

Abstract
Aim
Ipilimumab is a fully human, monoclonal antibody that blocks cytotoxic T-lymphocyte antigen-4. The objective of the present study was to characterize the clinical pharmacology profile of ipilimumab using a population pharmacokinetic (PPK) approach.
Methods
The PPK model was developed using 2095 ipilimumab serum concentration values from 499 patients with unresectable stage III or IV melanoma from four phase II studies, with ipilimumab doses ranging from 0.3 to 10 mg kg−1. The structural PK model was determined by developing a base PPK model. The effect of covariates on model parameters was assessed by a full covariate model, which incorporated all pre-specified covariate-parameter relationships into the base model. The final model was developed by backward elimination, followed by exclusion of covariates determined not to be of clinical relevance to ipilimumab, and was rigorously validated against both internal and external datasets.
Results
Ipilimumab PK was linear and time-invariant, with dose-proportional exposures over the available dose range, yielding a terminal half-life of approximately 15 days. Clearance of ipilimumab increased with increasing body weight and baseline serum lactate dehydrogenase concentrations, but was not affected by age, gender, concomitant budesonide, Eastern Cooperative Oncology Group performance status or prior systemic anticancer therapy. Furthermore, ipilimumab exposure was not affected by moderate renal impairment or mild hepatic impairment.
Conclusions
Ipilimumab concentration–time data were well described by a linear, two compartment, zero order i.v. infusion model. The model confirms that a body weight-normalized dosing regimen is appropriate for ipilimumab therapy in patients with advanced melanoma.
Keywords: cytotoxic T-lymphocyte antigen-4, dose–response relationship, drug effects, immunotherapy, monoclonal antibody
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Ipilimumab is an immuno-oncology monoclonal antibody that is currently approved in more than 40 countries for the treatment of unresectable or metastatic melanoma.
Exposure–response relationships of the efficacy and safety of ipilimumab have been reported, with exposure data having been determined by the current population pharmacokinetic (PPK) analyses.
WHAT THIS STUDY ADDS
This is the first peer-reviewed report of ipilimumab clinical PK.
The report describes the development, evaluation and application of a robust PPK model to support statements in clinical pharmacology sections in prescriber information (drug label).
The report also introduces an informative graphical assessment of covariate effects on PK parameters is presented.
Introduction
Ipilimumab is a first-in-class immuno-oncology monoclonal antibody that augments the ability of the adaptive immune system to target tumour cells. It specifically blocks CD80 and CD86 ligands on antigen-presenting cells (APCs) from binding to the cytotoxic T-lymphocyte antigen-4 (CTLA-4) receptor on activated T cells, thereby preventing the down-regulation of antitumour T cell activity [1–3]. Activation of T cells requires two signals: recognition of tumour antigen on APCs by T cell receptors, and the engagement of CD80 or CD86 by CD28 receptors on T cells. CTLA-4 has a higher affinity for CD80 and CD86 than does CD28, and a natural mechanism of regulating activated T cells is by disengagement of the second signal by expression of CTLA-4. Thus, ipilimumab sustains the proliferation and antitumour effect of T cells by blocking the binding of CD80 and CD86 to CTLA-4.
In two phase III clinical trials, ipilimumab improved overall survival (OS) in patients with advanced (unresectable stage III or IV) melanoma, one in previously treated patients with ipilimumab monotherapy at 3 mg kg−1 [4] and the other in previously untreated patients with ipilimumab at 10 mg kg−1 in combination with dacarbazine [5]. The most common treatment-related adverse events with ipilimumab were immune-related [4,5], which may reflect its immune-based mechanism of action. Ipilimumab has been approved for use in over 40 countries including the United States, the European Union, and Australia. A phase III study comparing the efficacy and safety of 3 vs. 10 mg kg−1 ipilimumab monotherapy in patients with advanced melanoma is ongoing [6].
Where approved, the ipilimumab clinical pharmacology sections of the prescriber information are largely based on the population pharmacokinetic (PPK) analyses results reported here [7,8]. Non-compartmental analysis (NCA) was not performed as the PK data were from patients with sparse sampling that were insufficient to determine PK parameters by NCA adequately. This report describes the development, validation, and application of the PPK model, including the assessment of the potential effect of intrinsic and extrinsic factors on ipilimumab PK and exposure. In addition, we introduce a covariate effect plot that enables a more intuitive assessment of the clinical relevance of these factors on ipilimumab PK than the conventional tabular listing. The PPK model was applied to assess the impact of the potentially clinically relevant covariates as well as renal and hepatic impairment on ipilimumab exposure. The PPK model was also applied to determine the ipilimumab exposures of individual subjects, which were used to characterize exposure–response relationships of efficacy and safety [9]. The exposure–response analyses found that higher steady-state trough concentrations were associated with increased tumour responses, longer survival, as well as increased rates of immune-related adverse events, thereby informing the benefit–risk assessment of the recommended ipilimumab dose regimen.
Methods
Data
The PPK model was developed and validated with data from phase II studies in patients with advanced melanoma. Specifically, the PPK model was developed using an index data set of 1767 ipilimumab serum concentration values from 420 patients in three phase II studies (CA184-007, CA184-008, and CA184-022) [10–12], and was validated with an external data set of 328 serum concentration values from 79 patients enrolled in a fourth phase II study (CA184-004) [13]. The baseline demographic, laboratory and disease status variables in the index and external validation data sets are summarized in Table 1.
Table 1.
