

Abstract
Aims
Infliximab, an anti-tumour necrosis factor-α monoclonal antibody, is indicated in rheumatoid arthritis (RA). Our objective was to evaluate the influence of the sources of infliximab pharmacokinetic variability in RA.
Methods
Eighty-four patients treated with infliximab for RA were included in a prospective noncomparative study. They were analysed between two consecutive infliximab infusions. Infliximab concentrations were measured before the infusion, 2 h, 1 and 4 weeks after the infusion and immediately before the next infusion. Infliximab concentrations were described using a two-compartment population pharmacokinetic model.
Results
The mean (interindividual standard deviation) estimated central volume of distribution was 2.3 l (36%) and systemic clearance was 0.019 l h−1 (37%). The central volume of distribution increased with bodyweight; it was doubled between 50 and 90 kg. Systemic clearance increased with pre-infusion C-reactive protein concentration by 20%, varying from 3 to 14 mg l−1, and was decreased by 30% when methotrexate was coadministered.
Conclusions
The influence of methotrexate and inflammation on infliximab clearance suggests that individual adjustment of infliximab doses according to disease activity may be useful in RA.
Keywords: inflammation, infliximab, monoclonal antibodies, pharmacokinetics, rheumatoid arthritis
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Infliximab pharmacokinetics in rheumatoid arthritis has never been described using population pharmacokinetic modelling.
Previous studies showed that inflammation and methotrexate cotreatment influence infliximab concentrations.
However, the influence of both factors on infliximab pharmacokinetic variability was never investigated.
WHAT THIS STUDY ADDS
This study is the first to describe infliximab pharmacokinetics in rheumatoid arthritis.
This study suggested that inflammation increases infliximab target-mediated clearance.
Methotrexate cotreatment may decrease infliximab clearance by decreasing inflammation.
Introduction
Infliximab is a chimeric immunoglobulin G1 (IgG1) monoclonal antibody targeting tumour necrosis factor-α (TNF-α). This biopharmaceutical has profoundly modified the treatment of several inflammatory diseases, such as rheumatoid arthritis (RA), ankylosing spondylitis, Crohn's disease, chronic ulcerative colitis, psoriatic arthritis and psoriasis.
In RA, the serum concentration of infliximab varies greatly between patients following administration [1]. This variability is relevant because there is a relationship between the infliximab concentration and the clinical response [1–4]. In indications other than RA, bodyweight, body surface area and antibodies toward infliximab (ATI) have been shown to influence infliximab pharmacokinetics [5–7]. In RA, concomitant treatment with methotrexate (MTX), a disease-modifying antirheumatic drug, is associated with high infliximab concentrations [8], whereas high pretherapeutic C-reactive protein (CRP) concentrations are associated with low infliximab trough concentrations [2,4]. To our knowledge, the influence of these factors on infliximab pharmacokinetics in RA patients has never been quantified using a population approach, although population modelling is a powerful approach to test and to quantify the influence of individual factors on interindividual pharmacokinetic variability.
The aim of this prospective study was to evaluate the effects of the sources of infliximab pharmacokinetic variability in RA patients by population modelling.
Methods
Patients
This prospective, noncomparative, open, multicentre study was approved by the ethics committee of Tours University Hospital and was conducted in accordance with the declaration of Helsinki. The FAKIR study (pharmacokinetics of infliximab in rheumatoid arthritis) has been registered on clinicaltrials.gov as NCT00840957. The study was designed to assess the pharmacokinetic variability of infliximab in rheumatoid arthritis patients using population modelling. Patients were recruited between December 2007 and September 2009 in six French centres, namely Tours, Orléans, Nantes, Rennes, Brest and Poitiers. The study details were explained to the patients, and all of them gave written informed consent. To be eligible, patients had to be adult and to fulfil the American College of Rheumatology (ACR) criteria for RA. They had to be treated with infliximab for at least 14 weeks before inclusion. The standard regimen for infliximab in RA patients is 3 mg kg−1 at week 0, 2 and 8 and then every 8 weeks. This dose could have been adjusted for some patients according to their clinical response, but no dose adjustment was allowed after the infusion previous to the inclusion in the study. The doses of MTX and prednisone had to be stable during the previous 4 weeks. Patients were treated in accordance with the guidelines of the French Society of Rheumatology for the use of infliximab [9].
Study design
For each included patient, the study started at the time of the routine visit for infliximab infusion and ended at the visit for the next infliximab infusion. The patient was also assessed at two intermediate visits, 1 and 4 weeks after the first visit.
