

Abstract
Aims
Although genetic, clinical and demographic factors have been shown to explain approximately half of the inter-individual variability in warfarin dose requirement in adults, less is known about causes of dose variability in children. This study aimed to identify and quantify major genetic, clinical and demographic sources of warfarin dose variability in children using modelling and simulation.
Methods
Clinical, demographic and genetic data from 163 children with a median age of 6.3 years (range 0.06–18.9 years), covering over 183 years of warfarin therapy and 6445 INR observations were used to update and optimize a published adult pharmacometric warfarin model for use in children.
Results
Genotype effects in children were found to be comparable with what has been reported for adults, with CYP2C9 explaining up to a four-fold difference in dose (CYP2C9 *1/*1 vs. *3/*3) and VKORC1 explaining up to a two-fold difference in dose (VKORC1 G/G vs. A/A), respectively. The relationship between bodyweight and warfarin dose was non-linear, with a three-fold difference in dose for a four-fold difference in bodyweight. In addition, age, baseline and target INR, and time since initiation of therapy, but not CYP4F2 genotype, had a significant impact on typical warfarin dose requirements in children.
Conclusions
The updated model provides quantitative estimates of major clinical, demographic and genetic factors impacting on warfarin dose variability in children. With this new knowledge more individualized dosing regimens can be developed and prospectively evaluated in the pursuit of improving both efficacy and safety of warfarin therapy in children.
Keywords: dose variability, paediatrics, pharmacogenetics, pharmacometrics, PKPD modelling, warfarin
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Genetic, clinical and demographic factors collectively make a significant contribution to the inter-individual variability in warfarin dose requirement in adults.
Less is known about the major causes of warfarin dose variability in children, including the relative importance of genetic, clinical and demographic factors.
WHAT THIS STUDY ADDS
This study shows that CYP2C9 and VKORC1 genotype, bodyweight, age, baseline and target INR, and time since initiation of therapy, but not CYP4F2 genotype, are significant causes of warfarin dose variability in children.
The model can provide the basis for development of more individualized warfarin dosing in children.
Introduction
Warfarin is the most commonly prescribed anticoagulant in both adults and children [1]. Although it has been in clinical use for over 60 years, warfarin therapy is still a clinical challenge due to the drug's narrow therapeutic range and large inter- and intra-individual variability in response to a fixed dose. Known factors contributing to the variability in dose requirement in adults include age, body size, co-morbidities, co-medications, vitamin K intake and genetic polymorphisms [2], with over 40% of the variability explained by polymorphisms in the two genes VKORC1 and CYP2C9 [3].
Less is known about the relative importance of genetic, clinical and demographic factors on warfarin dose variability in children, but data from small studies have started to emerge. Nowak-Göttl et al. [4], studying 34 warfarin-treated children with a median age of 15 years (range 1–19 years), found that age was the most important determinant, explaining 31.2% of the variability in dose requirement, while polymorphisms in VKORC1 and CYP2C9 only explained 2.8% and 0.5%, respectively. In contrast, Nguyen et al. [5] recently reported that, in 37 children with a mean age of 9.6 years (range 1.8–18.6 years), age explained only 12% and polymorphisms in VKORC1 and CYP2C9 accounted for 47% and 5% of the variability, respectively. A larger study by Moreau et al. [6] involving 83 children with a mean age of 8.4 years (range 0.2–18 years), reported that height explained 48.1% of the variability in dose requirement and polymorphisms in VKORC1 and CYP2C9 accounted for 18.2% and 2%, respectively. In the largest published pharmacogenetic study in children reported by Biss et al. [7], including 120 children with a median age of 11.7 years (range 1.7–18 years), height was found to account for 29.8%, VKORC1 for 26.6% and CYP2C9 for 12.8% of the variability in warfarin maintenance dose requirement. Hence, published results are inconclusive with regard to the relative importance of individual predictors.
Reported differences in the contribution of CYP2C9 and VKORC1 genotype to the variability in warfarin dose requirement in children could partly be explained by differences in minor allele frequencies between studies [8], but also heterogeneity in study design and analysis approaches [2], as well as the small sample sizes, which cause uncertainty in reported point estimates for rare covariate effects. True genotype–phenotype relationships may also be obscured when analyzing data from studies in infants and young children depending on the importance of ontogeny on enzyme activity levels [9]. For some of the polymorphically expressed CYPs, ontogeny has been shown to play a major role in explaining interindividual variability in enzyme activity in newborns and infants, and makes the genotype–phenotype relationship less evident in these age groups [9]. For the CYP2C9 enzyme published data indicate complete maturation around 6 months of age [10]. Corresponding information about the ontogeny of the VKORC1 enzyme is lacking.