Summary of baseline demographic and laboratory covariates*
| Covariate | PPK analyses index dataset n = 420 | PPK external validation dataset n = 79 |
|---|---|---|
| Continuous | Mean (SD) | |
| Age (years) | 57.75 (12.91) | 55.13 (14.75) |
| Body weight (kg) | 80.11 (16.87) | 78.71 (16.58) |
| eGFR (ml min−1 1.73−1 m−2)* | 86.66 (25.78) | 96.72 (27.65) |
| Direct bilirubin (mg dl−1) | 0.16 (0.15) | 0.17 (0.22) |
| Total bilirubin (mg dl−1) | 0.48 (0.27) | 0.53 (0.38) |
| Lactate dehydrogenase (IU l−1) | 326.74 (375.19) | 229.82 (227.92) |
| Alanine aminotransferase (IU l−1) | 23.65 (18.56) | 26.63 (18.32) |
| Categorical | n (%) | |
|---|---|---|
| Gender | ||
| Male | 263 (62.62) | 49 (62.03) |
| Female | 157 (37.38) | 30 (37.97) |
| Baseline ECOG performance status | ||
| 0 | 273 (65.00) | 50 (63.30) |
| 1 | 145 (34.52) | 29 (36.70) |
| 2 | 2 (0.48) | |
| Concomitant budesonide | ||
| No | 362 (86.19) | 79 (100) |
| Yes | 58 (13.81) | 0 (0) |
| Prior immunotherapy | ||
| No | 189 (45.00) | 32 (40.51) |
| Yes | 231 (55.00) | 47 (59.49) |
| Prior IL-2 therapy | ||
| No | 298 (70.95) | 47 (59.49) |
| Yes | 122 (29.05) | 32 (40.51) |
| Immunogenicity† | ||
| Positive | 18 (4.29) | 0 (0) |
| Negative | 402 (95.71) | 79 (100) |
| Prior systemic anticancer therapy | ||
| Previously untreated | 51 (12.14) | 28 (35.44) |
| Previously treated | 369 (87.86) | 51 (64.56) |
| HLA-A*201 genotype | ||
| Negative | 302 (60.64) | 38 (48.10) |
| Positive | 196 (39.36) | 41 (58.90) |
eGFR was computed using the following MDRD formula [13]: GFR(ml min−1 1.73 m−2) = 186.3 × SCr(mg dL−1)−1.154 × Age−0.203 × F2 × F3. Where, SCr is serum creatinine, F2 = 1.0 for men and 0.742 for women. F3 = 1.0 for non-African Americans and 1.212 for African Americans.
Immunogenicity positive referred to subjects who have at least one ADA positive at baseline or post-ipilimumab treatment, and immunogenicity negative referred to subjects who did not have any ADA negative.
Previously treated or untreated patients received ipilimumab at 10 mg kg−1 in CA184-007, with or without budesonide (investigated as a prophylactic agent for gastrointestinal toxicity) [10], and at 3 or 10 mg kg−1 in CA184-004 [13]. Heavily pretreated patients received ipilimumab at 10 mg kg−1 in CA184-008 [11], and patients who progressed on or were intolerant to prior therapy were randomized to ipilimumab at 0.3, 3 or 10 mg kg−1 in CA184-022 [12]. In all studies, ipilimumab was given every 3 weeks for up to four doses (induction phase), followed by maintenance therapy (every 12 weeks beginning at week 24) in eligible patients. PK samples were collected after the first and third induction doses. These studies were approved by the investigational review boards at the participating study sites and patients gave informed consent to participate.
An informative PK sampling scheme was specified in the phase II studies, based on an optimal sampling methodology [14]. PK samples were collected prior to the first and third induction doses on day 1 and day 43, and at the end of the 90 min infusion and 1 h after the end of infusion. Three additional PK samples were collected from each patient: between 3–7 days after the first dose, between 10–15 days after the third dose and prior to the fourth induction dose on day 64. A subset of patients in CA184-007 and CA184-008 had PK samples drawn on days 2, 3, 7, 15 and 21.
Ipilimumab serum concentrations were measured by a validated enzyme-linked immunosorbent assay, with a lower limit of quantification of 0.4 μg ml−1. The assay was accurate to ± 9.40% and the inter-assay and intra-assay coefficients of variation were 6.82% and 5.21%, respectively [15].
PPK model development
The PPK model was developed with the index dataset in three stages. First, a stable and parsimonious base model was developed to describe ipilimumab serum concentration–time data in advanced melanoma patients, without considering covariate effects [16]. Base model development consisted of determining three component models: a structural PK model, an inter-individual (IIV) model, and a residual variability model. Development of the appropriate structural PK model consisted of determining a functional form common to all patients that adequately describes their concentration–time profiles, including an assessment of linearity and time-invariance in model parameters.
The IIV model describes the inter-individual joint distribution of structural model PK parameters, under the assumption that they were log-normally distributed. The variance and covariance of IIV in clearance (CL) and volume of the central compartment (Vc) were estimated. The residual error model describes the random variability between observed and model-predicted concentration values for each patient. Log-normal and combined (additive and proportional) residual error models were evaluated. The stability of the base model was assessed by confirming that the condition number (ratio of the largest and smallest eigen values of the standard error variance-covariance matrix) was less than 1000, and that the correlations in the variance-covariance matrix of standard errors were less than 0.95 [17].
Second, a full covariate model was developed by incorporating the effect of all prespecified intrinsic and extrinsic covariate parameter relationships of interest into the base model. The selection of the covariates investigated was based upon their relevance to the clinical pharmacology profiling of ipilimumab. Covariate-parameter relationships were examined for the following baseline covariates: body weight (BW), age, gender, estimated glomerular filtration rate (eGFR), Eastern Cooperative Oncology Group (ECOG) performance status, baseline lactate dehydrogenase (LDH) concentrations, albumin (ALB), alanine aminotransferase (ALT), direct bilirubin (BIL), concomitant budesonide, prior systemic anticancer therapy and HLA-A*0201 genotype status (HLA). We included HLA because all patients in the registrational trial, MDX010-20, were HLA-A*0201 positive [4] and because PK data were not collected in this study. Furthermore, the effect of immunogenicity on clearance was assessed as a time-varying covariate to account for the possibility that anti-drug antibodies (ADA) are not present at all time-points in immunogenic patients. The effects of these covariates were estimated simultaneously in the full covariate model. This approach is preferable to forward selection of covariates, as the latter may introduce bias in the estimated values [18,19].
The relationship between the typical value of a parameter (PTV) and a continuous valued covariate (R) was tested using the following:
where P1 and P2 are fixed-effect parameters, and REF is the reference value of the covariate, which was selected to approximate the median value of the covariate. The relationship between the typical value of a parameter and a categorical time-invariant covariate (R) was characterized using:
where P1 and Pm are fixed effects parameters, and Im is the indicator variable.
Lastly, the final model was developed from the full covariate model by backward elimination of covariates followed by exclusion of covariates determined to not be of potential clinical relevance. Specifically, covariate effects were retained in the final model provided that they were statistically significant (likelihood ratio test, P < 0.001) and potentially clinically relevant (defined as covariates that changed parameter values by more than ± 20%).
