

Abstract
Aims
Levodopa-carbidopa intestinal gel (LCIG) provides continuous levodopa-carbidopa delivery through intrajejunal infusion. This study characterized the population pharmacokinetics of levodopa following a 16 h jejunal infusion of LCIG or frequent oral administration of levodopa-carbidopa tablets (LC-oral) in subjects with advanced Parkinson's disease (PD).
Methods
A non-linear mixed-effects model of levodopa pharmacokinetics was developed using serial plasma concentrations from an LCIG phase 1 study and a phase 3 double-blind, double-dummy study of the efficacy and safety of LCIG compared with LC-oral in advanced PD patients (n = 68 for model development; 45 on LCIG and 23 on LC-oral). The final model was internally evaluated using stochastic simulations and bootstrap and externally evaluated using sparse pharmacokinetic data from 311 subjects treated in a long term safety study of LCIG.
Results
The final model was a two compartment model with a transit compartment for absorption, first order elimination, bioavailability for LCIG (97%; confidence interval = 95% to 98%) relative to LC-oral, different first order transit absorption rate constants (LCIG = 9.2 h–1 vs. LC-oral = 2.4 h–1; corresponding mean absorption time of 7 min for LCIG vs. 25 min for LC-oral) and different residual (intra-subject) variability for LCIG (15% proportional error, 0.3 μg ml−1 additive error) vs. LC-oral (29% proportional error, 0.59 μg ml−1 additive error). Estimated oral clearance and steady-state volume of distribution for levodopa were 24.8 l h−1 and 131 l, respectively.
Conclusions
LCIG administration results in faster absorption, comparable levodopa bioavailability and significantly reduced intra-subject variability in levodopa concentrations relative to LC-oral administration.
Keywords: Duodopa, intestinal gel, levodopa, Parkinson's disease, population pharmacokinetics
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Levodopa and carbidopa combination is the primary standard treatment in Parkinson's disease.
Levodopa concentrations fluctuate significantly with oral treatment due to the unpredictable variability of gastric emptying and levodopa's short half-life. This contributes to the troublesome motor complications in advanced Parkinson's disease.
Levodopa-carbidopa intestinal gel (LCIG) was developed to overcome the limitations of oral treatment by providing continuous delivery through intrajejunal infusion.
WHAT THIS STUDY ADDS
This study characterized the population pharmacokinetics of levodopa following jejunal administration of LCIG or oral administration of levodopa-carbidopa to subjects with advanced Parkinson's disease.
Using the pharmacokinetic data from LCIG phase 1 and 3 clinical studies, this analysis compared levodopa bioavailability and the intra-subject variability in levodopa plasma concentrations for the two methods of administration in the target patient population.
Introduction
Parkinson's disease is the second most common neurodegenerative disorder after Alzheimer's disease [1]. The disease is characterized by progressive degeneration of the dopaminergic nigrostriatal system and depletion of dopamine, which results in the core motor symptoms of bradykinesia, rigidity, tremor, and postural instability [2]. Levodopa is the amino-acid precursor of dopamine and replenishes the depleted striatal dopamine. Carbidopa is administered with levodopa to inhibit its extracerebral decarboxylation, allowing more levodopa to cross the blood–brain barrier to target the striatal dopamine receptors [3]. The combination of levodopa and carbidopa is the primary standard of treatment in Parkinson's disease [4,5].
Patients with Parkinson's disease suffer from periods of ‘off’ time (periods of poor mobility, slowness, and stiffness) that may alternate with periods of ‘on’ time (periods of good motor system control without troublesome dyskinesia). In advanced stages of Parkinson's disease, patients treated with oral levodopa may develop troublesome motor fluctuations at the end of each dose (wearing off), unpredictable swings from mobility to immobility (‘on–off’ phenomenon), or levodopa-induced dyskinesia, which closely correlate with fluctuating plasma concentrations of levodopa and pulsatile availability of dopamine [6,7]. Plasma concentrations of orally administered levodopa fluctuate due to its short half-life and the unpredictable variability of gastric emptying [4]. Treatments that offer more continuous dopaminergic stimulation may decrease the risk of development of motor fluctuations and dyskinesias and clinical evidence suggests that chronic (continuous) infusion of levodopa dramatically ameliorates motor fluctuations [8–11].
Levodopa-carbidopa intestinal gel (LCIG) was developed to overcome the limitations of oral levodopa-carbidopa treatment. The LCIG System (Duodopa®) consists of a suspension of levodopa carbidopa monohydrate (4 : 1) in an aqueous gel (carboxymethyl cellulose) that is continuously delivered via a portable infusion pump to the proximal small intestine through a percutaneous endoscopic gastrostomy with jejunal extension (PEG-J). The delivery of LCIG directly to the proximal small intestine results in less variability in levodopa and carbidopa plasma concentrations and provides a continuous rather than intermittent stimulation of the dopaminergic receptors in the brain [9,11,12].
The objective of the present analysis was to characterize the population pharmacokinetics of levodopa following jejunal administration of LCIG or oral administration of immediate release levodopa-carbidopa (LC-oral) to subjects with advanced Parkinson's disease. Using the pharmacokinetic data from LCIG phase 1 and 3 clinical studies and a non-linear mixed-effects modelling approach, this analysis compares levodopa bioavailability and the intra-subject variability in levodopa plasma concentrations for the two methods of administration in the target patient population.
Methods
To characterize the population pharmacokinetics of levodopa following jejunal infusion of LCIG or frequent administration of LC-oral to subjects with advanced Parkinson's disease, levodopa pharmacokinetic data were combined from a phase 1 study and a phase 3 pivotal study of LCIG, where LC-oral was used as a comparator in the latter. Levodopa pharmacokinetic data from an open label phase 3 study of LCIG were used for external evaluation of the population model as described below.
Study designs
Studies included in model development
Study 1
This was a multicentre, open-label pharmacokinetic study of LCIG in subjects with advanced Parkinson's disease. The study design was previously described in detail [12]. Male and female subjects already on stable LCIG (AbbVie, North Chicago, IL, USA) regimens for more than 30 days were enrolled in the study. Nineteen subjects were enrolled in the study and one subject was prematurely discontinued prior to the pharmacokinetic assessment day. LCIG was administered to the jejunum with a portable infusion pump (CADD-Legacy® Duodopa, Smiths Medical, MN, USA) via a percutaneous endoscopic gastrostomy with jejunal extension tube. Subjects were confined in the clinic for 2 days and the infusion duration on each day was 16 h (day −1 and day 1). On day −1, night-time LC-oral was allowed after termination of infusion and up to 3 h prior to the start of the pump on the pharmacokinetic day (day 1).