Adult patients who possess CYP2C9 and/or VKORC1 polymorphisms have been shown to be more sensitive to warfarin and prone to supra-therapeutic INRs during warfarin initiation [3,11]. A recent study [12] reported that these polymorphisms also play an important role in the early response to warfarin therapy in children. Children with at least one CYP2C9 variant allele had higher mean peak INR values during the first week of therapy, and a higher proportion of INR values above target range during the first month of therapy, than children with wild-type CYP2C9. Children with the VKORC1 −1639 A/A genotype had higher mean peak INR values and a tendency to more INR values being above target range during the first month of therapy than those with the G/A or G/G genotype [12].
Pharmacometric models developed on adult data are increasingly being used to improve dose predictions in children [13,14]. Using a pharmacometric approach when analyzing clinical data can alleviate many of the difficulties encountered with data derived from studies in children. A model-based approach can benefit from the possibility to incorporate prior knowledge, and to pool data across studies. The approach also enables use of sparse and unstructured real-life data, rather than relying on data derived from stringent clinical trial settings [15]. It is also possible to bridge from an existing adult model to paediatrics through the use of physiological principles that acknowledge that adults and children are in some aspects similar and not discrete subpopulations [13,14,16]. Pharmacometric modelling and simulation are also advocated by regulatory bodies to support the selection of safe and efficacious dosing regimens for children [17,18].
We have previously developed a PKPD-based model that describes the relationship between warfarin dose and the International Normalized Ratio (INR) response in adults [19,20], and which takes into account variation in pharmacokinetics (PK) due to age and CYP2C9 genotype, and variation in pharmacodynamics (PD) due to VKORC1 genotype. By estimating allele effects rather than genotype effects, the model development is less sensitive to small sample sizes since individuals with two variant alleles are not required for estimation of rare genotype effects. The individual genotype effect is obtained by simply adding the estimated allele effects. We have theoretically adapted the adult model [20] for use in children [21] through allometric weight scaling of clearance and volume of distribution [22], and addition of a published maturation function for CYP2C9 enzyme activity [10] to handle the influence of enzyme ontogeny in neonates and infants [9]. External validation of the theoretically bridged model on data from 64 children 0–18 years old showed that the model overall performed well, but with a tendency to over predict INR response, or underestimate the dose, in children ≤2 years old [21].
The main objective of the present study was to identify and quantify major genetic, clinical and demographic sources of warfarin dose variability in children using modelling and simulation.
Methods
Patients and treatment data
Data from two observational warfarin studies in children with underlying heart disease [7,21] were used in the current analysis. The two studies are only briefly described here as relevant to this work. One study recruited 93 children at four hospitals in the UK and 27 children at one hospital in Canada; the other included 67 children from four hospitals in Sweden. Warfarin was administered using formulations available in the respective countries: 2.5 mg tablets or extemporaneous 0.1 mg or 0.3 mg capsules in Sweden, 1, 3 or 5 mg tablets in the UK, and 1, 2, 2.5, 3, 4, 5, 6, 7.5 or 10 mg tablets in Canada. Local ethics committees approved the studies and informed consent was obtained from patients 18 years or older or from parents of children below 18 years.
Longitudinal treatment data were collected from hospital records and patient charts, including information on treatment indication, baseline INR, target INR, warfarin doses, INRs, date of birth, height and total bodyweight. All children were genotyped for VKORC1 −1639 G > A (rs9923231), CYP2C9 *2 and *3 (rs1799853 and rs1057910) and CYP4F2 *3 (rs2108622). The patients' ethnic background was self-reported according to pre-specified categories.
One hundred and sixty-three of the 187 children enrolled had sufficiently detailed treatment records to be included in the analysis. Seventy-eight children had data available from treatment start. For the other 85 children a sufficiently long dosing history to ascertain pharmacokinetic steady-state was required before INR observations were included in the analysis. The assumption on steady-state was based on CYP2C9 genotype and predicted half-lives for S-warfarin, using the following set of rules: at least 7 (*1/*1), 10 (*1/*2), 14 (*1/*3 or *2/*2) or 21 days (*2/*3) of dosing information before inclusion of an INR (no *3/*3 included). The assumption was tested in a sensitivity analysis by doubling the treatment length, with no impact on the results, suggesting that the rules applied were robust.