PPK model evaluation and validation
The base and final model were evaluated by examining standard diagnostic plots of conditional weighted residuals and parameter distributions [14], and by visual predictive checks (VPC) against the index dataset. Furthermore, a rigorous validation of the final model was performed by VPC against the external validation dataset.
The VPC analysis was designed to assess the predictive performance of the PPK model with respect to the central tendency and extremes of the distribution of ipilimumab serum concentration values over the induction-dosing period. Specifically, VPC was performed by comparing the 90% prediction intervals of the median, 5th, and 95th percentiles of ipilimumab concentration–time profiles obtained by simulation (500 iterations) with the corresponding observed percentiles, by dose. The final model parameters were estimated with a combined analysis dataset consisting of the index and validation datasets prior to applying the model as described below.
PPK model application
The final model was applied to predict ipilimumab exposures of each patient in the combined analysis dataset using the model-estimated maximum a posteriori Bayesian individual PK parameter values. Specifically, the model was applied to predict ipilimumab peak and trough concentration values during the induction period, as well as steady-state peak, trough, and time-averaged serum concentrations (Cmax,ss, Cmin,ss and Cav,ss, respectively) that would be achieved with every 3 weeks dosing. The Cav,ss was determined by dividing the area under steady-state serum concentration–time curve (AUCss) with the dosing interval, and the correlations between the steady-state measures of exposure were calculated.
Cmin,ss is considered the most pharmacologically relevant summary measure of exposure, as it enables an assessment of doses that maximally block the binding of CD80 or CD86 to CTLA-4. The binding of CTLA-4 to CD80 and CD86 was determined by an in vitro assay to be maximally inhibited by 6 to 20 μg ml−1 and 1 to 3 μg ml−1 of ipilimumab, respectively. The appropriateness of BW-normalized dosing was assessed by examining the similarity in Cmin,ss values over the observed BW range, and the extent to which the doses of 0.3, 3 and 10 mg kg−1 maximally block the binding of CD80 and CD86 to CTLA-4. The potential clinical relevance of covariates with a greater than 20% effect on a PK parameter was assessed with respect to AUCss, as this is the most common measure of exposure.
The potential impact of renal and hepatic impairment on ipilimumab exposure was assessed with respect to AUCss. Specifically, the AUCss distributions of renally and hepatically impaired patients were compared with the AUCss distribution of patients with normal renal and hepatic function, respectively. Patients were categorized by renal function defined by the Kidney Outcome Quality Initiative (K/DOQI) Clinical Practice Guidelines for Chronic Kidney Disease (CKD) from the National Kidney Foundation in 2002 [20]: normal renal function (eGFR > 90 ml min 1.73−1 m−2), and mild impairment (eGFR between 60 and 90 ml min 1.73−1 m−2), moderate impairment (eGFR between 30 and 60 ml 1.73 m−2), and severe renal impairment (eGFR < 30 ml min 1.73−1 m−2). Patients were also categorized by hepatic impairment categories proposed by the US National Cancer Institute (NCI) [21]: baseline total bilirubin (TB), baseline aspartate aminotransferase (AST) < upper limit of normal (ULN), mild hepatic disease (TB>ULN to 1.5 × ULN or AST >ULN), and moderate hepatic disease (TB >1.5–3 × ULN, any AST).
The analysis datasets were prepared and summarized using SAS® Version 8.2 (SAS Institute Inc., Cary, NC, USA), and the model was developed with the nonmem® computer program Version VI, level 1.1, which was also used for model-based simulations. S-Plus® software Version 7.0 (Insightful Corporation, Seattle, WA, USA) was used for diagnostic plots and the graphical presentation of results.
Results
PPK model development
Ipilimumab PK was determined to be linear and time-invariant within the dose range. Following intravenous (i.v.) administration, ipilimumab undergoes biphasic elimination consisting of a rapid distribution phase with a geometric mean distribution half-life (t1/2,α) of 27.4 h and a slow elimination phase with a geometric mean elimination half-life (t1/2,β) of 14.7 days. Ipilimumab serum concentration–time data were well described by a linear two compartment model with zero order i.v. infusion and first order elimination. The model was parameterized in terms of CL, Vc, inter-compartmental clearance (Q) and volume of peripheral compartment (Vp). IIV in CL and Vc were characterized by log-normal distributions, but the IIV in Q and Vp were fixed to zero as these variances could not be reliably estimated. The residual error was described by a combined additive and proportional error model. The condition number of the base model was 4, and the maximum value of correlations between parameter estimates was 0.2, indicating the model was stable.
Assessment of inter-occasion variability on CL and diagnostic plots of conditional weighted residuals (CWRES) vs. time after first dose showed PK parameters were time-invariant. Conceptually, the elimination of monoclonal antibodies consists of a linear component due to non-specific catabolic processes, and a non-linear target-mediated drug disposition component [22,23]. The potential non-linearity of ipilimumab PK was assessed during base model development by incorporation of parallel linear and non-linear elimination. However, the additional non-linear term on CL did not improve the model based on the goodness-of-fit plots and non-significant reduction of objective function values, relative to the model with constant CL. Thus, there was no evidence of non-linearity in ipilimumab PK.
Results of the full model indicated that BW is the most influential covariate for CL and Vc (Figure 1). This finding is consistent with the non-specific reticulo-endothelial system-mediated mechanism of elimination of monoclonal antibodies [24,25]. The effects of other covariates on CL were within ± 20% (Figure 1), which indicates that they are unlikely to be clinically relevant. The estimated 22% increase in CL due to ADA was judged to not be clinically relevant, as less than 5% (18/402) of patients developed ADA, and ADA were transient in most. Given the substantial overlap of CL estimates, and the small number of ADA-positive patients, the immunogenicity effect was not considered clinically relevant.
Figure 1.

