2. doi: 10.1007/s10072-005-0386-9. [DOI] [PubMed] [Google Scholar]
- 37.Miller E. Multiple sclerosis. Adv Exp Med Biol. 2012;724:222–238. doi: 10.1007/978-1-4614-0653-2_17. [DOI] [PubMed] [Google Scholar]
- 38.Luessi F, Siffrin V, Zipp F. Neurodegeneration in multiple sclerosis: novel treatment strategies. Expert Rev Neurother. 2012;12:1061–1077. doi: 10.1586/ern.12.59. [DOI] [PubMed] [Google Scholar]
- 39.Engelhardt B. The blood-central nervous system barriers actively control immune cell entry into the central nervous system. Curr Pharm Des. 2008;14:1555–1565. doi: 10.2174/138161208784705432. [DOI] [PubMed] [Google Scholar]
- 40.Engelhardt B. Immune cell entry into the central nervous system: involvement of adhesion molecules and chemokines. J Neurol Sci. 2008;274:23–26. doi: 10.1016/j.jns.2008.05.019. [DOI] [PubMed] [Google Scholar]
- 41.Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, Giuliani F, Arbour N, Becher B, Prat A. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. 2007;13:1173–1175. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huppert J, Closhen D, Croxford A, White R, Kulig P, Pietrowski E, Bechmann I, Becher B, Luhmann HJ, Waisman A, Kuhlmann CR. Cellular mechanisms of IL-17-induced blood-brain barrier disruption. FASEB J. 2010;24:1023–1034. doi: 10.1096/fj.09-141978. [DOI] [PubMed] [Google Scholar]
- 43.Plosker G. Interferon PGL beta-1b: a review of its use in multiple sclerosis. CNS Drugs. 2011;25:67–88. doi: 10.2165/11206430-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 44.Beer K, Muller M, Hew-Winzeler AM, Bont A, Maire P, You X, Foulds P, Marlind J, Curtius D. The prevalence of injection-site reactions with disease-modifying therapies and their effect on adherence in patients with multiple sclerosis: an observational study. BMC Neurol. 2011;11:144. doi: 10.1186/1471-2377-11-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Balak DM, Hengstman GJ, Cakmak A, Thio HB. Cutaneous adverse events associated with disease-modifying treatment in multiple sclerosis: a systematic review. Mult Scler. 2012;18:1705–1717. doi: 10.1177/1352458512438239. [DOI] [PubMed] [Google Scholar]
- 46.Koch-Henriksen N, Sorensen PS, Bendtzen K, Flachs EM. The clinical effect of neutralizing antibodies against interferon-beta is independent of the type of interferon-beta used for patients with relapsing-remitting multiple sclerosis. Mult Scler. 2009;15:601–605. doi: 10.1177/1352458508101946. [DOI] [PubMed] [Google Scholar]
- 47.Rio J, Nos C, Tintore M, Tellez N, Galan I, Pelayo R, Comabella M, Montalban X. Defining the response to interferon-beta in relapsing-remitting multiple sclerosis patients. Ann Neurol. 2006;59:344–352. doi: 10.1002/ana.20740. [DOI] [PubMed] [Google Scholar]
- 48.Bertolotto A. Implications of neutralising antibodies on therapeutic efficacy. Journal of the neurological sciences. 2009;277(Suppl 1):S29–32. doi: 10.1016/S0022-510X(09)70009-7. [DOI] [PubMed] [Google Scholar]
- 49.Rice GP, Incorvaia B, Munari L, Ebers G, Polman C, D'Amico R, Filippini G. Interferon in relapsing-remitting multiple sclerosis. Cochrane Database Syst Rev. 2001;(4) doi: 10.1002/14651858.CD002002. CD002002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.La Mantia L, Munari LM, Lovati R. Glatiramer acetate for multiple sclerosis. Cochrane Database Syst Rev. 2010;(5) doi: 10.1002/14651858.CD004678.pub2. CD004678. [DOI] [PubMed] [Google Scholar]
- 51.La Mantia L, Vacchi L, Di Pietrantonj C, Ebers G, Rovaris M, Fredrikson S, Filippini G. Interferon beta for secondary progressive multiple sclerosis. Cochrane Database Syst Rev. 2012;(1) doi: 10.1002/14651858.CD005181.pub3. CD005181. [DOI] [PubMed] [Google Scholar]