On the pharmacokinetic day, all subjects received their individualized doses of LCIG. The total daily dose of LCIG was the sum of three components: morning (loading) dose, continuous (maintenance) dose and extra doses. At end of the infusion duration, the jejunal tubes were flushed with 3 ml of water to administer the drug product remaining in the tubing dead space. On the pharmacokinetic assessment day, the total mean (SD) LCIG dose of levodopa was 1580 (403) mg. The morning dose ranged from 4 to 11.5 ml (infused at a rate of 40 ml h–1), corresponding to 80 to 230 mg levodopa. The continuous dose infusion rate ranged from 2.7 to 6.1 ml h–1 (54 to 122 mg levodopa h–1). Extra doses were given if the patient became hypokinetic during the day. Use of extra doses of LCIG was discouraged during the pharmacokinetic sampling day, on which only two subjects received extra doses [two 2 ml (40 mg levodopa) extra doses for one subject and one 5 ml (100 mg levodopa) extra dose dose for another]. Serial blood samples were collected on the pharmacokinetic sampling day immediately prior to the initiation of LCIG infusion and at the following time points after the initiation of infusion: 5 min, immediately after the end of the morning dose, every 30 min up to 8 h (0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 5.5, 6, 6.5, 7, 7.5, 8 h), 12, 16 (immediately after flushing the tube), 17, 18 and 19 h.
Study 2
This was a 12 week, active-controlled, randomized, double-blind, double-dummy, parallel group, multicentre study of the efficacy, safety and tolerability of LCIG in the treatment of levodopa-responsive subjects with advanced Parkinson's disease. Subjects had to have persistent motor fluctuations, despite optimized treatment with oral levodopa-carbidopa in addition to other available anti-Parkinson's disease medications to be eligible for the study. The study design was previously described in detail [11]. Subjects were randomly assigned to treatment with either a) LCIG infusion plus placebo capsules or b) over-encapsulated immediate release levodopa/carbidopa 100/25 mg (LC-oral) plus placebo gel infusion. A total of 71 subjects were randomized to treatment in this study. Of those, 37 subjects were randomly assigned to LCIG infusion plus placebo capsules and 34 subjects were randomly assigned to LC-oral plus placebo gel infusion. The gel was delivered via a percutaneous endoscopic gastrostomy with jejunal extension tube. Dosing of the gel and the capsules was individually optimized by titration to optimal effect (double-titration of capsules and gel to maintain blinding) during the first 4 weeks of the study. Subjects then remained on their individualized doses for the remaining 8 weeks of the study. The study drugs (gel infusion and capsules) were administered over a 16 h period and consisted of a morning dose (infusion and oral capsules) and a continuous infusion of the gel, and a regimen of oral capsules. Additional doses of open-label oral immediate-release levodopa-carbidopa tablets were used to treat acute changes in the subject's Parkinson's disease symptoms. During the 8 h the subjects were not receiving the infusion, they received individualized regimens of oral immediate-release levodopa-carbidopa tablets for symptom control. The mean (SD) daily levodopa dose from the double-blind treatment across the entire study period was 1117 (474) mg day−1 for LCIG and 1351 (618) mg day−1 for LC-oral. The mean (SD) total daily dose of levodopa from all sources was 1164 (483) mg for the LCIG group and 1409 (617) mg for the LC-oral group.
For subjects enrolled early in the study (n = 20), planned pharmacokinetic sampling was on weeks 4 and 12 as follows: at 12, 16, 17, and 18 h post-initiation of intestinal gel infusion on days 28 and 84, and prior to initiation of intestinal gel infusion and after start of infusion at the following time points: 5 min, end of morning dose, and 1, 1.33, 1.67, 2, 2.33, 2.67, 4, 4.33, 4.67, 8, 8.33 and 8.67 h on days 29 and 85.
For subjects enrolled later in the study (n = 51), planned pharmacokinetic sampling was only during week 6 to reduce the burden of the study procedures. For those subjects, pharmacokinetic samples were planned as follows: prior to initiation of intestinal gel infusion and after start of infusion at the following time points: 1, 2, 4, and 8 h on study day 43. Some subjects had additional blood samples collected at 12 and 16 h post-infusion initiation on study day 42.
Study utilized in external evaluation of the model
Study 3
This was a phase 3, open-label, multicentre study of the safety, tolerability and efficacy of LCIG administered for 12 months in subjects with levodopa-responsive advanced Parkinson's disease with severe motor fluctuations despite optimized treatment with available Parkinson's disease medications. A total of 354 subjects were allocated to treatment in this study. The study started with a nasojejunal test period (2 to 14 days) during which subjects received LCIG via a nasojejunal tube. This was followed by percutaneous endoscopic gastrostomy (PEG) surgery and post-PEG long term treatment period through the PEG-J tube. The study design was previously described in detail [13]. The LCIG dose was individually optimized by titration at the start of the nasojejunal phase and again at the start of the PEG-J phase. Once the treatment via PEG-J tube administration was optimized, the 12 month (52 weeks) post-PEG-J long term treatment period began, during which dosing of LCIG was to be changed according to clinical condition. Individually-optimized dosing of LCIG was delivered over a 16 h period, administered as a morning bolus followed by continuous infusion, and if needed, intermittent extra doses (patient-initiated based on symptom experience). Oral levodopa-carbidopa was allowed at night as supplemental night medication after disconnecting the pump or as rescue medication during the day if unable to use the extra dose pump infusion. The overall mean daily dose of levodopa from week 4 to endpoint was 1551 to 1572 mg day−1 from LCIG and 1614 to 1621 mg day−1 from all sources. One blood sample for pharmacokinetic analysis was collected during the post-PEG long term treatment period on days 28, 84, 168, and 378 (four total planned samples per subject).
Bioanalysis
Processing of the pharmacokinetic samples and analysis of levodopa plasma concentrations using liquid chromatography with tandem mass spectrometric detection (LC-MS/MS) were previously described in detail [12]. The analytical method was validated over a concentration range of 10 to 5000 ng ml−1. Across studies, the %CV for levodopa analytical precision was ≤ 11% and the bias was within −6.4 to 4.8%.
Population pharmacokinetic analysis dataset
Sixty-eight male and female subjects with advanced Parkinson's disease who participated in studies 1 or 2 (18 subjects from study 1 and 50 subjects from study 2) and who had available pharmacokinetic data and dosing history information (recorded) during the pharmacokinetic sampling study days were included in the model development. Of the 68 subjects, 45 subjects received LCIG and 23 subjects received LC-oral. Sparse pharmacokinetic data from adult male and female subjects (n = 311) with advanced Parkinson's disease who participated in study 3 and who had plasma concentration and dosing history information during the pharmacokinetic sampling study days (recorded or imputed as described below) were included in the model external evaluation using stochastic simulations.