Covariate effects on ipilimumab CL and Vc (estimated with the full model), relative to the parameter values of a reference subject (male, body weight of 80 kg, aged 60 years, eGFR of 80 ml min−1 1.73−1 m−2, direct bilirubin of 0.12 mg dl−1, alanine aminotransferase of 19 IU l−1, albumin at 4.2 g dl−1, LDH at 206 IU l−1, ECOG performance status of 0, HLA-A*0201 negative and no prior systemic anticancer therapy, immunogenicity, or concomitant budesonide). The open/shaded area of the boxes represents the range of covariate effects from the median to the 5th and 95th percentile of the continuous covariate. The dashed vertical lines represent 80% and 120% of the reference subject parameter value.  , estimate (95% CI): continuous (P95);
, estimate (95% CI): continuous (P95);  , estimate (95% CI): continuous (P05);
, estimate (95% CI): continuous (P05);  , estimate (95% CI): categorial;
, estimate (95% CI): categorial;  , estimate (continuous values > references)
, estimate (continuous values > references)
Only BW and LDH were retained as covariates in the final model. CL was not affected by age (range 26–86 years), gender, hepatic function (as measured by albumin, direct bilirubin and alkaline phosphatase), concomitant budesonide, renal function (estimated GFR), ECOG performance status, HLA-A*0201 status and prior systemic anticancer therapy. CL increased with increasing BW and baseline LDH, and Vc increased with increasing BW. The covariate effects of BW and LDH on typical values (model estimated geometric mean) of CL and Vc were described by:
and
where CLREF and Vc,REF are typical values (model-estimated geometric mean) of CL and Vc at the reference values of BW and LDH (80 kg and 206 IU l−1, respectively), and CLBW, CLLDH, Vc,BW are model parameters. The value for LDH was log-transformed due to its right-skewed distribution. The reference values of BW and LDH were selected to be approximately median values of variables in the PPK dataset. The IIV of CL and Vc in the final model were reduced by 24% and 52%, respectively, compared with the base model. Parameter estimates are provided in Table 2.
Table 2.
Final PPK model parameter estimates
| Parameter [units] | Estimate* | 95% confidence interval† |
|---|---|---|
| Structural model parameters | ||
| CLREF (l h−1) | 0.0150 | 0.0143, 0.0156 |
| VcREF (l) | 4.15 | 4.08, 4.26 |
| QREF (l h−1) | 0.0411 | 0.0378, 0.0517 |
| VpREF (l) | 3.11 | 2.95, 3.46 |
| CLBW | 0.642 | 0.423, 0.819 |
| VcBW | 0.708 | 0.616, 0.832 |
| CLLDH | 1.13 | 0.653, 1.49 |
| Inter-individual variability model parameters | ||
| ω2CL | 0.125 (0.354) | 0.0965, 0.161 |
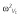
|
0.0223 (0.149) | 0.0192, 0.036 |
|
0.0254 (0.452) | 0.0096, 0.0351 |
| Residual error model parameters | ||
| Proportional error (%) | 15.7 | 13.7, 16.6 |
| Additive error (μg ml−1) | 0.244 | 0.005, 0.510 |
The final parameter estimates were obtained from the final PPK model with combined dataset.
Estimate values in parentheses are standard deviations for estimated variances and correlations for estimated covariances. Reference subject: 80 kg and 206 IU l−1, respectively.
Confidence interval values are taken from bootstrap calculations (1970 successful out of a total of 2000). BW, body weight; CL, clearance; LDH, lactate dehydrogenase; Q, inter-compartmental clearance; REF, reference value; Vc, volume of central compartment; Vp, volume of peripheral compartment.
PPK model evaluation and validation
We evaluated the PPK model using standard diagnostic plots, including model predictions vs. observations, residuals vs. model predictions, and residuals vs. time. The diagnostic plots showed that the model described the observed data well, and that the assumptions about random variability were reasonably satisfied (not shown). Internal model validation demonstrated that the final PPK model adequately described ipilimumab concentration–time profile for 0.3 to 10 mg kg−1 (Figure 2A), and the external validation confirmed the accuracy of the model parameter estimates (Figure 2B). The external model validation was performed on dose-normalized concentrations, as data were insufficient to determine the 5th and 95th percentiles by dose. Overall, the median, 5th and 95th percentiles of the observed concentration–time profiles are within the corresponding 90% prediction intervals, demonstrating that the model adequately predicts the shape and variability of ipilimumab concentration–time data. One patient with CL below three times interquartile range (lower extreme of clearance distribution), and more than five-fold lower than the second lowest CL value, was excluded from the subsequent model application.
Figure 2.
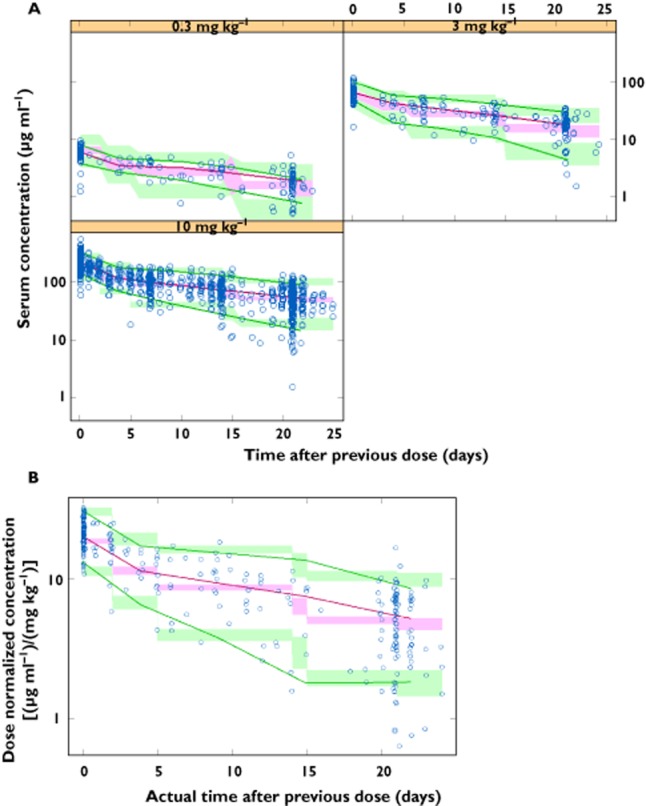
(A) Internal validation (CA184-007, CA184-008 and CA184-022), by dose level, and (B) external validation (CA184-004), dose normalized. Observed and predicted median, 5th, and 95th percentile (P05 and P95) concentrations (90% prediction interval, PI) vs. relative time from previous dose of ipilimumab.  , observed;
, observed;  , observed median;
, observed median;  , observed P05/P95;
, observed P05/P95;  , 90% PI median;
, 90% PI median;  , 90% PI P05/P95
, 90% PI P05/P95
PPK model application
Minimal systemic accumulation was evident by an accumulation index of approximately 1.5-fold, and steady-state was reached by the end of the third dosing interval. The model-predicted steady-state Cmax,ss, Cmin,ss and Cav,ss were highly correlated (Pearson correlation coefficient > 0.8). Model-predicted trough concentration after the first ipilimumab dose (Cmin1) exceeded the target trough concentration of 3 μg ml−1 by ∼99% and 100% of patients at the 3 and 10 mg kg−1 doses, respectively. Although only approximately 2% of patients receiving 3 mg kg−1 ipilimumab had Cmin1 values above 20 μg ml−1, approximately 90% had Cmin1 greater than 6 μg ml−1 (Figure 3). The 20 μg ml−1 and 3 μg ml−1 target concentrations have been used as the cut-off for maximal binding for CD80 and CD86, respectively. However there is considerable (>three-fold) uncertainty in the range of concentrations obtained from in vitro cell-based assay that are required for maximal binding (6 to 20 μg ml−1 and 1 to 3 μg ml−1, respectively).