The measured effect of warfarin therapy is the change in INR from baseline, i.e. if baseline INR is 1.0 and the measured INR is 2.8, the treatment effect is a ΔINR of 1.8. Baseline INR was only available in 35 children and ranged from 1.0–1.7, with a median of 1.2. Attempts were made to estimate baseline INR for those children where this was missing, but the data were not informative enough to separate baseline and EC50 when estimated simultaneously. Instead we used the observed baseline when available, and fixed it to the median value of 1.2 for children with missing baseline values.
Model development
All modelling was performed in nonmem, version 7 (ICON, ICON Development Solutions, Ellicott City, MD, USA), using the first order conditional estimation method with interaction [23]. Analyses were carried out using an additive residual error model on log-transformed data, and with interindividual variability implemented using exponential models. Model selection was guided by (i) the decrease in the objective function value (the minimization criteria in nonmem), (ii) the condition number obtained from nonmem, (iii) graphical goodness-of-fit analysis in Xpose [24], (version 4.3.5 [xpose.sourceforge.net]) and R (version 2.14.2 [http://www.r-project.org]), (iv) the estimated uncertainty in parameter estimates as reported by the standard error obtained from nonmem, (v) simulation based diagnostics (prediction corrected visual predictive checks (pc-VPCs)) and (vi) plausibility of parameter estimates. The difference in the objective function value between two hierarchical models is approximately χ2-distributed, meaning that a nominal difference of 3.84 corresponds to a P value <0.05 for one degree of freedom. Criteria for final model selection included: (i) a statistically significant improvement over a competing simpler model, (ii) a condition number that was reasonably low (<1000), (iii) reasonable sized standard errors, and with 95% confidence intervals not including zero or unity (whatever applicable), (iv) pc-VPCs showing good agreement between observed and model predicted INRs and (v) biologically plausible estimates of model parameters.
Base model
This work is a further development of our published adult PKPD-based warfarin model with S-warfarin as the only exposure predictor for INR response [20], or rather a theoretically scaled version of this model for use in children [21]. Scaling to children was achieved by adding bodyweight to the adult model using allometric scaling [22], and addition of a published maturation function for CYP2C9 enzyme activity [10]. Both models are adapted according to the KPD-framework, as described by Jacqmin et al. [25]. This means that the models operate without PK observations, but make use of available information about variability in PK on a population level, e.g. the known influence of CYP2C9 genotype on the typical clearance (CL) of S-warfarin. A drawback compared with a full PKPD-model is that it becomes more difficult to separate remaining sources of variability into PK and PD, respectively, and hence less mechanistic interpretation can be made from a KPD-model.
Equation 1 provides the general formula for the prediction of clearance (CL) of S-warfarin as a function of age (CYP2C9 enzyme ontogeny) and size (bodyweight), and equation 2 provides the formula for the prediction of volume of distribution (V) of S-warfarin as a function of size (bodyweight):
| (1) |
| (2) |
CLi and Vi represent the typical parameter values in an individual with the bodyweight BWi, and CLs and Vs represent typical parameter values in an individual with the bodyweight BWs. AGEi is the post-natal age expressed in years of an individual with the bodyweight BWi. Using the published maturation function, CYP2C9 enzyme activity in a typical child is mature and reaches adult values at the age of 6 months.
Figure 1 provides a schematic picture and a brief description of the theoretically scaled warfarin model [21], which served as the base model in this analysis.
Figure 1.