- 52.Rojas JI, Romano M, Ciapponi A, Patrucco L, Cristiano E. Interferon Beta for primary progressive multiple sclerosis. Cochrane Database Syst Rev. 2010;(1) doi: 10.1002/14651858.CD006643.pub3. CD006643. [DOI] [PubMed] [Google Scholar]
- 53.Clerico M, Faggiano F, Palace J, Rice G, Tintore M, Durelli L. Recombinant interferon beta or glatiramer acetate for delaying conversion of the first demyelinating event to multiple sclerosis. Cochrane Database Syst Rev. 2008;(2) doi: 10.1002/14651858.CD005278.pub3. CD005278. [DOI] [PubMed] [Google Scholar]
- 54.Comi G, Martinelli V, Rodegher M, Moiola L, Bajenaru O, Carra A, Elovaara I, Fazekas F, Hartung HP, Hillert J, King J, Komoly S, Lubetzki C, Montalban X, Myhr KM, Ravnborg M, Rieckmann P, Wynn D, Young C, Filippi M, Pre C. Effect of glatiramer acetate on conversion to clinically definite multiple sclerosis in patients with clinically isolated syndrome (PreCISe study): a randomised, double-blind, placebo-controlled trial. Lancet. 2009;374:1503–1511. doi: 10.1016/S0140-6736(09)61259-9. [DOI] [PubMed] [Google Scholar]
- 55.Carter NJ, Keating GM. Glatiramer acetate: a review of its use in relapsing-remitting multiple sclerosis and in delaying the onset of clinically definite multiple sclerosis. Drugs. 2010;70:1545–1577. doi: 10.2165/11204560-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 56.Fox EJ. Management of worsening multiple sclerosis with mitoxantrone: a review. Clin Ther. 2006;28:461–474. doi: 10.1016/j.clinthera.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 57.Edan G, Comi G, Le Page E, Leray E, Rocca MA, Filippi M, Mitoxantrone F-I. Interferon-beta-1b Trial G. Mitoxantrone prior to interferon beta-1b in aggressive relapsing multiple sclerosis: a 3-year randomised trial. J Neurol Neurosurg Psychiatry. 2011;82:1344–1350. doi: 10.1136/jnnp.2010.229724. [DOI] [PubMed] [Google Scholar]
- 58.Hartung HP, Gonsette R, Konig N, Kwiecinski H, Guseo A, Morrissey SP, Krapf H, Zwingers T Mitoxantrone in Multiple Sclerosis Study G. Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet. 2002;360:2018–2025. doi: 10.1016/S0140-6736(02)12023-X. [DOI] [PubMed] [Google Scholar]
- 59.Martinelli Boneschi F, Rovaris M, Capra R, Comi G. Mitoxantrone for multiple sclerosis. Cochrane Database Syst Rev. 2005;(4) doi: 10.1002/14651858.CD002127.pub2. CD002127. [DOI] [PubMed] [Google Scholar]
- 60.Pucci E, Giuliani G, Solari A, Simi S, Minozzi S, Di Pietrantonj C, Galea I. Natalizumab for relapsing remitting multiple sclerosis. Cochrane Database Syst Rev. 2011;(10) doi: 10.1002/14651858.CD007621.pub2. CD007621. [DOI] [PubMed] [Google Scholar]
- 61.Lanzillo R, Quarantelli M, Bonavita S, Ventrella G, Lus G, Vacca G, Prinster A, Orefice G, Tedeschi G, Brescia Morra V. Natalizumab vs interferon beta 1a in relapsing-remitting multiple sclerosis: a head-to-head retrospective study. Acta Neurol Scand. 2011;26:306–314. doi: 10.1111/j.1600-0404.2011.01622.x. [DOI] [PubMed] [Google Scholar]
- 62.Bloomgren G, Sperling B, Cushing K, Wenten M. Assessment of malignancy risk in patients with multiple sclerosis treated with intramuscular interferon beta-1a: retrospective evaluation using a health insurance claims database and postmarketing surveillance data. Ther Clin Risk Manag. 2012;8:313–321. doi: 10.2147/TCRM.S31347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hellwig K, Gold R. Progressive multifocal leukoencephalopathy and natalizumab. J Neurol. 2011;258:1920–1928. doi: 10.1007/s00415-011-6116-8. [DOI] [PubMed] [Google Scholar]
- 64.Pelletier D, Hafler DA. Fingolimod for multiple sclerosis. N Engl J Med. 2012;366:339–347. doi: 10.1056/NEJMct1101691. [DOI] [PubMed] [Google Scholar]