Figure 3.
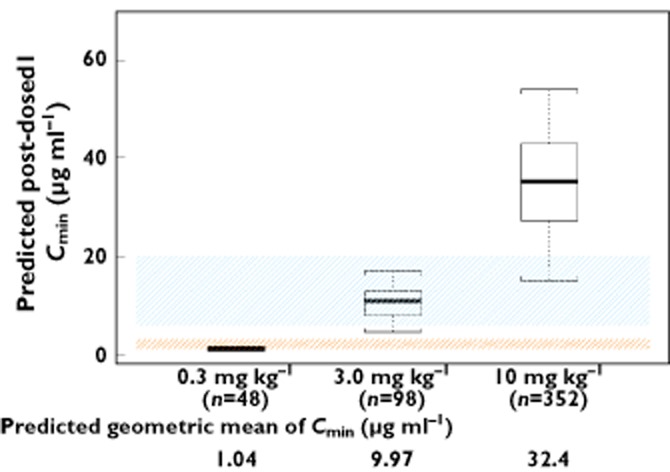
Distribution of post-dose 1 trough concentrations (Cmin1) by dose. The box plots represent median (bold line), 25th, and 75th percentiles of the Cmin1 distribution. The whiskers represent 5th and 95th percentiles of the distribution. The shaded areas indicate the target trough concentration ranges that maximally inhibit the binding of CTLA-4 to B7.1 (6–20 μg ml−1) and B7.2 (1–3 μg ml−1)
The appropriateness of BW-normalized dosing was confirmed by a visual examination of the relationship between Cmin,ss and BW (Figure 4). This showed that Cmin,ss was relatively uniform over the BW range in the dataset, which is representative of advanced melanoma patients. These results support a BW-normalized dose regimen for ipilimumab in advanced melanoma.
Figure 4.
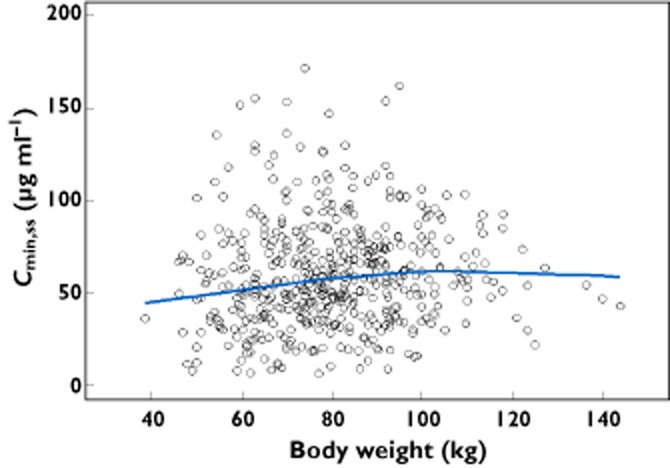
Ipilimumab Cminss
vs. body weight for a body weight-based regimen.  , individual estimates;
, individual estimates;  , locally weighted smooth line
, locally weighted smooth line
The relationships between AUCss and LDH, and between AUCss and renal or hepatic impairment, are shown in Figure 5. The AUCss tends to decrease with increasing LDH (Figure 5A), as expected from the covariate effect of LDH on CL. However, this decrease is not considered clinically significant, based on available safety and efficacy data [8]. Furthermore, the AUCss in patients with mild or moderate renal impairment is similar to that of other patients (Figure 5B), as is the AUCss in patients with mild hepatic impairment (Figure 5C). Thus, no dose adjustment is needed for patients with pre-existing mild hepatic impairment, or mild or moderate renal impairment. The relationship between Cmin,ss and these covariates is similar to AUCss, given the high correlation between these exposure measures.
Figure 5.