Data
Infliximab concentrations and antibodies toward infliximab
All samples were centralized and assayed in Tours at the CePiBAc (centre pilote de suivi biologique des anticorps thérapeutiques – pilot centre for therapeutic antibody monitoring). Blood samples were collected at each visit to measure serum infliximab concentrations. At the time of the first visit, samples were collected before and 2 h after the end of the infusion. Infliximab concentrations were measured using a validated enzyme-linked immunosorbent assay [10]. The limit of detection was 0.014 mg l−1 and the lower (LLOQ) and upper limits of quantification [ULOQ; between-assay accuracy coefficient of variation % (CV%)] were 0.04 mg l−1 (9.8%) and 4.5 mg l−1 (5.3%), respectively. Antibodies toward infliximab were assayed using a double-antigen enzyme-linked immunosorbent assay based on their capture by infliximab-coated microplates and their detection with peroxidase-conjugated infliximab. The assay was standardized using a mouse monoclonal antibody against human IgG. Owing to the interference of circulating infliximab, only sera with infliximab concentrations <2 mg l−1 were tested. The positive threshold of detection was 0.07 mg l−1. A patient was considered ATI positive (ATI+) if ATI were detected in any sample; however, due to potential interference with rheumatoid factors, results were considered to be false positives if the serum drawn after infusion (and thus with high infliximab concentration) also gave a positive result.
Other laboratory analyses
At the time of the first visit, erythrocyte sedimentation rate and CRP concentration were measured before infliximab infusion. Erythrocyte sedimentation rate was measured locally in the laboratories of the recruiting centres, whereas CRP determination was centralized in the laboratory of biochemistry of Tours university hospital.
Clinical end-points
At the times of first and last visits, treatment efficacy was assessed by a trained rheumatologist by the measurement of the disease activity score (DAS28) [11], number of tender joints and number of swollen joints.
Pharmacokinetic analysis
Software
Pharmacokinetic data were analysed by a population approach using the nonlinear mixed-effects program MONOLIX 3.1 software, which combines the stochastic expectation-maximization (SAEM) algorithm and a Markov chain Monte-Carlo procedure for likelihood maximization. This software showed satisfactory performance in difficult analyses. To ensure the best possible convergence, a large number of iterations (1000 for K1 and 250 for K2) was used. The terms K1 and K2 refer to the SAEM procedure of Monolix, called ‘iteration kernels’. During K1, the sequence of step sizes is constant, which allows the exploration of the parameter space. During K2, the step sizes decrease to ensure convergence. Five Markov chains were used, and simulated annealing was used to improve the convergence of the SAEM algorithm towards the global maximum of the likelihood. Each run was performed three times to ensure that estimated parameters and likelihood remained stable. The random seed was changed between each of the three runs.
Structural model design
Infliximab concentrations were described using compartmental pharmacokinetic models. One, two and three mammillary models with first-order distribution constants were tested. Linear and nonlinear (Michaelis–Menten) eliminations were also tested. Structural models were compared using Akaike's information criterion (AIC), defined as: AIC = OFV + 2.p, where OFV is the value of the objective function and p is the number of model parameters to estimate. The use of AIC is based on the parsimony between a best fit to the data and a limited number of parameters. The OFV was −2.ln-likelihood (–2LL). The model with the lowest AIC was selected.
Interindividual model
The interindividual variability of pharmacokinetic parameters was described using an exponential model: θi = θTV × exp(ηi), where θi is the estimated individual parameter, θTV is the typical value of the parameter and ηi is the random effect for the ith patient. The values of ηi were assumed to be normally distributed, with mean 0 and variance ω2. For each parameter, ω2 was fixed to 0 if ω2 or ηi could not be estimated properly.
Error model
Additive, proportional and mixed additive–proportional models were tested. For example, the combined additive–proportional model was implemented as follows: YO,ij = YP,ij × (1 + εprop,ij) + εadd,ij, where YO,ij and YP,ij are observed and predicted jth measurements for the ith patient, respectively, and εprop,ij and εadd,ij are proportional and additive errors, which are assumed to follow a Gaussian distribution with mean 0 and variances σprop2 and σadd2, respectively.
Covariates
The influence of the following covariates was tested in the population excluding the ATI+ patients: (i) binary covariates, i.e. sex (SX), association with methotrexate and/or corticosteroids; and (ii) continuous covariates, i.e. age, bodyweight (WT), height, disease duration, infliximab treatment duration, CRP concentration and DAS28, which is the disease activity score on 28 joints [11].
The influence of a binary covariate (CAT) on θTV was implemented as ln(θTV) = ln(θCAT=0) + βCAT=1, where θCAT=0 is the value of θ for an arbitrary reference category and βCAT=1 is the value of θTV for the other category. Continuous covariates (COV) were centred on their median as follows: θi = θ0 × (COV/med(COV))βcov, where θ0 is value of θ for a median subject, βCOV quantifies the influence of COV on θ and med(COV) is the median value of COV in the population.
Model comparison and covariate selection
Interindividual, residual and covariate models were compared using OFV and AIC. From pairs of nested models, the one with the lowest OFV was chosen. This was assessed by a likelihood ratio test (LRT), in which the difference in OFV between two models (ΔOFV) is assumed to follow a χ2 distribution. The influence of patient characteristics (covariates) was assessed in two steps, as follows.