Dosing records on the pharmacokinetic sampling days and the days immediately preceding pharmacokinetic sampling days were included in the analysis dataset. These dosing records were deemed sufficient to characterize the pharmacokinetics of levodopa since levodopa has a very short half-life (less than 2 h). All dosing records for any formulation containing levodopa-carbidopa (morning, continuous and extra-doses of LCIG, doses associated with LCIG tube flush, night-time, rescue and unclassified LC tablets and LC-oral capsules) were included in the analysis datasets. Dosing records included the amounts, dosing times, rates of infusion and infusion duration (when applicable, doses associated with tube flush were considered bolus). Actual dosing times, durations and sampling times were used in the analysis.
Missing continuous infusion durations (for two occasions in the model development dataset) were assumed to be 16 h, per protocol. No other imputation of dosing information was conducted for the model development dataset. For the external evaluation dataset, missing dosing information on the pharmacokinetic sampling days was imputed from the days prior to pharmacokinetic sampling since dosing was to be stable during this period (309 out of the 311 subjects had imputed dosing at least on one occasion). Missing body weight values for two subjects in the external evaluation dataset were imputed with 70 kg (which approximated the median body weight in the dataset). All levodopa concentrations in the development and evaluation datasets were above the limit of quantitation.
Model development
The population pharmacokinetic model was developed using the non-linear mixed-effects modelling software nonmem (version 7.2; Icon Development Solutions, Ellicott City, MD, USA). The first order conditional estimation method (FOCE) with INTERACTION and a user-defined model (ADVAN6 nonmem Subroutine) were used for the analysis. One and two compartment structural models were evaluated and the two compartment model was found to fit the data better. The two compartment model was parameterized in terms of clearance (CL), volume of central (Vc) and peripheral (Vp) compartments, inter-compartmental clearance (Q) and first order absorption rate constant (Ka). Addition of parameters to the base model was conducted in a stepwise manner. Inclusion of either a lag-time or a transit compartment (between the dosing and central compartments) was evaluated. Inclusion of a relative bioavailability factor (Frel) where Frel was set to 1 for the oral LC formulations (LC-oral capsules and night-time, rescue, or unclassified LC tablets) as the reference and Frel was estimated for LCIG (morning doses, continuous infusion, extra-doses and doses associated with tube flush) was evaluated. Estimation of a different transit rate constant for absorption (Ktr) for the LC-oral treatment was assessed.
Body weight was evaluated as a covariate for CL, Vc, Vp and Q and age was evaluated as a covariate for CL using a power model as described with the following equation
| (1) |
where TVPl is the typical value of evaluated parameter P for a subject with covariate value of l (Covl), PF is a normalization factor (70 kg for body weight and 60 years for age). Accordingly, Pθ2 is a constant that determines the non-linear relationship between the parameter and the normalized covariate value and Pθ1 is the typical parameter value for a subject with 70 kg body weight or 60 years of age. For body weight, an estimated Pθ2 or a fixed value of 0.75 for clearance parameters and 1 for volume parameters (allometric models) were evaluated.
Gender and entacapone [a catechol-O-methyl transferase (COMT) inhibitor; COMT is an enzyme involved in metabolism of levodopa] concomitant use were evaluated as covariates for levodopa clearance via binary indicator variables (0 or 1) as follows:
| (2) |
where CLθ1 is the typical clearance value for the reference group for each binary covariate (males or subjects who did not concomitantly use entacapone) and CLθ2 is the ratio of the clearance of females (CovIND = 1) to males (CovIND = 0) or ratio of clearance of subjects who concomitantly used entacapone (CovIND = 1) to those who did not use entacapone (CovIND = 0) on the pharmacokinetic sampling days.
The majority of subjects in the analysis dataset were of White race. Therefore, race could not be robustly evaluated as a covariate on clearance. Inter-subject variability in CL, Vc and Ktr (or Ka in the starting models) was estimated using an exponential error model as follows:
| (3) |
where ηi is the proportional difference between the parameter estimate of the ith subject (Pi) and the typical population parameter value (TVP) and ηi is assumed to be normally distributed with a mean of 0 and a variance of ω2. Covariance between inter-subject variability in the pharmacokinetic parameter was evaluated using the BLOCK statement in nonmem. The residual random error was modeled using a combined proportional and additive error models as follows:
| (4) |
where Cij is the measured plasma concentration of the ith individual at time j, Ĉij is the corresponding model predicted concentration, and ε1ij and ε2ij are the proportional and additive components, respectively, of the residual random error. Each of the residual error components was assumed to be independently normally distributed with a mean of 0 and variances of σ2: εn ∼ N (0, 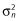 ).
).
Different residual error variances for subjects treated with LCIG (study 1 and arm 1 of study 2) vs. subjects treated with LC-oral (arm 2 of study 2) were evaluated.
Several criteria were used to evaluate the improvement in the model performance and to select the final model. The Likelihood Ratio Test was used for comparing rival hierarchical models where a decrease in nonmem objective function (–2 log likelihood) of 7.88 points was necessary to consider the improvement in model performance statistically significant at α = 0.005 and 1 degree of freedom [14]. The Akaike information criterion (AIC) was used for comparing rival non-hierarchical models [15]. Other selection criteria used included improved goodness of fit and residual plots, increased precision in parameter estimation and reduced variance of inter-subject and residual errors.
Model internal evaluation
Bootstrap evaluation
The robustness of the final model was evaluated using non-parametric bootstrap. In this procedure, subjects were randomly sampled with replacement from the original dataset (with no stratification) to form 1000 new datasets each having the same number of subjects as the original dataset. The final model was then fitted to the bootstrap datasets and all the model parameters were estimated. The median and 95% confidence interval (defined by the 2.5th and 97.5th percentiles) for each parameter were then calculated from the successfully converging runs (regardless of success of the covariance step) and compared with the point estimates from the original dataset.