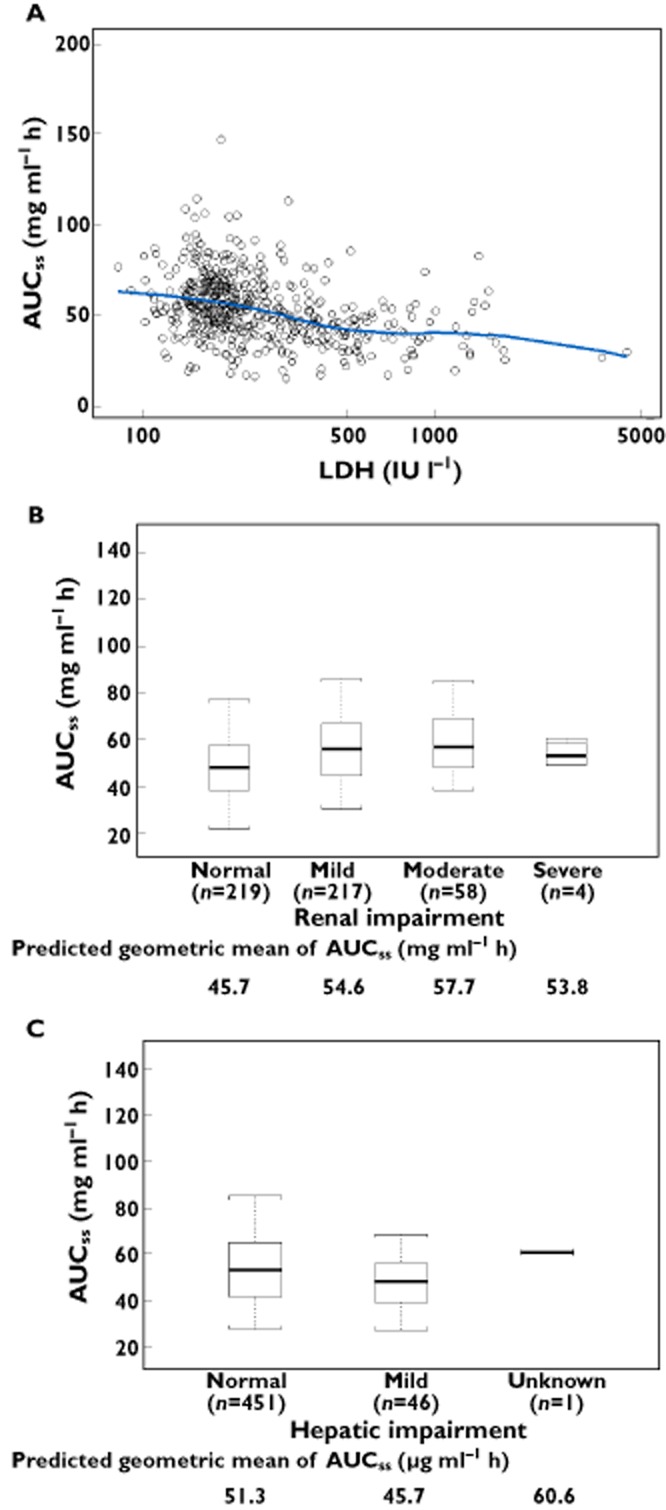
Ipilimumab AUCss
vs. (A) baseline LDH levels, (B) renal function status and (C) NCI liver dysfunction group. The box plots represent median (bold line), 25th, and 75th percentiles of the AUCss distribution. The whiskers represent 5th and 95th percentiles of the distribution.  , individual estimates;
, individual estimates;  , locally weighted smooth line
, locally weighted smooth line
Discussion
This report is the first comprehensive characterization of ipilimumab PK, accomplished by PPK analysis of data from patients with advanced melanoma. The magnitude of the effects of intrinsic and extrinsic factors on ipilimumab PK was estimated, and the combined effects on ipilimumab exposure in specific subpopulations were assessed. Ipilimumab concentration–time data were well described by a linear, two-compartment, zero order i.v. infusion model. The linearity of the ipilimumab PK model was established by assessing the goodness-of-fit and diagnostic plots of the linear model, as well as by determining that incorporation of a parallel non-linear term for drug elimination did not improve the goodness-of-fit [23]. Diagnostic plots with respect to time established that ipilimumab PK were time-invariant. The IIV in all model parameters was investigated in the development of the base model, and IIV in only CL and Vc was retained as the IIV in other parameters were not identifiable or could not be reliably estimated. The simplification of the IIV structure enabled the establishment of a stable base model (as determined by condition number <1000 and correlation <0.95 in the parameter standard error matrix), which was essential for the robust estimation of covariate effects on PK parameters in the full model.
The full model approach to estimate the effect of pre-specified covariate-parameter relationships provides an unbiased estimate of these effects, and is preferred to the forward inclusion method of selecting covariates [18,19]. The full model was simplified by backward elimination of covariate effects not clinically or statistically significant to obtain a parsimonious final model. The final PPK model was validated against both internal and external datasets. Validation with an external dataset not used in model development is considered the most rigorous [16,26]. These validations showed that the PPK model predicted the central tendency (median) and extremes (5th and 95th percentiles) of the concentration–time profile.
The only covariate-parameter relationships retained in the final model were BW CL and Vc, and log-transformed LDH CL. Incorporation of these covariates in the final model explained approximately 24% and 52% of the base model variability of CL and Vc, respectively. The typical values of CL and Vc increased with increasing BW, and the rate of increase supports BW-normalized dosing of ipilimumab. An extensive analysis of whether the recommended dose should be fixed or BW-normalized found that the latter provided more uniform exposures over the range of BW when either of the power model coefficients of CL and Vc are greater than 0.5 [27]. The recommended BW-normalized dosing of ipilimumab is justified as the BW power coefficients of CL and Vc in the final PPK model were greater than 0.5. The suitability of BW-normalized dosing was confirmed by the relatively uniform values of Cmin,ss over a wide BW range.
Other baseline covariates, including age, gender, eGFR, direct bilirubin, AST, albumin hepatic function (as assessed by NCI Criteria [20]), concomitant budesonide, performance status, HLA-A*0201 status, prior systemic anticancer therapy and immunogenicity, had no clinically significant effects on ipilimumab PK. Consistent with our results, analysis of data from the phase II studies showed that efficacy and safety were independent of HLA-A*0201 status [28]. The absence of a relationship between CL and eGFR is consistent with physiology, as the large size of the ipilimumab molecule (148 kDa) is expected to prevent it from being filtered through the glomerulus and eliminated via the kidney. It is also established that monoclonal antibodies are cleared via the reticulo-endothelial system, and not via liver metabolism like many small molecules [29]. We found no evidence of decreased ipilimumab clearance in patients with normal-to-mild hepatic impairment.
The magnitude of covariate effects on CL and Vc in the full model was presented as a forest plot, which is the approach recently recommended by the US FDA and others [30], as it provides a more intuitive and accessible assessment of the effect of covariates [30–32] than conventional tabular reporting [33–35]. In particular, the magnitude of continuous covariate effects is not readily apparent from a tabular presentation of parameter estimates, because the magnitude of effect is dependent upon the distribution of the continuous covariate and the parameter value. Forest plot representation of covariate effects shows the magnitude of the effect of continuous covariates over a broad range (5th to 95th percentile) of values. This representation also enables assessment of the potential clinical relevance of a covariate – those with less than a 20% effect on PK model parameters are unlikely to be relevant.
The covariate-effect plot can show the effect of covariates on PK model parameters, but is insufficient to profile drug clinical pharmacology as it does not provide a direct assessment of the relationship between exposure and subpopulations defined by sets of covariate values. Drug exposure is dependent upon the dosing regimen, as well as on the combined effect of all covariates with an effect on one or more PK model parameters. This is important when an exposure measure is determined by more than one PK model parameter, and when several correlated covariates affect the same model parameter. For ipilimumab, CL and Vc increase with increasing BW, and BW-normalized dosing achieved relatively uniform values of Cmin,ss across a wide range of BWs. Similarly, the distributions of AUCss by renal function reflects the totality of the effect of dose, and all covariates that have an effect on CL, not just the effect of eGFR on CL. PPK model-based analysis provided a more robust assessment in these special populations with large numbers of patients in the target population compared with traditional NCA analysis where the assessment is based on relatively small patient numbers.