- 65.Khatri B, Barkhof F, Comi G, Hartung HP, Kappos L, Montalban X, Pelletier J, Stites T, Wu S, Holdbrook F, Zhang-Auberson L, Francis G, Cohen JA. Comparison of fingolimod with interferon beta-1a in relapsing-remitting multiple sclerosis: a randomised extension of the TRANSFORMS study. Lancet Neurol. 2011;10:520–529. doi: 10.1016/S1474-4422(11)70099-0. [DOI] [PubMed] [Google Scholar]
- 66.Roskell NS, Zimovetz EA, Rycroft CE, Eckert BJ, Tyas DA. Annualized relapse rate of first-line treatments for multiple sclerosis: a meta-analysis, including indirect comparisons versus fingolimod. Curr Med Res Opin. 2012;28:767–780. doi: 10.1185/03007995.2012.681637. [DOI] [PubMed] [Google Scholar]
- 67.Sweetser MT, Dawson KT, Bozic C. Manufacturer's response to case reports of PML. N Engl J Med. 2013;368:1659–1661. doi: 10.1056/NEJMc1300283. [DOI] [PubMed] [Google Scholar]
- 68.Claussen MC, Korn T. Immune mechanisms of new therapeutic strategies in MS: teriflunomide. Clin Immunol. 2012;142:49–56. doi: 10.1016/j.clim.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 69.He D, Xu Z, Dong S, Zhang H, Zhou H, Wang L, Zhang S. Teriflunomide for multiple sclerosis. Cochrane Database Syst Rev. 2012;(12) doi: 10.1002/14651858.CD009882.pub2. CD009882. [DOI] [PubMed] [Google Scholar]
- 70.Papadopoulou A, Kappos L, Sprenger T. Teriflunomide for oral therapy in multiple sclerosis. Expert Rev Clin Pharmacol. 2012;5:617–628. doi: 10.1586/ecp.12.56. [DOI] [PubMed] [Google Scholar]
- 71.Palmer AM. Efficacy and safety of teriflunomide in treatment of multiple sclerosis. Signs and Symptoms. 2013;(2):444–457. [Google Scholar]
- 72.Ghoreschi K, Bruck J, Kellerer C, Deng C, Peng H, Rothfuss O, Hussain RZ, Gocke AR, Respa A, Glocova I, Valtcheva N, Alexander E, Feil S, Feil R, Schulze-Osthoff K, Rupec RA, Lovett-Racke AE, Dringen R, Racke MK, Rocken M. Fumarates improve psoriasis and multiple sclerosis by inducing type II dendritic cells. J Exp Med. 2011;208:2291–2303. doi: 10.1084/jem.20100977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K, Tornatore C, Sweetser MT, Yang M, Sheikh SI, Dawson KT investigators DS. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367:1098–1107. doi: 10.1056/NEJMoa1114287. [DOI] [PubMed] [Google Scholar]
- 74.Fox RJ, Miller DH, Phillips JT, Hutchinson M, Havrdova E, Kita M, Yang M, Raghupathi K, Novas M, Sweetser MT, Viglietta V, Dawson KT investigators CS. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012;367:1087–1097. doi: 10.1056/NEJMoa1206328. [DOI] [PubMed] [Google Scholar]
- 75.Kappos L, Gold R, Miller DH, MacManus DG, Havrdova E, Limmroth V, Polman CH, Schmierer K, Yousry TA, Eraksoy M, Meluzinova E, Dufek M, Yang M, Dawson K, O'Neill GN. Effect of BG-12 on contrast-enhanced lesions in patients with relapsing–remitting multiple sclerosis: subgroup analyses from the phase 2b study. Mult Scler. 2012;18:314–321. doi: 10.1177/1352458511421054. [DOI] [PubMed] [Google Scholar]
- 76.Brown J, William L, Coles AJ. Alemtuzumab: evidence for its potential in relapsing-remitting multiple sclerosis. Drug Des Dev Ther. 2013;7:131–138. doi: 10.2147/DDDT.S32687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cohen JA, Coles AJ, Arnold DL, Confavreux C, Fox EJ, Hartung HP, Havrdova E, Selmaj KW, Weiner HL, Fisher E, Brinar VV, Giovannoni G, Stojanovic M, Ertik BI, Lake SL, Margolin DH, Panzara MA, Compston DA investigators C-MI. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. 2012;380:1819–1828. doi: 10.1016/S0140-6736(12)61769-3. [DOI] [PubMed] [Google Scholar]