A univariate step, in which the influence of each factor on pharmacokinetic parameters associated with interindividual variability was tested. Covariates were separately included into the base model. Covariates showing a significant influence (α < 0.1) were included in the model (full model).
A multivariate step, in which a backward stepwise elimination was performed; the covariates of the full model were removed one by one. Covariates whose removal resulted in a statistically significant increase in the OFV (α < 0.01) were retained in the model.
Model goodness of fit and evaluation
The goodness of fit was assessed for each model by plotting population-predicted (PRED) and individually predicted (IPRED) concentrations vs. observed concentrations (DV) and IPRED and DV vs. time. Population predictions were obtained using typical parameters, which include explained variability (i.e. population estimates and covariates), whereas individually predicted concentrations were obtained using individual parameters, which include both explained and unexplained (i.e. the random effects ηi for each pharmacokinetic parameter). In addition, the distribution of residuals was evaluated by graphical inspection of population (PWRES) and individual weighted residual distributions (IWRES), visual predictive checks (VPC) and normalized prediction distribution errors (NPDE) [12]. These residuals should follow a standard normal distribution to confirm a satisfactory fit of the model to the data and (ii) to allow a χ2 distribution for LRT tests.
Results
Patients
Eighty-four patients treated with infliximab and who were assumed to be at steady state were included (Table 1). A total of 412 serum samples were available for analysis. Median (range) pre-infusion dose, dosing interval and infliximab concentrations were 3.6 mg kg−1 (2.5–6.8), 8.0 weeks (5.0–13.0) and 1.3 mg l−1 (<0.014–12.0), respectively. Antibodies toward infliximab were detected in the pre-infusion serum of three patients (nos 34, 55 and 79); two of these three patients were also treated with methotrexate and one was not. Of note, the infliximab concentrations for these three patients were below the limit of detection within 4 weeks after infliximab administration (data not shown). As the number of ATI+ patients was too small to add ATI status as a covariate and the pharmacokinetic profiles of ATI+ patients were very different from those of ATI-negative (ATI–) patients, the pharmacokinetic model was developed without the three ATI+ patients. The final model was then applied to all patients (including ATI+ patients) and ATI status was tested.
Table 1.
Summary of patient characteristics
| Characteristics | Patients (n = 84) |
|---|---|
| Women | 62 (74) |
| Age (years) | 58 [27–84] |
| Bodyweight (kg) | 65 [27–84] |
| Disease duration (years) | 12 [1–35] |
| Infliximab treatment duration (years) | 5.5 [0.3–8.2] |
| Dose (mg kg−1) | 3.6 [2.5–6.8] |
| Dosing interval (weeks) | 8.0 [5.0–13.0] |
| Pre-infusion infliximab concentration (mg l−1) | 1.3 [<0.014–12.0] |
| Methotrexate cotreatment | 69 (82) |
| Prednisone cotreatment | 51 (61) |
| DAS28 at inclusion | 2.96 [0.78–6.72] |
| ESR at inclusion (mm) | 14 [2–70] |
| CRP concentration at inclusion (mg l−1) | 3.2 [0.20–85.9] |
| ATI positive | 3 (4) |
Results are presented as the absolute number (%) or as the median [range]. Abbreviations are as follows: ATI, antibodies toward infliximab; CRP, C-reactive protein; DAS28, disease activity score on 28 joints; ESR, erythrocyte sedimentation rate.
Pharmacokinetic analysis excluding ATI+ patients
Base pharmacokinetic model
The best description of infliximab concentrations was obtained using a two-compartment model with first-order distribution and elimination constants. The structural pharmacokinetic model was as follows:
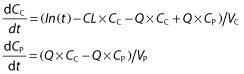 |
where In(t) (in milligrams per litre per hour) is the infliximab infusion rate, CC (in milligrams per litre) and CP (in milligrams per litre) are infliximab concentrations in the central and peripheral compartments, respectively, VC (in litres) and VP (in litres) are volumes of distribution of central and peripheral compartments, respectively, and CL (in litres per hour) and Q (in litres per hour) are systemic and distribution clearances, respectively. Parameters describing a third compartment or a nonlinear elimination were not identifiable. The VC, CL, VP and Q, shrinkages were 19, 3, 23 and 90%, respectively. The shrinkage for Q, ωQ, was removed from the model. The best error model was mixed additive–proportional. Plots of predicted vs. observed concentrations (Figure 1) and a visual predictive check plot (Figure 2) showed that the model agreed well with the data. A slight overprediction of concentrations was observed for concentrations close to the limit of detection (Figure 1), with a bias in population (PWRES) and individual weighted residuals (IWRES; Figure 3). However, the normalized prediction distribution error (NPDE) plots showed that there was no obvious model misspecification (Figure 3). The pharmacokinetic parameters were estimated with good accuracy (Table 2).
Figure 1.

Observed vs. population model-predicted concentrations (PRED; A), individual predicted values (IPRED; B), log-PRED (C) and log-IPRED (D)
Figure 2.