The PPK model was applied to predict Cmin1 (not measured in the phase II studies), to support selection of phase III study doses. Although only approximately 2% of patients who received 3 mg kg−1 ipilimumab had Cmin1 values above 20 μg ml−1, approximately 90% had Cmin1 greater than 6 μg ml−1 (the lower limit needed to maximally inhibit the binding of CTLA-4 to CD80). The 20 μg ml−1 and 3 μg ml−1 target concentrations have been conservatively used as the cut-off for maximal binding. However, there is substantial (>three-fold) uncertainty in the range of concentrations required for maximal binding. Moreover, the in vitro assay was performed with a cultured cell line that overexpresses CTLA-4 and with fluorophore-conjugated human CD80 and CD86 fusion proteins, and thus the relevance to in vivo human expression of CTLA-4 is unknown. Therefore, these target concentrations were only used to provide non-clinical justification for studying higher doses and cannot be used to make clinical comparisons between 3 and 10 mg kg−1 doses. The benefit–risk of ipilimumab at 3 vs. 10 mg kg−1 is being evaluated in a randomized phase III study in patients with advanced melanoma [6].
In conclusion, the PPK model adequately described ipilimumab PK data in patients with advanced melanoma. The PPK analysis demonstrated that a BW-normalized dosing regimen is appropriate for ipilimumab, and dose adjustments are not required for mild-to-moderate renal impairment or mild hepatic impairment. Data were not available to extend these findings to severe renal impairment or moderate-to-severe hepatic impairment.
Acknowledgments
We thank Neelima Thanneer of Bristol-Myers Squibb for preparing the PPK datasets and data summaries. Editorial and writing assistance was provided by StemScientific, funded by Bristol-Myers Squibb.
Competing Interests
All authors have completed the unified Competing Interest Form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: YF, EM, SMP, DB, and AR are currently employed by the study sponsor (Bristol-Myers Squibb). DD is currently employed by Vertex Pharmaceuticals, with no stock ownership or options in Bristol-Myers Squibb.
References
- 1.Melero I, Hervas-Stubbs S, Glennie M, Pardoll DM, Chen L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat Rev Cancer. 2007;7:95–106. doi: 10.1038/nrc2051. [DOI] [PubMed] [Google Scholar]
- 2.Fong L, Small EJ. Anti-cytotoxic T-lymphocyte antigen-4 antibody: the first in an emerging class of immunomodulatory antibodies for cancer treatment. J Clin Oncol. 2008;26:5275–5283. doi: 10.1200/JCO.2008.17.8954. [DOI] [PubMed] [Google Scholar]
- 3.Hoos A, Ibrahim R, Korman A, Abdallah K, Berman D, Shahabi V, Chin K, Canetta R, Humphrey R. Development of ipilimumab: contribution to a new paradigm for cancer immunotherapy. Semin Oncol. 2010;37:533–546. doi: 10.1053/j.seminoncol.2010.09.015. [DOI] [PubMed] [Google Scholar]
- 4.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbé C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robert C, Thomas L, Bondarenko I, O'Day S, Weber J, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ, Davidson N, Richards J, Maio M, Hauschild A, Miller WH, Jr, Gascon P, Lotem M, Harmankaya K, Ibrahim R, Francis S, Chen TT, Humphrey R, Hoos A, Wolchok JD. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517–2526. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 6.ClinicalTrial.gov. Phase 3 trial in subjects with metastatic melanoma comparing 3 mg/kg ipilimumab versus 10 mg/kg ipilimumab. Available at http://clinicaltrials.gov/show/NCT01515189 (last accessed 19 June 2013)
- 7.US Food and Drug Administration. Yervoy. Available at http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Label_ApprovalHistory#labelinfo (last accessed 13 June 2013)
- 8.European Medicines Agency. Yervoy. Available at http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002213/human_med_001465.jsp&mid=WC0b01ac058001d124 (last accessed 13 June 2013)
- 9.Feng Y, Roy A, Masson E, Chen TT, Humphrey R, Weber J. Exposure-response relationships of the efficacy and safety of ipilimumab in patients with advanced melanoma. Clin Cancer Res. 2013;19:3977–3986. doi: 10.1158/1078-0432.CCR-12-3243. [DOI] [PubMed] [Google Scholar]
- 10.Weber J, Thompson JA, Hamid O, Minor D, Amin A, Ron L, Ridolfi R, Assi H, Maraveyas A, Berman D, Siegel J, O'Day SJ. A randomized, double-blind, placebo-controlled, phase II study comparing the tolerability and efficacy of ipilimumab administered with or without prophylactic budesonide in patients with unresectable stage III or IV melanoma. Clin Cancer Res. 2009;15:5591–5598. doi: 10.1158/1078-0432.CCR-09-1024. [DOI] [PubMed] [Google Scholar]
- 11.O'Day SJ, Maio M, Chiarion-Sileni V, Gajewski TF, Pehamberger H, Bondarenko IN, Queirolo P, Lundgren L, Mikhailov S, Roman L, Verschraegen C, Humphrey R, Ibrahim R, de Pril V, Hoos A, Wolchok JD. Efficacy and safety of ipilimumab monotherapy in patients with pretreated advanced melanoma: a multicenter single-arm phase II study. Ann Oncol. 2010;21:1712–1717. doi: 10.1093/annonc/mdq013. [DOI] [PubMed] [Google Scholar]
- 12.Wolchok JD, Neyns B, Linette G, Negrier S, Lutzky J, Thomas L, Waterfield W, Schadendorf D, Smylie M, Guthrie T, Jr, Grob JJ, Chesney J, Chin K, Chen K, Hoos A, O'Day SJ, Lebbé C. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010;11:155–164. doi: 10.1016/S1470-2045(09)70334-1. [DOI] [PubMed] [Google Scholar]
- 13.Hamid O, Schmidt H, Nissan A, Ridolfi L, Aamdal S, Hansson J, Guida M, Hyams DM, Gómez H, Bastholt L, Chasalow SD, Berman D. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J Transl Med. 2011;9:204–219. doi: 10.1186/1479-5876-9-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. WinPOPT: home. [online]. Available at http://www.winpopt.com/index.htm (last accessed 13 June 2013)