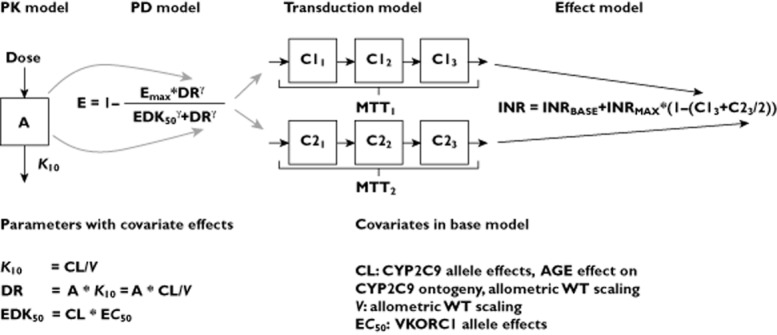
Schematic picture of the bridged warfarin model [21] used as base model in this study and a list of covariate effects included in the base model. The model consists of a one compartment PK model that describes the relationship between warfarin dose and S-warfarin concentrations (PK), and an inhibitory Emax-model to describe inhibition of the vitamin K cycle (PD), with S-warfarin as the only predictor of the anticoagulant response. Included also is a transduction model that captures the time delay between the inhibition of the vitamin K cycle, and the decrease in functioning coagulation factors (II, VII, IX and X), and finally an effect model that translates the decrease in functioning coagulation factors to an increase in INR. Compartment A represents amount of warfarin in the body, K10 the rate constant governing drug elimination from the body but also distribution to the site of action, here denoted DR for dose rate. EDK50, is the dose rate that results in 50% of the maximum effect Emax and MTT is the mean transit time through the transduction-model
Covariate selection
The base model included CYP2C9 genotype, bodyweight and age as predictors of CL, bodyweight as predictor of V and VKORC1 genotype as predictor of EC50. Additional covariates tested when adapting and optimizing the model for children included age, gender, bodyweight, indication, time since treatment start, dose, and CYP4F2 genotype (tested as allele effects). Covariates were screened for using scatter plots of individual parameter estimates (K10 and EC50) against individual covariates, and by generalized additive modelling (GAM) using Xpose [24]. Nested models were compared statistically using a likelihood ratio test on the differences in objective function value (OFV).
Model evaluation
VPCs were used to evaluate the predictive performance of candidate models. The principle of a VPC is to assess graphically whether simulations from a model are able to reproduce both the central trend (the median curve) and the variability (outer percentiles) in the observed data when plotted against an independent variable (e.g. time, age or bodyweight). A prediction corrected VPC (pc-VPC) is a modification of the standard VPC that is appropriate to apply on data collected in studies with an adaptive design [26] as in the case with warfarin. All pc-VPCs were constructed based on 500 simulated replicates of the original dataset design.
Predictions of maintenance dose
The final model was used to predict the warfarin maintenance dose for typical children aged 2, 8 and 14 years old, adopting the approach described by Jönsson & Karlsson [27]. Briefly, this entails (i) defining a target steady-state INR response, in our case 2.5, and (ii) using the model to estimate the dose required to achieve the target response, conditioned on the population (mean) parameter estimates.
Results
Patient and treatment data
Of 187 children enrolled in the two warfarin studies [7,21], 163 had sufficiently detailed data records to be included in this analysis. Demographic information, including treatment indication, target INR and genotype frequencies for VKORC1 −1639 G > A (rs9923231), CYP2C9 *2 and *3 (rs1799853 and rs1057910) and CYP4F2 *3 (rs2108622) are shown in Table 1. All genotypes were in Hardy–Weinberg equilibrium. Age and weight for the entire study period ranged from 0.06–18.9 years and 3.6–100.1 kg, respectively. The median dose from all phases of therapy was 2.5 mg (range, 0.125–12.5 mg) or 0.112 mg kg−1 (range, 0.0156–0.585 mg kg−1), respectively. Self-reported ethnic origin included 76.7% White, 7.4% Asian, 4.9% Black African and 11% other. Treatment data varied in length from 3 weeks up to 10 years per child, comprising a total of 6445 INR observations covering more than 183 treatment years. Figure 2 provides a raw data plot of all INR observations per subject, showing the age span for which treatment data were available. Coherent treatment records that included data after a child had turned 18 years old were also included in the analysis to increase the amount of data per individual. There were in total 55 INR observations from seven subjects that were between 18 and 19 years old.
Table 1.
Patient and clinical characteristics
| Gender, n (% male/female) | 105/58 (64/36) |
| Median age, years (range) | 6.3 (0.066–19) |
| Median weight, kg (range) | 20 (3.6–100.1) |
| CYP2C9 genotype, n (%) | |
| *1/*1 | 116 (71.2) |
| *1/*2 | 20 (12.3) |
| *1/*3 | 23 (14.1) |
| *2/*2 | 2 (1.2) |
| *2/*3 | 2 (1.2) |
| *3/*3 | – |
| VKORC1 genotype, n (%) | |
| G/G | 57 (35.0) |
| A/G | 79 (48.5) |
| A/A | 27 (16.5) |
| CYP4F2 genotype, n (%) | |
| C/C | 87 (53.4) |
| C/T | 66 (40.5) |
| T/T | 10 (6.1) |
| Treatment indication, n (%) | |
| Fontan procedure | 75 (46.0) |
| Prosthetic heart valve | 34 (20.8) |
| Dilated cardiomyopathy | 15 (9.2) |
| Coronary aneurysm | 11 (6.7) |
| Other | 28 (17.3) |
| Target INR, n (%) | |
| 2.0–3.0 | 119 (73.0) |
| 2.5–3.5 | 32 (19.6) |
| Other (lower range <2 or upper range >3.5) | 8 (4.8) |
| Missing | 4 (2.5) |
| INR observations, n (%) assayed with | |
| Owren based method | 4298 (66.7) |
| Quick based method | 2147 (33.3) |
| Total number of children, n (%) | 163 |
| Swedish study [21] | 64 (39.3) |
| UK/Canadian study [7] | 99 (60.7) |
Figure 2.