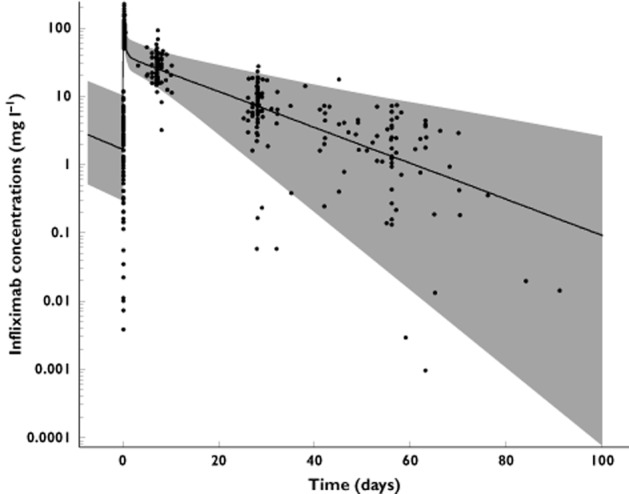
Visual predictive check for infliximab pharmacokinetics. Filled circles are infliximab concentrations, continuous line is median prediction and grey shaded area indicates 5–95% confidence interval
Figure 3.

Population (left) and individual residuals (middle), and normalized prediction distribution errors (NPDE; right). Plots are vs. time (top), vs. estimated concentrations (middle) and vs. distribution (bottom)
Table 2.
Estimated pharmacokinetic parameters
| Parameter (units) | Estimate | RSE (%) | r2 (%) | ΔLL |
|---|---|---|---|---|
| VC (l) | 2.3 | 5 | – | – |
| WT on VC | 1.2 | 22 | 34 | –17.9 |
| CL (l h−1) | 0.019 | 13 | – | – |
| MTX on CL | –0.36 | 32 | 20 | –9.7 |
| CRP concentration on CL | 0.12 | 34 | 16 | –10.5 |
| VP (l) | 3.6 | 4 | – | – |
| Q (l h−1) | 0.18 | 1 | – | – |
| ωVc (%) | 36 | 10 | – | – |
| ωCL (%) | 37 | 9 | – | – |
| ωVp (%) | 30 | 11 | – | – |
| ωQ (%) | – | – | – | – |
| σadd (mg l−1) | 0.15 | 16 | – | – |
| σprop (%) | 16 | 7 | – | – |
Abbreviations of the parameters are explained in the text. The RSE (%) was obtained as follows: RSE = (estimate/standard error) × 100. Other abbreviations are as follows: CRP, C-reactive concentration at inclusion; ΔLL, difference in ln-likelihood for each covariate; MTX, methotrexate cotreatment; r, coefficient of determination; WT, bodyweight.
Covariate testing and final model
The univariate step indicated that VC was influenced by SX, WT and methotrexate cotreatment, and that CL was influenced by methotrexate cotreatment, pre-infusion DAS28, and pre-infusion CRP concentration. In the final model, VC increased with WT (r2 = 34%, ΔLL = −17.9, P < 0.01; Figure 4), and CL increased with pre-infusion CRP concentration (r2 = 16%, ΔLL = −9.7, P < 0.01) and decreased when methotrexate was coadministered (r2 = 20%, ΔLL = −10.5, P < 0.01; Figure 4). The central volume was halved between 50 and 90 kg, and CL was increased by 20% for CRP varying from 3 to 14 mg l−1 and was decreased by 30% with methotrexate. For a median subject not treated with methotrexate, typical distribution (T1/2α) and elimination half-lives (T1/2β) were 0.2 and 9.3 days, respectively. In a median subject cotreated with methotrexate, typical values were 0.2 and 13 days, respectively.
Figure 4.

Log values of pharmacokinetic parameters (top) and random effects (ETA; bottom). Central volume of distribution is influenced by bodyweight (WT on VC), and systemic clearance is influenced by C-reactive protein concentration at inclusion (CRP on CL) and by methotrexate cotreatment (MTX on CL)
Pharmacokinetic analysis including ATI+ patients
Finally, the model was run with all the 84 patients, including ATI+ patients. Estimated pharmacokinetic parameters and the influence of covariates were similar compared with the analysis without ATI+ patients. Notably, VC was still found to increase with WT (P < 0.01), and CL was still found to increase with pre-infusion CRP concentration (P < 0.01) and to be lower when methotrexate was administered (P < 0.01). In addition, CL was very much higher (around fivefold) in ATI+ patients (P < 0.01) than in other patients.