- 78.Coles AJ, Twyman CL, Arnold DL, Cohen JA, Confavreux C, Fox EJ, Hartung HP, Havrdova E, Selmaj KW, Weiner HL, Miller T, Fisher E, Sandbrink R, Lake SL, Margolin DH, Oyuela P, Panzara MA, Compston DA investigators C-MI. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet. 2012;380:1829–1839. doi: 10.1016/S0140-6736(12)61768-1. [DOI] [PubMed] [Google Scholar]
- 79.Cossburn M, Pace AA, Jones J, Ali R, Ingram G, Baker K, Hirst C, Zajicek J, Scolding N, Boggild M, Pickersgill T, Ben-Shlomo Y, Coles A, Robertson NP. Autoimmune disease after alemtuzumab treatment for multiple sclerosis in a multicenter cohort. Neurology. 2011;77:573–579. doi: 10.1212/WNL.0b013e318228bec5. [DOI] [PubMed] [Google Scholar]
- 80.Bielekova B, Catalfamo M, Reichert-Scrivner S, Packer A, Cerna M, Waldmann TA, McFarland H, Henkart PA, Martin R. Regulatory CD56(bright) natural killer cells mediate immunomodulatory effects of IL-2Ralpha-targeted therapy (daclizumab) in multiple sclerosis. Proc Natl Acad Sci U S A. 2006;103:5941–5946. doi: 10.1073/pnas.0601335103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu J, Wang L, Zhan S, Tan J, Xia Y. Daclizumab for relapsing remitting multiple sclerosis. Cochrane Database Syst Rev. 2010;(4) doi: 10.1002/14651858.CD008127.pub2. CD008127. [DOI] [PubMed] [Google Scholar]
- 82.Kappos L, Li D, Calabresi PA, O'Connor P, Bar-Or A, Barkhof F, Yin M, Leppert D, Glanzman R, Tinbergen J, Hauser SL. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet. 2011;378:1779–1787. doi: 10.1016/S0140-6736(11)61649-8. [DOI] [PubMed] [Google Scholar]
- 83.Giacomini PS, Bar-Or A. Laquinimod in multiple sclerosis. Clin Immunol. 2012;142:38–43. doi: 10.1016/j.clim.2011.02.021. [DOI] [PubMed] [Google Scholar]
- 84.Comi G, Jeffery D, Kappos L, Montalban X, Boyko A, Rocca MA, Filippi M. Placebo-controlled trial of oral laquinimod for multiple sclerosis. N Engl J Med. 2012;366:1000–1009. doi: 10.1056/NEJMoa1104318. [DOI] [PubMed] [Google Scholar]
- 85.Bruck W, Zamvil SS. Laquinimod, a once-daily oral drug in development for the treatment of relapsing-remitting multiple sclerosis. Expert Rev Clin Pharmacol. 2012;5:245–256. doi: 10.1586/ecp.12.12. [DOI] [PubMed] [Google Scholar]
- 86.Rammohan KW, Shoemaker J. Emerging multiple sclerosis oral therapies. Neurology. 2010;74(Suppl 1):S47–53. doi: 10.1212/WNL.0b013e3181c97f89. [DOI] [PubMed] [Google Scholar]
- 87.Miller DH, Weber T, Grove R, Wardell C, Horrigan J, Graff O, Atkinson G, Dua P, Yousry T, Macmanus D, Montalban X. Firategrast for relapsing remitting multiple sclerosis: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2012;11:131–139. doi: 10.1016/S1474-4422(11)70299-X. [DOI] [PubMed] [Google Scholar]
- 88.Palmer A. Immunomodulatory medicines for multiple sclerosis: Progress and prospects. Drug Dev Res. 2011;72:664–672. [Google Scholar]
- 89.Leray E, Yaouanq J, Le Page E, Coustans M, Laplaud D, Oger J, Edan G. Evidence for a two-stage disability progression in multiple sclerosis. Brain. 2010;133:1900–1913. doi: 10.1093/brain/awq076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Waxman SG. Axonal conduction and injury in multiple sclerosis: the role of sodium channels. Nat Rev Neurosci. 2006;7:932–941. doi: 10.1038/nrn2023. [DOI] [PubMed] [Google Scholar]
- 91.Filippi M, Rocca MA. MR imaging of gray matter involvement in multiple sclerosis: implications for understanding disease pathophysiology and monitoring treatment efficacy. AJNR Am J neuroradiol. 2010;31:1171–1177. doi: 10.3174/ajnr.A1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fisniku LK, Altmann DR, Cercignani M, Tozer DJ, Chard DT, Jackson JS, Miszkiel KA, Schmierer K, Thompson AJ, Miller DH. Magnetization transfer ratio abnormalities reflect clinically relevant grey matter damage in multiple sclerosis. Mult Scler. 2009;15:668–677. doi: 10.1177/1352458509103715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tallantyre EC, Bo L, Al-Rawashdeh O, Owens T, Polman CH, Lowe J, Evangelou N. Greater loss of axons in primary progressive multiple sclerosis plaques compared to secondary progressive disease. Brain. 2009;132:1190–1199. doi: 10.1093/brain/awp106. [DOI] [PubMed] [Google Scholar]