Visual predictive check
The adequacy of the final model was evaluated using standard visual predictive check (VPC) as well as prediction-corrected VPC (pcVPC) [16]. The final model parameters were used to simulate 500 replicates of the observed data using nonmem. For calculation of summary statistics and graphical display, observed and simulated concentration data were categorized by rounded time after dosing. Time after dosing was defined as the time relative to start of morning infusion of LCIG or time relative to administration of the immediately preceding dose of LC-oral. Time points with low number of concentrations were combined with adjacent time points. Simulated negative plasma concentrations (because of the additive component of the error model) were replaced with zero. In the standard VPC, the observed data as well as calculated statistics [median, 5th percentile (P5) and 95th percentile (P95)] of observed concentrations were compared graphically to the 95% confidence intervals for the median, P5 and P95 of simulated concentrations. The 95% confidence intervals for the median, P5 and P95 of simulated concentrations were calculated from the 2.5th percentile and 97.5th percentiles of each parameter across simulated replicates. In the pcVPC, to normalize for the variability in independent variables (dose, time and body weight) in the graphical display of the predictive performance of the model, the observations and simulations were normalized based on the typical population predictions as follows and previously described [16].
| (5) |
where Yij is the observed or simulated plasma concentration for subject i at time j, pcYij is the prediction-corrected Yij, PREDij is the typical model prediction (PRED) for Yij, PREDbin is the median value of all the PREDs in the bin within which Yij is graphically presented. The PRED-corrected observed and simulated values were then summarized and displayed graphically as described above for the standard VPC.
Model external evaluation
The final model parameters were used to simulate 500 replicates of study 3 pharmacokinetic data using nonmem. Study 3 was not used for the model development. Therefore, this represents an external evaluation of the predictive ability of the model for LCIG. The external evaluation provides no information on the ability of the model to predict data for LC-oral since LC-oral was not evaluated in study 3. Calculation of summary statistics and graphical display of observed and simulated data for study 3 was conducted as described above under visual predictive check. PRED corrections for the observed and simulated concentrations were also conducted.
TIBCO Spotfire S + ® 8.1 for Windows was used for statistical calculations and for graphical display of the results.
Results
Demographics and subject disposition
Summary of the demographic data for the subjects included in the development and evaluation of the levodopa pharmacokinetic model are presented in Table 1. Among the subjects included in model development, 12 subjects reported concomitant use of entacapone on the pharmacokinetic sampling days. For model development and internal evaluation, 1182 levodopa plasma concentrations were available from 68 subjects. For external evaluation of the model, 1041 levodopa plasma concentrations were available from 311 subjects. Four out of the 1041 concentrations were later excluded from the external evaluation because they were clearly erroneous.
Table 1.
Demographic data summary for subjects included in model development and evaluation
| Demographic Characteristic | Model development dataset | External evaluation dataset | ||
|---|---|---|---|---|
| LCIG (n = 45) | LC-oral (n = 23) | Total (n = 68) | LCIG (n = 311)* | |
| Gender, n (%) | ||||
| Male | 28 (62.2) | 14 (60.8) | 42 (61.7) | 177 (57.0) |
| Female | 17 (37.7) | 9 (39.1) | 26 (38.2) | 134 (43.0) |
| Race, n (%) | ||||
| White | 44 (97.7) | 21 (91.3) | 65 (95.5) | 289 (92.9) |
| Other | 1 (2.2) | – | 1 (1.4) | 2 (0.643) |
| Asian | – | 2 (8.6) | 2 (2.9) | 20 (6.4) |
| Age (years) | ||||
| Mean (SD) | 64.3 (9.6) | 64.7 (6.9) | 64.4 (8.7) | 64.2 (9.0) |
| Range | 39–83 | 45–79 | 39–83 | 38–83 |
| Weight (kg) | ||||
| Mean (SD) | 72.8 (16.7) | 74.5 (21.1) | 73.3 (18.1) | 70.8 (15.5) |
| Range | 44.7–135 | 55–148 | 44.7–148 | 39.7–123 |
| BMI (kg m−2) | ||||
| Mean (SD) | 24 (4.58) | 26.1 (7.14) | 24.7 (5.62) | 24.8 (4.6) |
| Range | 17.7–41.7 | 17.7–45.7 | 17.7–45.7 | 15.9–49.9 |
Sparse pharmacokinetic samples (up to four total/subject) were available for subjects in the external evaluation dataset. BMI, body mass index.
Levodopa pharmacokinetic model
The levodopa pharmacokinetic model building history and the associated changes in nonmem objective function are presented in supporting information Table S1. The base model was a two compartment model with a transit compartment for absorption (between the dosing and central compartments) and first order elimination. The model was parameterized in terms of clearance (CL), volume of central compartment (Vc), volume of peripheral compartment (Vp), inter-compartmental clearance (Q) and first-order transit rate constant for absorption (Ktr). Inter-subject variability was estimated for CL, Vc and Ktr using an exponential model. The residual variability was estimated using a combined additive and proportional error models. The transit compartment model was selected over the lag-time model since the transit compartment model has one less parameter and transit models are generally more numerically stable than lag-time models. The two models (lag time and transit) provided comparable objective functions once relevant covariates were incorporated (supporting information Table S1). Inclusion of formulation (LCIG vs. oral LC) as a covariate on bioavailability significantly improved the model fit (ΔOF = −28.60, P < 0.005). A relative bioavailability factor (Frel) was included in the model where Frel was set to 1 for the oral LC formulations (LC-oral capsules and night-time, rescue, or unclassified LC tablets) as the reference and Frel was estimated for LCIG (morning doses, continuous infusion, extra doses and doses associated with tube flush). The estimated relative bioavailability for LCIG in the final model was 97% (95% bootstrap confidence interval: 95 to 98%). Estimation of a different transit rate constant for absorption (Ktr) for the LC-oral treatment arm of study 2 resulted in further significant reduction in the nonmem objective function (ΔOF = −13.14, P < 0.005). Inclusion of body weight (normalized to 70 kg) as a covariate for Vc using an allometric model with a fixed exponent of 1 improved the model fit (ΔOF = −9.13, P < 0.005). Inclusion of body weight as a covariate on levodopa clearance did not improve the model fit to any appreciable extent (3.5 and 5.9 points reduction in nonmem objective function for fixed 0.75 or estimated allometric exponent for relationship between levodopa clearance and body weight, respectively, P > 0.01). Additionally, levodopa clearance was not found to be statistically significantly correlated with gender of the subject or concomitant use of the catechol-O-methyl transferase inhibitor, entacapone (P > 0.01). Age almost reached significance for inclusion as a covariate for levodopa clearance (ΔOF = –7.65, P = 0.0057). Estimating different residual error variances for subjects treated with LCIG (Study 1 and treatment arm 1 of study 2) vs. subjects treated with LC-oral (arm 2 of study 2) significantly improved the model fit (ΔOF = −189, Table 5). Finally, estimating covariance between inter-subject variability in CL and Ktr resulted in further reduction in the objective function (ΔOF = −7.93, P < 0.005, Table 5). Using a full covariance matrix for inter-subject variability in CL, Vc and Ktr did not result in further significant improvement in the model fit.