Discussion
To our knowledge, this is the first study using population pharmacokinetic modelling to describe infliximab pharmacokinetics in RA patients. This approach has been used previously to describe infliximab pharmacokinetics in ankylosing spondylitis [7,13] and in inflammatory bowel diseases [5,6]. In the study of ankylosing spondylitis by Xu et al., infliximab concentration data were sparse, with only trough and peak samples available [7]. In a different approach, St Clair et al. described infliximab pharmacokinetics in RA using noncompartmental methods [1]. Surprisingly, in our study, clearance of infliximab (0.46 l day−1) was higher than reported values in ankylosing spondylitis (around 0.25 l day−1 [7,13]) and in inflammatory bowel diseases (0.37 l day−1 [5,6]).
In our study, infliximab concentrations were satisfactorily described by a two-compartment model, and pharmacokinetic parameters were reliably estimated. The increase of the central volume of distribution of infliximab with bodyweight is in agreement with previous studies in other conditions [5–7,13]. We also observed that methotrexate cotreatment was associated with an increase of infliximab clearance. This is consistent with other studies in RA, which report increased serum infliximab concentrations when methotrexate was coadministered with infliximab [8].
In addition to these previously reported covariates, we observed an increase in infliximab clearance with higher CRP concentration before infusion. The patients were expected to be at steady state because infliximab doses were unchanged since the previous infusion before inclusion. A concentration–response relationship of infliximab has been reported in RA [1–4]. Therefore, the relationship between pre-infusion CRP concentration and infliximab clearance may be explained by a higher patient exposure to infliximab during the dosing interval being associated with lower CRP concentration at the end of the dosing interval.
However, the relationship between infliximab clearance and pre-infusion CRP may also be explained by target-mediated drug disposition, a mechanism of elimination frequently reported for monoclonal antibodies [14,15]. Production of TNF-α leads to increased CRP concentrations [16,17], so serum CRP may be considered as an indirect marker of the TNF-α concentration. In patients with substantial inflammation, infliximab clearance is high because of its capture by TNF-α. In two studies analysing RA patients when infliximab treatment is initiated, an inverse relationship of this type was indeed observed between pretherapeutic CRP and infliximab trough concentrations [2,4]. Our population modelling confirms that differences in inflammation (disease activity) explain part of the interindividual variability in infliximab pharmacokinetics (Figure 5). The relationship between pre-infusion CRP and infliximab clearance in our population may reflect both the anti-inflammatory effect of previous infliximab infusion and the target-mediated clearance of infliximab. However, to be realizable, the full version of this model would require the estimation of target-mediated clearance, which would be estimable using target-mediated drug disposition models. In practice, to be used to describe the infliximab dose–concentration–effect relationship, this model would require pharmacokinetic data with nonlinear elimination, measurements of free TNF-α concentrations and, even better, measurements of infliximab–TNF-α complexes. Unfortunately, no nonlinear elimination was ever observed in patients with inflammatory diseases treated with infliximab, and relevant measurements of TNF-α have been difficult to obtain and, moreover, to interpret.
Figure 5.
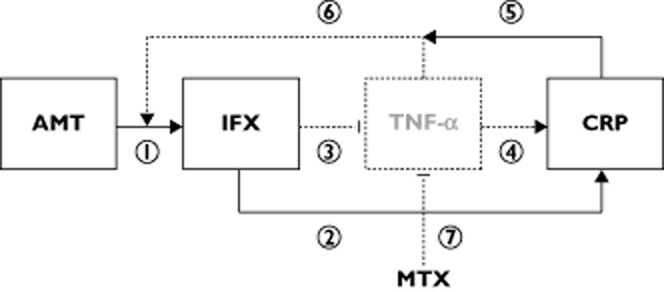
Dose–concentration–response relationship for infliximab in rheumatoid arthritis. Observed and latent relationships are represented in bold and dotted arrows or boxes, respectively. The boxes represent dose (AMT), infliximab concentrations (IFX), unobserved (latent) tumour necrosis factor-α (TNF-α) concentrations and C-reactive protein (CRP) concentrations. Arrows represent relationships between compartments, which are as follows: (1) the dose–concentration relationship (pharmacokinetic); (2) the observed concentratio CRP relationship; (3) the latent concentration–TNF-α relationship; (4) the latent TNF-α–CRP relationship; (5) the observed reciprocal influence of CRP on pharmacokinetics; (6) the latent target-antigen influence of TNF-α on infliximab pharmacokinetics; and (7) the inflammatory effect of methotrexate (MTX)
Interestingly, systemic inflammation, hence the TNF-α ‘burden’, is higher in RA than in ankylosing spondylitis [18]. This difference may explain the higher infliximab clearance in RA (0.46 l day−1) than in ankylosing spondylitis patients, which is around 0.25 days [7,13]. Methotrexate, acting as an anti-inflammatory drug, contributes to a significant decrease in TNF-α levels. Therefore, methotrexate may influence infliximab pharmacokinetics in RA patients by decreasing its target-mediated clearance. In cases of ankylosing spondylitis, the antigen burden is lower than in cases of RA, such that methotrexate may have a more limited effect on target-mediated drug disposition; this may explain the nonsignificant effects of methotrexate on infliximab pharmacokinetics in ankylosing spondylitis patients [13,19]. The differences between infliximab clearance in RA and inflammatory bowel diseases might be also explained by differences of antigenic burden expression between these diseases.