- 94.Tallantyre EC, Bo L, Al-Rawashdeh O, Owens T, Polman CH, Lowe JS, Evangelou N. Clinico-pathological evidence that axonal loss underlies disability in progressive multiple sclerosis. Mult Scler. 2010;16:406–411. doi: 10.1177/1352458510364992. [DOI] [PubMed] [Google Scholar]
- 95.Tallantyre EC, Morgan PS, Dixon JE, Al-Radaideh A, Brookes MJ, Morris PG, Evangelou N. 3 Tesla and 7 Tesla MRI of multiple sclerosis cortical lesions. J Magn Reson Imaging. 2010;32:971–977. doi: 10.1002/jmri.22115. [DOI] [PubMed] [Google Scholar]
- 96.Tardif CL, Collins DL, Eskildsen SF, Richardson JB, Pike GB. Segmentation of cortical MS lesions on MRI using automated laminar profile shape analysis. Med Image Comput Comput Assist Interv Int Conf Med Image Comput Comput Assist Interv. 2010;13:181–188. doi: 10.1007/978-3-642-15711-0_23. [DOI] [PubMed] [Google Scholar]
- 97.Calabrese M, Filippi M, Gallo P. Cortical lesions in multiple sclerosis. Nat Rev Neurol. 2010;6:438–444. doi: 10.1038/nrneurol.2010.93. [DOI] [PubMed] [Google Scholar]
- 98.Calabrese M, De Stefano N, Atzori M, Bernardi V, Mattisi I, Barachino L, Morra A, Rinaldi L, Romualdi C, Perini P, Battistin L, Gallo P. Detection of cortical inflammatory lesions by double inversion recovery magnetic resonance imaging in patients with multiple sclerosis. Arch Neurol. 2007;64:1416–1422. doi: 10.1001/archneur.64.10.1416. [DOI] [PubMed] [Google Scholar]
- 99.Schutzer SE, Angel TE, Liu T, Schepmoes AA, Xie F, Bergquist J, Vecsei L, Zadori D, Camp DG, 2nd, Holland BK, Smith RD, Coyle PK. Gray matter is targeted in first-attack multiple sclerosis. PLoS ONE. 2013;8:e66117. doi: 10.1371/journal.pone.0066117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lucchinetti CF, Popescu BF, Bunyan RF, Moll NM, Roemer SF, Lassmann H, Bruck W, Parisi JE, Scheithauer BW, Giannini C, Weigand SD, Mandrekar J, Ransohoff RM. Inflammatory cortical demyelination in early multiple sclerosis. N Engl J Med. 2011;365:2188–2197. doi: 10.1056/NEJMoa1100648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med. 2000;6:67–70. doi: 10.1038/71555. [DOI] [PubMed] [Google Scholar]
- 102.Smith T, Groom A, Zhu B, Turski L. Autoimmune encephalomyelitis ameliorated by AMPA antagonists. Nat Med. 2000;6:62–66. doi: 10.1038/71548. [DOI] [PubMed] [Google Scholar]
- 103.Kapoor R, Furby J, Hayton T, Smith KJ, Altmann DR, Brenner R, Chataway J, Hughes RA, Miller DH. Lamotrigine for neuroprotection in secondary progressive multiple sclerosis: a randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Neurol. 2010;9:681–688. doi: 10.1016/S1474-4422(10)70131-9. [DOI] [PubMed] [Google Scholar]
- 104.Vergo S, Craner MJ, Etzensperger R, Attfield K, Friese MA, Newcombe J, Esiri M, Fugger L. Acid-sensing ion channel 1 is involved in both axonal injury and demyelination in multiple sclerosis and its animal model. Brain. 2011;134:571–584. doi: 10.1093/brain/awq337. [DOI] [PubMed] [Google Scholar]
- 105.Arun T, Tomassini V, Sbardella E, de Ruiter MB, Matthews L, Leite MI, Gelineau-Morel R, Cavey A, Vergo S, Craner M, Fugger L, Rovira A, Jenkinson M, Palace J. Targeting ASIC1 in primary progressive multiple sclerosis: evidence of neuroprotection with amiloride. Brain. 2013;136:106–115. doi: 10.1093/brain/aws325. [DOI] [PubMed] [Google Scholar]