Diagnostic plots for the final model are presented in Figure 1A–1D. Scatter plots of population predicted vs. observed levodopa concentration (Figure 1A) and the individual predicted vs. observed levodopa concentrations (Figure 1B) showed symmetric distribution around the line of identity and good agreement between observed and individual predicted concentrations, indicating that the model fit the data well at the population and individual subjects levels. Scatter plots of the conditional weighted residuals [17] vs. population predicted levodopa concentrations or time did not show any systematic bias in the model fit.
Figure 1.

Diagnostic plots of the final levodopa pharmacokinetic model. (A) observed vs. population predicted plasma concentration; (B) observed vs. individual predicted plasma concentrations; (C) conditional weighted residuals (CWRES) vs. population predicted concentrations; (D) conditional weighted residuals (CWRES) vs. time. Solid lines represent lines of identity in A and B and zero conditional residuals in C and D. Dashed lines represent ± 3 SD in C and D
The shrinkage of inter-subject variability (ETA shrinkage) [18] for CL/F, Vc/F and Ktr was 8%, 25% and 24%, respectively. The shrinkage of the proportional and additive intra-subject variability (Epsilon Shrinkage) was 6% for LCIG and 5% for LC-oral.
The estimated levodopa pharmacokinetic parameters and their associated variability are presented in Table 2.
Table 2.
Parameter estimates for the final levodopa population pharmacokinetic model and results of the bootstrap evaluation
| Parameter | Point estimate (%RSE)* | Bootstrap Median [95% CI]† |
|---|---|---|
| Ktr (h–1) | ||
| LCIG | 9.2 (19) | 9.7 [6.0 to 14.3] |
| LC-oral | 2.4 (30) | 2.2 [0.85 to 5.2] |
| CL/F (l h−1) | 24.8 (5) | 24.4 [20.4 to 26.8] |
| Vc/F (l) | 58.5 (11)* WT(kg)/70 | 56.0 [36.6 to 71.0]* WT(kg)/70 |
| Q/F (l h−1) | 6.8 (22) | 7.9 [4.1 to 17.2] |
| Vp/F (l) | 72.9 (49) | 80.3 [22.9 to 408] |
| Frel | ||
| LCIG | 0.97 (1) | 0.97 [0.95 to 0.98] |
| LC-oral | 1 Fixed | 1 Fixed |
| ISVKtr (%) | 88 (16) | 79 [36 to 113] |
| ISVCL (%) | 32 (9) | 32 [27 to 39] |
| ISVVc (%) | 61 (20) | 58 [31 to 97] |
| CorrelationKtr,CL (%) | −47 (16) | −45 [–75 to −3.7] |
| Proportional error (%) | ||
| LCIG | 15 (15) | 15 [9 to 23] |
| LC-oral | 29 (14) | 29 [0.1 to 37] |
| Additive error SD (μg ml−1) | ||
| LCIG | 0.30 (20) | 0.29 [0.07 to 0.44] |
| LC-oral | 0.59 (15) | 0.59 [0.30 to 0.81] |
nonmem point estimate and the associated % relative standard error (% RSE).
The median and 95% confidence interval (2.5th and 97.5th percentiles) calculated from the parameter estimates of the successfully converging runs (977) of the 1000 bootstrap datasets. Additive Error SD, additive residual error standard deviation (σadd); CI, confidence interval; ISV, inter-subject variability calculated as ω * 100; Proportional error, proportional residual error calculated as σprop*100.
Model evaluation
Internal evaluation
The final model converged for 977 out of the 1000 bootstrap datasets (of those, covariance step was complete for 856 datasets). The median and 95% confidence intervals (calculated as the 2.5th and 97.5th percentiles) of the bootstrap parameter estimates are presented in Table 2. The median parameter estimates from the bootstrap datasets were comparable with the point estimates from the original dataset. Additionally, the bootstrap calculated uncertainty in the fixed effects parameter estimates were generally in agreement with the uncertainty in the estimates from the original dataset.
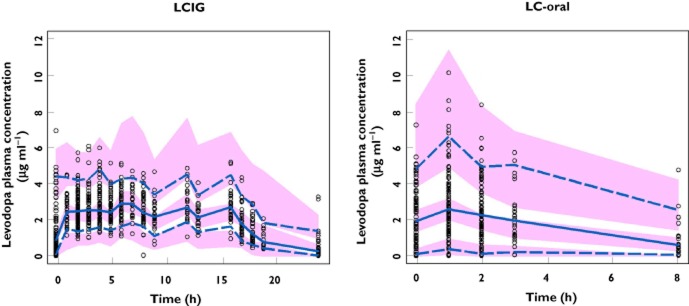
The standard VPC for the final model stratified by treatment is presented in Figure 2. The prediction-corrected VPC is presented in Figure 3. Using the final model, there was good agreement between the observed and simulated levodopa plasma concentrations with the medians, 5th and 95th percentiles of observed levodopa plasma concentrations falling within the simulated confidence intervals for these parameters during the majority of the time course, particularly after normalizing for the variability in the independent variables (doses, time of sampling, body weight differences) using the PRED correction (Figure 3).
Figure 2.
Standard visual predictive check stratified by treatment for the final pharmacokinetic model. ○, observed concentration;  , observed median;
, observed median;  , observed P5 and P95;
, observed P5 and P95;  , 95% CI for simulated median, P5 and P95
, 95% CI for simulated median, P5 and P95
Figure 3.
Prediction-corrected visual predictive check stratified by treatment for the final pharmacokinetic model. ○, observed;  , observed median;
, observed median;  , observed P5 and P95;
, observed P5 and P95;  , 95% CI for simulated median, P5 and P95
, 95% CI for simulated median, P5 and P95
External evaluation
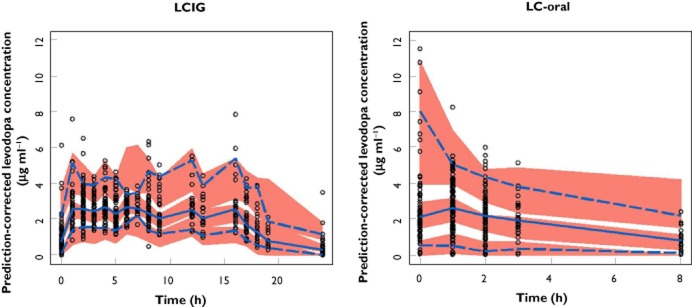
The observed and final model predicted levodopa plasma concentrations for study 3 are presented in Figure 4. Study 3 was not included in the dataset used for model development. Therefore, Figure 4 represents an external evaluation of the predictive ability of the final model for LCIG. There was good agreement between the observed and simulated levodopa plasma concentrations for the external evaluation dataset with the medians, 5th and 95th percentiles of observed levodopa plasma concentrations falling within the simulated confidence intervals for these parameters during the majority of the time points, particularly after normalizing for the variability in the independent variables using the PRED correction (Figure 5).