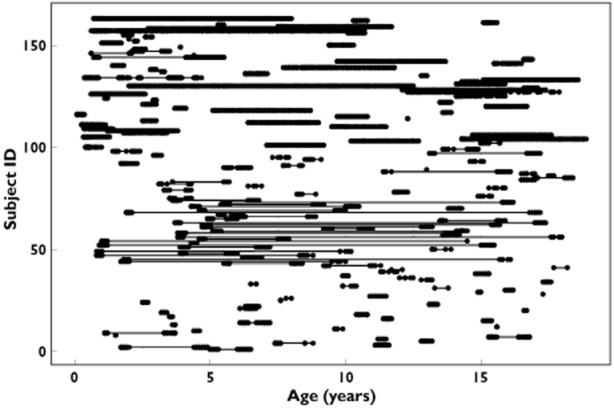
Raw data plot of INR observations included in the analysis, and where the dots along the x-axis show the age span for which observations were available per child. INR observations from several treatment periods in the same child are connected with a line. ID 1–99 represent children from the British/Canadian study and ID 100–163 represent children from the Swedish study
Final model
The theoretically scaled warfarin model [21] was used as the base model in this analysis. All model parameters in the base model, except the Hill factor in the Emax-model, were re-estimated on the paediatric dataset. Data supported addition of two new covariates to the base model: an exponential age-effect on the elimination rate constant K10, and an exponential effect of time since treatment start on EC50, the parameter describing sensitivity to warfarin.
The principal impact on K10 and EC50, respectively, are shown in Equations 3 and 4:
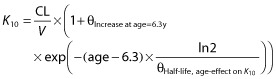 |
(3) |
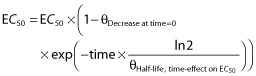 |
(4) |
where θIncrease at age=6.3y describes the relative increase in K10 in a 6.3 year old child and 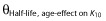 the half-life of the age-effect on K10, and θDecrease at time = 0 describes the decrease in EC50 at treatment start (time = 0) and
the half-life of the age-effect on K10, and θDecrease at time = 0 describes the decrease in EC50 at treatment start (time = 0) and 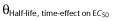 the half-life of the time-effect on EC50. This means that the model predicts that the elimination rate of warfarin (K10) is higher than that predicted from bodyweight (allometric weight scaling of CL and V) and age (maturation function on CL). For example, at the age of 2 years the model predicts a K10 that would result in a half-life of S-warfarin (t1/2 = ln2/K10) approximately five times shorter than that predicted without this additional age-effect. With the time effect on EC50, the model predicts that children are more sensitive to warfarin at the start of therapy, with an approximate 60% reduction of EC50. This effect showed a half-life of 170 h (7 days), which means that the effect disappears after approximately 4 weeks (four half-lives) of therapy.
the half-life of the time-effect on EC50. This means that the model predicts that the elimination rate of warfarin (K10) is higher than that predicted from bodyweight (allometric weight scaling of CL and V) and age (maturation function on CL). For example, at the age of 2 years the model predicts a K10 that would result in a half-life of S-warfarin (t1/2 = ln2/K10) approximately five times shorter than that predicted without this additional age-effect. With the time effect on EC50, the model predicts that children are more sensitive to warfarin at the start of therapy, with an approximate 60% reduction of EC50. This effect showed a half-life of 170 h (7 days), which means that the effect disappears after approximately 4 weeks (four half-lives) of therapy.