Our model showed that patients without methotrexate cotreatment and/or with high inflammatory activity may present a higher clearance of infliximab and may therefore be underexposed to treatment [2,4]. In addition, patients with low infliximab concentrations present a higher risk of developing ATI than other patients [20]. Therefore, a patient with high inflammatory disease may be at risk of both insufficient clinical response and development of ATI. Our model could be used to calculate the dose that should be administered.
Our study has some limitations. First, the interindividual variability in distribution clearance was not included in the model because shrinkage (underestimation of variability) was observed. This is a common phenomenon with monoclonal antibodies [5–7,21], and closely spaced sampling is required during the first day following the infusion to overcome this problem. Second, a slight overestimation of concentrations was observed in some patients near the lower limit of the range. This overestimation suggests either a potential bias in parameter estimates due to observation below the (0.04 mg l−1) or a nonlinear elimination of infliximab at low concentrations, presumably related to target-mediated disposition. However, the bias in parameter estimates should be limited, because concentrations <LLOQ are relatively seldom (27 of 412, i.e. 4.6% of total observations), and because the additive component of the error model (σadd = 0.15 mg l−1; Table 2) compensates for the shortage of information due to LLOQ. A model including nonlinear elimination was tested, but its parameters were not identifiable. Third, among the 84 patients included, only three were positive for ATI, a prevalence lower than previously reported [2,20,22,23]. This may reflect the cross-sectional design of our study. Given that our patients had been treated for a mean of 5.5 years, a large majority of them were good responders; patients who develop ATI are more likely to discontinue treatment and therefore be under-represented in a study with this design (Table 1). Fourth, because there were only three ATI+ patients, their inclusion in the population analysis could have influenced the statistical analysis and possibly led to erroneous conclusions. However, the results were similar with or without these patients in the population analysis data set. Finally, the number of ATI+ patients in this population was too small to investigate the reported decreased risk of ATI development associated with methotrexate [8].
Our study used a rich data set to describe infliximab pharmacokinetics in patients with RA. Methotrexate cotreatment and CRP concentrations at inclusion influenced infliximab clearance, suggesting that inflammatory activity, in association with the TNF-α target-antigen burden, affects infliximab pharmacokinetics. Patients with a high disease activity may therefore have lower exposure to infliximab than patients with low disease activity and, consequently, may benefit from increased infliximab doses [24], which may be calculated using our model.
Acknowledgments
The authors thank Dr Nicole Gando-Loembe and Stéphanie Gérard for recruiting patients; Dr Bruno Giraudeau for his methodological advice in the study design; Yoann Desvignes for technical support with the study protocol and data management; Hélène Bansard, Fanny Teasdale, Françoise Gouais and Fabienne Chapacou for blood sampling; Coraline Gadras and Céline Vignault for blood sample management; Anne-Claire Duveau for technical assistance with infliximab assays; Audrey Farnault for technical assistance for assays of antibody toward infliximab and Laura Heraty for her kind assistance with the manuscript. This work was promoted by the Regional University Hospital Center of Tours and supported by grants from the French Ministry for Health and Sport within the framework of the ‘Programme Hospitalier de Recherche Clinique 2007’. The Regional University Hospital of Tours received a FEDER (Fonds européen de développement regional – European funding for regional development) for its CePiBAc (Centre pilote de suivi biologique des anticorps thérapeutiques – Pilot centre for therapeutic antibodies monitoring). This work was a collaborative venture by HUGO (Hôpitaux Universitaires du Grand Ouest – Western France University Hospitals Network).
Competing Interests
ED has received fees for serving as a speaker for BMS; she has been personally invited to attend international congresses by Roche, Chugai, UCB, BMS and Abbott. BLG has received fees for serving as a speaker for Roche and Abbott; he has been personally invited to attend international congresses by Pfizer and Abbott; he received funding for scientific research from MSD and Roche. AC has received fees for serving as a speaker for Amgen; she has been personally invited to attend international congresses by MSD and Roche. VD-P has participated on behalf of her institution in clinical trials sponsored by Abbott, Roche, Chugai, BMS, Pfizer and UCB; she has been personally invited to attend international congresses by UCB, Chugai, Roche and Abbott. ES-G has participated on behalf of her institution in clinical trials sponsored by Roche and BMS, and her hospital received grants for research from Roche in 2010 and 2011; she has acted as a consultant and given lectures on behalf of her institution for Roche, Abbott, BMS and Pfizer; she has been personally invited to attend international congresses by BMS, Roche and Abbott. PG has participated on behalf of his institution in clinical trials sponsored by Abbott, BMS, Janssen, Lilly, MSD, Pfizer, Roche and UCB; he has acted as a consultant and given lectures for Abbott, BMS, Janssen, MSD, Pfizer and UCB; he has been personally invited to attend international congresses by Abbott, MSD and Pfizer. GP is a consultant for Laboratoires Français du Fractionnement et des Biotechnologies (LFB) and Pierre-Fabre Laboratories; his research team has received finance from Roche Pharma, Chugai, Pfizer, Novartis and Janssen. DM has participated on behalf of his institution in clinical trials sponsored by Abbott, Roche, BMS, Pfizer, UCB and MSD; his hospital received a grant for research from Abbott in 2004; he has acted as a consultant and given lectures on behalf of his institution for MSD and Pfizer; he has been personally invited to attend international congresses by MSD, Roche, BMS and Abbott. The other authors have no competing interests to declare.