Figure 4.
External evaluation of the predictive performance of the final pharmacokinetic model using study 3. Observed and simulated levodopa concentrations are depicted vs. time. ○, observed concentration; , observed median;
, observed median;  , observed 5th and 95th percentiles;
, observed 5th and 95th percentiles;  , 95% CI for simulated median, 5th and 95th percentiles
, 95% CI for simulated median, 5th and 95th percentiles
Figure 5.
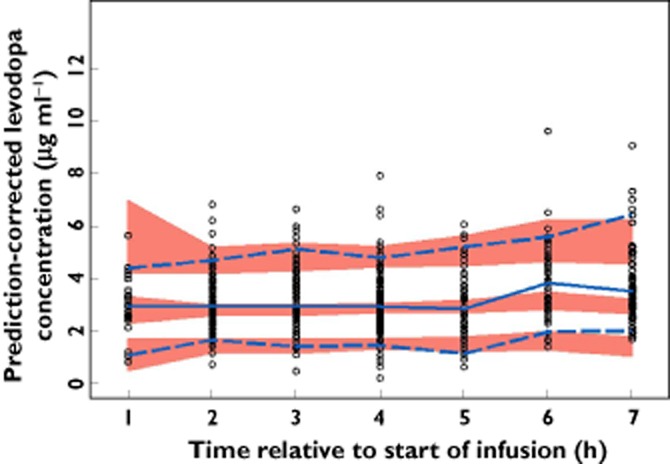
Prediction-corrected observed and simulated levodopa concentrations for study 3 vs. time. The typical prediction correction normalizes for the differences in independent variables within a time bin allowing for better visualization of the model predictive performance. ○, observed;  , observed median;
, observed median;  , observed 5th and 95th percentiles;
, observed 5th and 95th percentiles;  , 95% CI for simulated median, 5th and 95th percentiles
, 95% CI for simulated median, 5th and 95th percentiles
Discussion
The present analysis characterized the population pharmacokinetics of levodopa following jejunal infusion of LCIG or frequent oral administration of immediate release levodopa-carbidopa (LC-oral) tablets in subjects with advanced Parkinson's disease. The analysis utilized combined levodopa intensive pharmacokinetic data from 68 subjects who participated in a phase 1 pharmacokinetic study or a phase 3 double-blind double-dummy efficacy study of LCIG. The developed model compares levodopa absorption, bioavailability and intra-subject variability for the two treatment modalities in the target patient population for LCIG use.
Results from the present analysis demonstrate that LCIG has comparable bioavailability to the oral levodopa-carbidopa 100/25 mg immediate release (IR) tablets (over-encapsulated Sinemet tablets used as active control in the LCIG phase 3 study). The bioavailability estimate for levodopa from LCIG relative to LC-oral was 97% (95% bootstrap confidence interval of 95% to 98%). Comparable levodopa bioavailability for LCIG and LC-oral is in agreement with direct delivery of levodopa to the proximal small intestine with LCIG infusion, where the expression of the large neutral amino acid transporter, the absorptive carrier of levodopa, is highest [12]. No previous studies have been conducted to characterize directly the absolute bioavailability of levodopa from LCIG or the bioavailability of LCIG relative to immediate release oral levodopa-carbidopa. With standard LC-oral (4 : 1 ratio) tablets, Yeh et al. reported an absolute levodopa bioavailability of 84 ± 13% in healthy young subjects and 99 ± 21% in elderly subjects [19]. A pooled analysis from 20 patients across three small LCIG studies by Westin et al. suggested that LCIG has an absolute bioavailability of 88% with duodenal delivery [20]. The analysis by Westin et al. had the limitation of fixing levodopa disposition parameters to values reported in the literature for intravenous infusion in different patients. It is also noteworthy that the majority of patients who contributed to the dataset analyzed by Westin et al. received LCIG through a nasoduodenal tube, not through PEG-J as in the current clinical practice. Overall, results from the present analysis (directly) and the analysis by Westin et al. (indirectly) demonstrate that delivery of LCIG to the proximal small intestine (jejunum or duodenum) results in comparable bioavailability to oral levodopa-carbidopa.
The results of the present analysis also suggest that LCIG is absorbed faster than LC-oral, consistent with skipping residence in the stomach with LCIG jejunal delivery. The first order absorption transit rate constant was estimated to be 9.2 h–1 for LCIG and 2.4 h–1 for LC-oral (corresponding mean absorption time of 7 min for LCIG vs. 25 min for LC-oral; estimated from the entire pharmacokinetic dataset regardless of food intake for both treatments). In a typical patient simulation, this difference corresponds to a 30 min earlier tmax for a 200 mg morning infusion of LCIG compared with a 200 mg oral dose (tmax 45 min for LCIG vs. 75 min for LC-oral).
Both one compartment [21,22] and two compartment [23,24] structural models have been previously used to describe the pharmacokinetics of levodopa administered with a decarboxylase inhibitor. In the present analysis, a two compartment model described levodopa disposition better than a one compartment model. Body weight was a statistically significant covariate for levodopa Vc (Vc allometrically scaled on 70 kg normalized body weight with an exponent of 1). Inclusion of body weight as a covariate for levodopa clearance did not improve the model fit to any appreciable extent. Similarly, Jorga et al. reported body weight as a significant covariate for levodopa volume of distribution, but not clearance [22]. Chan et al. included body weight as a covariate on all levodopa disposition parameters in their analysis as a standard approach supported by the principles of allometry [24]. However, the results from Chan et al. also suggest that body weight explained more of the between-subject variability in levodopa's Vc than in levodopa's total clearance. In our analysis, introduction of body weight as a covariate on all the clearance and volume parameters resulted in more deviation of the remaining inter-subject variability (η) for clearance from the normality assumption (data not shown). Therefore, body weight was retained as a covariate for the volume of distribution only in the final model.
Gender was not found to be a statistically significant covariate for levodopa clearance in the present analysis. This is in agreement with the analysis by Chan et al. [24]. Jorga et al. suggested that gender is not a statistically significant covariate for levodopa clearance in subjects without motor fluctuations but it is a statistically significant covariate for levodopa clearance in subjects with motor fluctuations [22]. The presence of a gender-related effect on levodopa pharmacokinetics has been in evaluated in several other studies using non-compartmental approaches and conflicting evidence has been reported [25–27]. It has been suggested that the apparent gender-related difference in exposure was partly because of incorrectly normalizing for body weight difference between males and females in some analyses [27]. Based on the present analysis and previously reported population analyses of levodopa [22,23], there is no clinically meaningful gender-related difference in levodopa clearance.