- 15.Weber JS, O'Day S, Urba W, Powderly J, Nichol G, Yellin M, Snively J, Hersh E. Phase I/II study of ipilimumab for patients with metastatic melanoma. J Clin Oncol. 2008;26:5950–5956. doi: 10.1200/JCO.2008.16.1927. [DOI] [PubMed] [Google Scholar]
- 16.Sherwin CM, Kiang TK, Spigarelli MG, Ensom MH. Fundamentals of population pharmacokinetic modelling: validation methods. Clin Pharmacokinet. 2012;51:573–590. doi: 10.1007/BF03261932. [DOI] [PubMed] [Google Scholar]
- 17.Montgomery DC, Peck EA, Vining GG. Introduction to Linear Regression Analysis. 4th edn. New York: Wiley-Interscience; 2006. [Google Scholar]
- 18.Gastonguay M. Full covariate models as an alternative to methods relying on statistical significance for inferences about covariate effects: a review of methodology and 42 case studies. PAGE Conference, June 2011, Athens, Greece. Available at http://www.page-meeting.org/?abstract=2229 (last accessed 13 June 2013)
- 19.Harrell FE., Jr . Regression Modeling Strategies: With Applications to Linear Models, Logistic Regression, and Survival Analysis. New York: Springer; 2010. [Google Scholar]
- 20.Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D for Modification of Diet in Renal Disease Study Group. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Ann Intern Med. 1999;130:461–470. doi: 10.7326/0003-4819-130-6-199903160-00002. [DOI] [PubMed] [Google Scholar]
- 21.Ramalingam SS, Kummar S, Sarantopoulos J, Shibata S, LoRusso P, Yerk M, Holleran J, Lin Y, Beumer JH, Harvey RD, Ivy SP, Belani CP, Egorin MJ. Phase I study of vorinostat in patients with advanced solid tumors and hepatic dysfunction: a National Cancer Institute Organ Dysfunction Working Group study. J Clin Oncol. 2010;28:4507–4512. doi: 10.1200/JCO.2010.30.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mager DE, Jusko WJ. General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn. 2001;28:507–532. doi: 10.1023/a:1014414520282. [DOI] [PubMed] [Google Scholar]
- 23.Gibiansky L, Gibiansky E, Kakkar T, Ma P. Approximations of the target-mediated drug disposition model and identifiability of model parameters. J Pharmacokinet Pharmacodyn. 2008;35:573–591. doi: 10.1007/s10928-008-9102-8. [DOI] [PubMed] [Google Scholar]
- 24.Mould DR, Green B. Pharmacokinetics and pharmacodynamics of monoclonal antibodies: concepts and lessons for drug development. BioDrugs. 2010;24:23–39. doi: 10.2165/11530560-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 25.Keizer RJ, Huitema AD, Schellens JH, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49:493–507. doi: 10.2165/11531280-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 26.FDA Guidance for industry: population pharmacokinetics. 1999. Available at http://www.fda.gov/downloads/ScienceResearch/SpecialTopics/WomensHealthResearch/UCM133184.pdf (last accessed 20 June 2013)
- 27.Bai S, Jorga K, Xin Y, Jin D, Zheng Y, Damico-Beyer LA, Gupta M, Tang M, Allison DE, Lu D, Zhang Y, Joshi A, Dresser MJ. A guide to rational dosing of monoclonal antibodies. Clin Pharmacokinet. 2012;51:119–135. doi: 10.2165/11596370-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 28.Wolchok JD, Weber JS, Hamid O, Lebbé C, Maio M, Schadendorf D, de Pril V, Heller K, Chen TT, Ibrahim R, Hoos A, O'Day SJ. Ipilimumab efficacy and safety in patients with advanced melanoma: a retrospective analysis of HLA subtype from four trials. Cancer Immun. 2010;10:9–14. [PMC free article] [PubMed] [Google Scholar]
- 29.Newsome BW, Ernstoff MS. The clinical pharmacology of therapeutic monoclonal antibodies in the treatment of malignancy; have the magic bullets arrived? Br J Clin Pharmacol. 2008;66:6–19. doi: 10.1111/j.1365-2125.2008.03187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Menon-Andersen D, Yu B, Madabushi R, Bhattaram V, Hao W, Uppoor RS, Mehta M, Lesko L, Temple R, Stockbridge N, Laughren T, Gobburu JV. Essential pharmacokinetic information for drug dosage decisions: a concise visual presentation in the drug label. Clin Pharmacol Ther. 2011;90:471–474. doi: 10.1038/clpt.2011.149. [DOI] [PubMed] [Google Scholar]
- 31.Rawa P, Gastonguay MR, Tensfeldt TG, Faessel TM. Population pharmacokinetic analysis of varenicline in adult smokers. Br J Clin Pharmacol. 2009;68:669–681. doi: 10.1111/j.1365-2125.2009.03520.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riggs MM, Bergsma TT, Rogers JA, Gastonguay MR, Subramanian GM, Chen C, Devalaraja M, Corey AE, Sun H, Yu J, Stein DS. Population pharmacokinetics and exposure-response of albinterferon alfa-2B. J Clin Pharmacol. 2012;52:475–486. doi: 10.1177/0091270011399576. [DOI] [PubMed] [Google Scholar]
- 33.Xu XS, Smit JW, Lin R, Stuyckens K, Terlinden R, Nandy P. Population pharmacokinetics of tapentadol immediate release (IR) in healthy subjects and patients with moderate or severe pain. Clin Pharmacokinet. 2010;49:671–682. doi: 10.2165/11535390-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 34.Mueck W, Lensing AW, Agnelli G, Decousus H, Prandoni P, Misselwitz F. Rivaroxaban: population pharmacokinetic analyses in patients treated for acute deep-vein thrombosis and exposure simulations in patients with atrial fibrillation treated for stroke prevention. Clin Pharmacokinet. 2011;50:675–686. doi: 10.2165/11595320-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 35.Mueck W, Eriksson BI, Bauer KA, Borris L, Dahl OE, Fisher WD, Gent M, Haas S, Huisman MV, Kakkar AK, Kälebo P, Kwong LM, Misselwitz F, Turpie AG. Population pharmacokinetics and pharmacodynamics of rivaroxaban – an oral, direct factor Xa inhibitor–in patients undergoing major orthopaedic surgery. Clin Pharmacokinet. 2008;47:203–216. doi: 10.2165/00003088-200847030-00006. [DOI] [PubMed] [Google Scholar]