References
- 1.St Clair EW, Wagner CL, Fasanmade AA, Wang B, Schaible T, Kavanaugh A, Keystone EC. The relationship of serum infliximab concentrations to clinical improvement in rheumatoid arthritis: results from ATTRACT, a multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002;46:1451–1459. doi: 10.1002/art.10302. [DOI] [PubMed] [Google Scholar]
- 2.Bendtzen K, Geborek P, Svenson M, Larsson L, Kapetanovic MC, Saxne T. Individualized monitoring of drug bioavailability and immunogenicity in rheumatoid arthritis patients treated with the tumor necrosis factor alpha inhibitor infliximab. Arthritis Rheum. 2006;54:3782–3789. doi: 10.1002/art.22214. [DOI] [PubMed] [Google Scholar]
- 3.Mulleman D, Chu Miow Lin D, Ducourau E, Emond P, Ternant D, Magdelaine-Beuzelin C, Valat JP, Paintaud G, Goupille P. Trough infliximab concentrations predict efficacy and sustained control of disease activity in rheumatoid arthritis. Ther Drug Monit. 2010;32:232–236. doi: 10.1097/FTD.0b013e3181cc6fef. [DOI] [PubMed] [Google Scholar]
- 4.Wolbink GJ, Voskuyl AE, Lems WF, de Groot E, Nurmohamed MT, Tak PP, Dijkmans BA, Aarden L. Relationship between serum trough infliximab levels, pretreatment C reactive protein levels, and clinical response to infliximab treatment in patients with rheumatoid arthritis. Ann Rheum Dis. 2005;64:704–707. doi: 10.1136/ard.2004.030452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fasanmade AA, Adedokun OJ, Ford J, Hernandez D, Johanns J, Hu C, Davis HM, Zhou H. Population pharmacokinetic analysis of infliximab in patients with ulcerative colitis. Eur J Clin Pharmacol. 2009;65:1211–1228. doi: 10.1007/s00228-009-0718-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ternant D, Aubourg A, Magdelaine-Beuzelin C, Degenne D, Watier H, Picon L, Paintaud G. Infliximab pharmacokinetics in inflammatory bowel disease patients. Ther Drug Monit. 2008;30:523–529. doi: 10.1097/FTD.0b013e318180e300. [DOI] [PubMed] [Google Scholar]
- 7.Xu Z, Seitz K, Fasanmade A, Ford J, Williamson P, Xu W, Davis HM, Zhou H. Population pharmacokinetics of infliximab in patients with ankylosing spondylitis. J Clin Pharmacol. 2008;48:681–695. doi: 10.1177/0091270008316886. [DOI] [PubMed] [Google Scholar]
- 8.Maini RN, Breedveld FC, Kalden JR, Smolen JS, Davis D, Macfarlane JD, Antoni C, Leeb B, Elliott MJ, Woody JN, Schaible TF, Feldmann M. Therapeutic efficacy of multiple intravenous infusions of anti-tumor necrosis factor alpha monoclonal antibody combined with low-dose weekly methotrexate in rheumatoid arthritis. Arthritis Rheum. 1998;41:1552–1563. doi: 10.1002/1529-0131(199809)41:9<1552::AID-ART5>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 9.Fautrel B, Pham T, Mouterde G, Le Loet X, Goupille P, Guillemin F, Ravaud P, Cantagrel A, Dougados M, Puechal X, Sibilia J, Soubrier M, Mariette X, Combe B. Recommendations of the French Society for Rheumatology regarding TNFalpha antagonist therapy in patients with rheumatoid arthritis. Joint Bone Spine. 2007;74:627–637. doi: 10.1016/j.jbspin.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 10.Ternant D, Mulleman D, Degenne D, Willot S, Guillaumin JM, Watier H, Goupille P, Paintaud G. An enzyme-linked immunosorbent assay for therapeutic drug monitoring of infliximab. Ther Drug Monit. 2006;28:169–174. doi: 10.1097/01.ftd.0000189901.08684.4b. [DOI] [PubMed] [Google Scholar]
- 11.van Gestel AM, Prevoo ML, van 't Hof MA, van Rijswijk MH, van de Putte LB, van Riel PL. Development and validation of the European League Against Rheumatism response criteria for rheumatoid arthritis. Comparison with the preliminary American College of Rheumatology and the World Health Organization/International League Against Rheumatism Criteria. Arthritis Rheum. 1996;39:34–40. doi: 10.1002/art.1780390105. [DOI] [PubMed] [Google Scholar]