Earlier studies suggested modest reduction of levodopa clearance with increase in age [28,29]. The population analyses by Jorga et al. and Chan et al. did not find age as a significant covariate for explaining inter-subject variability in levodopa clearance [22,23]. In the present analysis, age was not included as a covariate for clearance in the final model but it almost reached the set criterion for inclusion in the model (P = 0.0057). Finally, no statistically significant relationship was found between concomitant use of the catechol-O-methyl transferase inhibitor, entacapone, and levodopa clearance. However, few subjects in the analysis dataset reported concomitant use of entacapone on the pharmacokinetic sampling days. Therefore, the available limited information does not allow for a meaningful conclusion.
The estimated value of levodopa oral clearance in the present analysis (24.8 l h−1, 95% bootstrap confidence interval 20.4 to 26.8 l h−1) is in middle of the previously reported range in the literature (7.7–38.5 l h−1 as summarized by Chen et al. [23]). Levodopa steady-state volume of distribution (Vc + Vp) (131 l, 95% bootstrap confidence interval of 63.6 to 461 l) is at the upper end of the reported range of 15.4 to 124 l across different analysis methodologies [23]. The between subject variability in levodopa oral clearance and volume of distribution in the present analysis (32 and 61%, respectively) are in line with previously reported values by Jorga et al. for oral administration (26 to 33% for levodopa clearance and 42 to 80% for volume of distribution).
In the final model, administration of LCIG was estimated to be associated with approximately half the intra-subject variability in levodopa concentrations compared with administration of oral levodopa-carbidopa in subjects with advanced Parkinson's disease. The estimated proportional residual error (first component of intra-subject variability) was 15% for LCIG vs. 29% for LC-oral. The estimated standard deviation of the additive residual error in levodopa concentrations (second component of intra-subject variability) was 0.3 μg ml−1 for LCIG vs. 0.59 μg ml−1 for LC-oral. These results are consistent with previous findings by Nyholm et al. which demonstrated that the average intra-subject coefficient of variation, calculated using standard statistical methods, for the plasma levodopa concentrations was 14% during continuous duodenal infusion of LCIG vs. 34% during oral therapy (P < 0.01) [9].
In summary, we developed a population model that describes levodopa pharmacokinetics with LCIG jejunal infusion or LC-oral administration in subjects with advanced Parkinson's disease. The analysis results demonstrate that levodopa bioavailability is comparable for LCIG infusion and LC-oral administration. LCIG jejunal infusion results in approximately half the intra-subject variability in levodopa concentrations observed with LC-oral administration. LCIG is absorbed faster than LC-oral, consistent with direct delivery of LCIG to the jejunum. The developed model was robust and replicated the features of the data from which it was built in simulations. In addition, the model was able to predict adequately levodopa plasma concentrations for a study of LCIG that was not utilized in model development. The model will be a useful research tool and can help optimize dosing with LCIG.
Competing Interests
Both authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: the studies presented in this manuscript were sponsored by AbbVie. AbbVie contributed to the study design, research, and interpretation of data, writing, reviewing, and approving the publication. Drs Othman and Dutta are employees of AbbVie and are shareholders of AbbVie and Abbott Laboratories. The authors declare no other relationships or activities that could appear to have influenced the submitted work.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Table S1 Levodopa population pharmacokinetic model building history and associated changes in nonmem Objective Function (OF)
References
- 1.Nussbaum RL, Ellis CE. Alzheimer's disease and Parkinson's disease. N Engl J Med. 2003;348:1356–1364. doi: 10.1056/NEJM2003ra020003. [DOI] [PubMed] [Google Scholar]
- 2.Hornykiewicz O. Basic research on dopamine in Parkinson's disease and the discovery of the nigrostriatal dopamine pathway: the view of an eyewitness. Neurodegener Dis. 2008;5:114–117. doi: 10.1159/000113678. [DOI] [PubMed] [Google Scholar]
- 3.Calne DB, Reid JL, Vakil SD, Rao S, Petrie A, Pallis CA, Gawler J, Thomas PK, Hilson A. Idiopathic Parkinsonism treated with an extracerebral decarboxylase inhibitor in combination with levodopa. Br Med J. 1971;3:729–732. doi: 10.1136/bmj.3.5777.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nutt JG. Pharmacokinetics and pharmacodynamics of levodopa. Mov Disord. 2008;23(Suppl. 3):S580–584. doi: 10.1002/mds.22037. [DOI] [PubMed] [Google Scholar]
- 5.Agid Y, Ahlskog E, Albanese A, Calne D, Chase T, De Yebenes J, Factor S, Fahn S, Gershanik O, Goetz C, Koller W, Kurth M, Lang A, Lees A, Lewitt P, Marsden D, Melamed E, Michel PP, Mizuno Y, Obeso J, Oertel W, Olanow W, Poewe W, Pollak P, Tolosa E. Levodopa in the treatment of Parkinson's disease: a consensus meeting. Mov Disord. 1999;14:911–913. doi: 10.1002/1531-8257(199911)14:6<911::aid-mds1001>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 6.Fahn S. How do you treat motor complications in Parkinson's disease: medicine, surgery, or both? Ann Neurol. 2008;64(Suppl. 2):S56–64. doi: 10.1002/ana.21453. [DOI] [PubMed] [Google Scholar]
- 7.Shoulson I, Glaubiger GA, Chase TN. On-off response. Clinical and biochemical correlations during oral and intravenous levodopa administration in parkinsonian patients. Neurology. 1975;25:1144–1148. doi: 10.1212/wnl.25.12.1144. [DOI] [PubMed] [Google Scholar]
- 8.Olanow CW, Obeso JA, Stocchi F. Continuous dopamine-receptor treatment of Parkinson's disease: scientific rationale and clinical implications. Lancet Neurol. 2006;5:677–687. doi: 10.1016/S1474-4422(06)70521-X. [DOI] [PubMed] [Google Scholar]
- 9.Nyholm D, Askmark H, Gomes-Trolin C, Knutson T, Lennernas H, Nystrom C, Aquilonius SM. Optimizing levodopa pharmacokinetics: intestinal infusion versus oral sustained-release tablets. Clin Neuropharmacol. 2003;26:156–163. doi: 10.1097/00002826-200305000-00010. [DOI] [PubMed] [Google Scholar]
- 10.Nilsson D, Nyholm D, Aquilonius SM. Duodenal levodopa infusion in Parkinson's disease – long-term experience. Acta Neurol Scand. 2001;104:343–348. doi: 10.1034/j.1600-0404.2001.00153.x. [DOI] [PubMed] [Google Scholar]
- 11.Olanow CW, Kieburtz K, Odin P, Espay AJ, Standaert DJ, Fernandez HH, Vanagunas A, Othman AA, Widnell KL, Robieson WZ, Pritchett Y, Chatamra K, Benesh J, Lenz RA, Antonini A LCIG Horizon Study Group. Continuous intrajejunal infusion of levodopa-carbidopa intestinal gel for patients with advanced Parkinson's disease: a randomised, controlled, double-blind, double-dummy study. Lancet Neurol. 2014;13:141–149. doi: 10.1016/S1474-4422(13)70293-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nyholm D, Odin P, Johansson A, Chatamra K, Locke C, Dutta S, Othman AA. Pharmacokinetics of levodopa, carbidopa, and 3-O-methyldopa following 16-hour Jejunal infusion of Levodopa-Carbidopa Intestinal Gel in advanced Parkinson's disease patients. AAPS J. 2013;15:316–323. doi: 10.1208/s12248-012-9439-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fernandez HH, Vanagunas A, Odin P, Espay AJ, Hauser RA, Standaert DG, Chatamra K, Benesh J, Pritchett Y, Hass SL, Lenz RA. Levodopa-carbidopa intestinal gel in advanced Parkinson's disease open-label study: interim results. Parkinsonism Relat Disord. 2013;19:339–345. doi: 10.1016/j.parkreldis.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sheiner LB, Ludden TM. Population pharmacokinetics/dynamics. Annu Rev Pharmacol Toxicol. 1992;32:185–209. doi: 10.1146/annurev.pa.32.040192.001153. [DOI] [PubMed] [Google Scholar]
- 15.Beal SL, Sheiner LB. NONMEM Users Guide, Part V. San Francisco, CA: Division of Clinical Pharmacology, University of California; 1979. –1992. [Google Scholar]
- 16.Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011;13:143–151. doi: 10.1208/s12248-011-9255-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hooker AC, Staatz CE, Karlsson MO. Conditional weighted residuals (CWRES): a model diagnostic for the FOCE method. Pharm Res. 2007;24:2187–2197. doi: 10.1007/s11095-007-9361-x. [DOI] [PubMed] [Google Scholar]
- 18.Savic RM, Karlsson MO. Importance of shrinkage in empirical bayes estimates for diagnostics: problems and solutions. AAPS J. 2009;11:558–569. doi: 10.1208/s12248-009-9133-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yeh KC, August TF, Bush DF, Lasseter KC, Musson DG, Schwartz S, Smith ME, Titus DC. Pharmacokinetics and bioavailability of Sinemet CR: a summary of human studies. Neurology. 1989;39:25–38. [PubMed] [Google Scholar]
- 20.Westin J, Nyholm D, Palhagen S, Willows T, Groth T, Dougherty M, Karlsson MO. A pharmacokinetic-pharmacodynamic model for duodenal levodopa infusion. Clin Neuropharmacol. 2011;34:61–65. doi: 10.1097/WNF.0b013e31820b570a. [DOI] [PubMed] [Google Scholar]
- 21.Triggs EJ, Charles BG, Contin M, Martinelli P, Cortelli P, Riva R, Albani F, Baruzzi A. Population pharmacokinetics and pharmacodynamics of oral levodopa in parkinsonian patients. Eur J Clin Pharmacol. 1996;51:59–67. doi: 10.1007/s002280050161. [DOI] [PubMed] [Google Scholar]
- 22.Jorga K, Banken L, Fotteler B, Snell P, Steimer JL. Population pharmacokinetics of levodopa in patients with Parkinson's disease treated with tolcapone. Clin Pharmacol Ther. 2000;67:610–620. doi: 10.1067/mcp.2000.106795. [DOI] [PubMed] [Google Scholar]
- 23.Chan PL, Nutt JG, Holford NH. Importance of within subject variation in levodopa pharmacokinetics: a 4 year cohort study in Parkinson's disease. J Pharmacokinet Pharmacodyn. 2005;32:307–331. doi: 10.1007/s10928-005-0039-x. [DOI] [PubMed] [Google Scholar]
- 24.Chan PL, Nutt JG, Holford NH. Modeling the short- and long-duration responses to exogenous levodopa and to endogenous levodopa production in Parkinson's disease. J Pharmacokinet Pharmacodyn. 2004;31:243–268. doi: 10.1023/b:jopa.0000039566.75368.59. [DOI] [PubMed] [Google Scholar]
- 25.Kompoliti K, Adler CH, Raman R, Pincus JH, Leibowitz MT, Ferry JJ, Blasucci L, Caviness JN, Leurgans S, Chase WM, Yones LC, Tan E, Carvey P, Goetz CG. Gender and pramipexole effects on levodopa pharmacokinetics and pharmacodynamics. Neurology. 2002;58:1418–1422. doi: 10.1212/wnl.58.9.1418. [DOI] [PubMed] [Google Scholar]
- 26.Martinelli P, Contin M, Scaglione C, Riva R, Albani F, Baruzzi A. Levodopa pharmacokinetics and dyskinesias: are there sex-related differences? Neurol Sci. 2003;24:192–193. doi: 10.1007/s10072-003-0125-z. [DOI] [PubMed] [Google Scholar]
- 27.Chen C, Cowles VE, Sweeney M, Stolyarov ID, Illarioshkin SN. Pharmacokinetics of levodopa/carbidopa delivered from gastric-retentive extended-release formulations in patients with Parkinson's disease. J Clin Pharmacol. 2012;52:1069–1077. doi: 10.1177/0091270011409232. [DOI] [PubMed] [Google Scholar]
- 28.Contin M, Riva R, Martinelli P, Albani F, Baruzzi A. Effect of age on the pharmacokinetics of oral levodopa in patients with Parkinson's disease. Eur J Clin Pharmacol. 1991;41:463–466. doi: 10.1007/BF00626370. [DOI] [PubMed] [Google Scholar]
- 29.Robertson DR, Wood ND, Everest H, Monks K, Waller DG, Renwick AG, George CF. The effect of age on the pharmacokinetics of levodopa administered alone and in the presence of carbidopa. Br J Clin Pharmacol. 1989;28:61–69. doi: 10.1111/j.1365-2125.1989.tb03506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Levodopa population pharmacokinetic model building history and associated changes in nonmem Objective Function (OF)