- 12.Comets E, Brendel K, Mentre F. Computing normalised prediction distribution errors to evaluate nonlinear mixed-effect models: the npde add-on package for R. Comput Methods Programs Biomed. 2008;90:154–166. doi: 10.1016/j.cmpb.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 13.Ternant D, Mulleman D, Lauféron F, Vignault C, Ducourau E, Wendling D, Goupille P, Paintaud G. Influence of methotrexate on infliximab pharmacokinetics and pharmacodynamics in ankylosing spondylitis. Br J Clin Pharmacol. 2012;73:55–65. doi: 10.1111/j.1365-2125.2011.04050.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gibiansky L, Gibiansky E. Target-mediated drug disposition model: approximations, identifiability of model parameters and applications to the population pharmacokinetic-pharmacodynamic modeling of biologics. Expert Opin Drug Metab Toxicol. 2009;5:803–812. doi: 10.1517/17425250902992901. [DOI] [PubMed] [Google Scholar]
- 15.Mager DE, Jusko WJ. General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn. 2001;28:507–532. doi: 10.1023/a:1014414520282. [DOI] [PubMed] [Google Scholar]
- 16.Maini RN, Elliott MJ, Brennan FM, Williams RO, Chu CQ, Paleolog E, Charles PJ, Taylor PC, Feldmann M. Monoclonal anti-TNF alpha antibody as a probe of pathogenesis and therapy of rheumatoid disease. Immunol Rev. 1995;144:195–223. doi: 10.1111/j.1600-065x.1995.tb00070.x. [DOI] [PubMed] [Google Scholar]
- 17.Elliott MJ, Maini RN. Anti-cytokine therapy in rheumatoid arthritis. Baillieres Clin Rheumatol. 1995;9:633–652. doi: 10.1016/s0950-3579(05)80306-1. [DOI] [PubMed] [Google Scholar]
- 18.Gratacos J, Collado A, Filella X, Sanmarti R, Canete J, Llena J, Molina R, Ballesta A, Munoz-Gomez J. Serum cytokines (IL-6, TNF-alpha, IL-1 beta and IFN-gamma) in ankylosing spondylitis: a close correlation between serum IL-6 and disease activity and severity. Br J Rheumatol. 1994;33:927–931. doi: 10.1093/rheumatology/33.10.927. [DOI] [PubMed] [Google Scholar]
- 19.Mulleman D, Lauféron F, Wendling D, Ternant D, Ducourau E, Paintaud G, Goupille P. Infliximab in ankylosing spondylitis: alone or in combination with methotrexate? A pharmacokinetic comparative study. Arthritis Res Ther. 2011;13:R82. doi: 10.1186/ar3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ducourau E, Mulleman D, Paintaud G, Chu Miow Lin D, Lauféron F, Ternant D, Watier H, Goupille P. Antibodies toward infliximab are associated with low infliximab concentration at treatment initiation and poor infliximab maintenance in rheumatic diseases. Arthritis Res Ther. 2011;13:R105. doi: 10.1186/ar3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mould DR, Baumann A, Kuhlmann J, Keating MJ, Weitman S, Hillmen P, Brettman LR, Reif S, Bonate PL. Population pharmacokinetics-pharmacodynamics of alemtuzumab (Campath) in patients with chronic lymphocytic leukaemia and its link to treatment response. Br J Clin Pharmacol. 2007;64:278–291. doi: 10.1111/j.1365-2125.2007.02914.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pascual-Salcedo D, Plasencia C, Ramiro S, Nuno L, Bonilla G, Nagore D, Ruiz Del Agua A, Martinez A, Aarden L, Martin-Mola E, Balsa A. Influence of immunogenicity on the efficacy of long-term treatment with infliximab in rheumatoid arthritis. Rheumatology (Oxford) 2011;50:1445–1452. doi: 10.1093/rheumatology/ker124. [DOI] [PubMed] [Google Scholar]
- 23.Wolbink GJ, Vis M, Lems W, Voskuyl AE, de Groot E, Nurmohamed MT, Stapel S, Tak PP, Aarden L, Dijkmans B. Development of antiinfliximab antibodies and relationship to clinical response in patients with rheumatoid arthritis. Arthritis Rheum. 2006;54:711–715. doi: 10.1002/art.21671. [DOI] [PubMed] [Google Scholar]
- 24.Mulleman D, Ducourau E, Paintaud G, Ternant D, Watier H, Goupille P. Should anti-TNF-alpha drug levels and/or anti-drug antibodies be assayed in patients treated for rheumatoid arthritis? Joint Bone Spine. 2012;79:109–112. doi: 10.1016/j.jbspin.2011.11.004. [DOI] [PubMed] [Google Scholar]