When the two additional covariates were included stepwise in a multivariate model, both remained significant. Following a confirmatory backward stepping approach, both remained significant, with a ΔOFV of 189 and 150 upon removal of the age-effect on K10 and the time-effect on EC50, respectively. Also the objective function value dropped from −10586.8 in the base model to −10906.1 in the final model, i.e. a ΔOFV of −319.3. The final model parameters are reported in Table 2, together with parameter estimates from the base model and adult parameter values [21] as reference. The proportion of unexplained variability in K10 dropped from 160% in the base model to 124% in the final model, and the residual error in INR response from 25.4% to 24.9%. ETA shrinkage in K10 and EC50 was 37.1% and 8.6%, respectively. As shrinkage in K10 was relatively high, post hoc plots were only used for guidance and all covariate effects were tested formally using the likelihood ratio test. The condition number was 250, which is reasonable and not indicative of over-parameterization.
Table 2.
Model parameters (%RSE) for (I) base model estimated in adults [21], (II) base model re-estimated in children and (III) final covariate model in children
| (I) Base model with parameters estimated in adults | (II) Base model with parameters estimated in children | (III) Final covariate model in children | |
|---|---|---|---|
| Objective function value | −10586.8 | −10906.1 | |
| Structural model parameters | |||
| EC50 per VKORC1 | |||
| - G allele (mg l−1) | 1.62 (10) | 2.05 (4.6) | 2.02 (4.3) |
| - A allele (mg l−1) | 0.770 (9.5) | 0.91 (10.4) | 0.997 (9.8) |
| MTT1 (h) | 22.6 (3.3) | 42.7 (18.7) | 37.4 (0.6) |
| MTT2-MTT1 (h) | 87.8 (5.8) | 6.2 (295) | 156 (6.6) |
| MTT2 (h)* | 110 | 48.9 | 193 |
| Hill factor (γ), Emax model | 1.37 (4.9) | 1.37 FIX | 1.37 FIX |
| Age effect on K10 | |||
| - Increase at age 6.3 years (%) | 0.879 (35.5) | ||
| - Half-life, age-effect on K10 (years) | 0.488 (3.9) | ||
| Time effect on EC50 | |||
| - Decrease at time 0 (%) | 61.7 (0.7) | ||
| - Half-life, time-effect on EC50 (h) | 170 (6.8) | ||
| Interindividual variability parameters (%) | |||
| ωEC50 | 32 (4.3) | 36 (6.6) | 33.9 (6.4) |
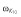
|
42 (8.3) | 159 (9.9) | 124 (11.3) |
| Residual error parameters (%) | |||
| σINR | 18 (1.7) | 25.4 (0.3) | 24.9 (0.3) |
MTT2 was not estimated but derived from MTT1 and MTT2-MTT1.
Attempts were made to re-estimate CYP2C9 allele effects on CL in children. However, due to the nature of the KPD model, with no input of drug concentrations, primary PK parameters such as CL and V were unidentifiable. The quantitative CYP2C9 allele effects on CL were therefore assumed to be the same in children as previously estimated for adults [20], an assumption that seems valid at least for children older than 6 months when the enzyme is expected to be fully matured. However, VKORC1 allele effects on drug sensitivity were successfully re-estimated in children. The quantitative effect was found to be very similar in children and adults, with a two-fold difference in dose between children with the G/G and A/A genotype. This indicates that the VKORC1 enzyme is also matured at an early age. The potential influence of genetic variation in the CYP4F2 gene has not been tested in the adult warfarin KPD-model. The gene codes for the CYP4F2 enzyme, which is involved in the metabolism of vitamin K, and carriers of a variant allele have been suggested to require a higher warfarin dose [28–30]. When CYP4F2 genotype was tested as a predictor for dose variability in children, there was a trend suggesting a 9% higher dose per T allele, but with a symmetrical 95% confidence interval ranging from a 3% lower to a 21% higher dose per T allele, hence including zero. In addition, the drop in OFV (−4.2) was only borderline significant, and resulted in a model with a condition number exceeding 2500, which is indicative of over-parameterization. Taken together, this did not support the retention of CYP4F2 genotype in the final model.
Internal model evaluation
Figure 3 shows pc-VPCs of the final model, both with and without stratification on age, and with stratification on VKORC1 genotype shown in Figure 4. Additional VPCs with stratification on CYP2C9 genotype or CYP4F2 genotype, or without stratification but with bodyweight or age as the independent variable, are included as Supporting Information Figures S1 and S2. Overall the model performed well in predicting the INR response in children, irrespective of age, bodyweight, CYP2C9, VKORC1 or CYP4F2 genotype. Observed INR values (Figure 4 and Supporting Information Figure S1) also confirmed previous findings that children with variant alleles have higher mean peak INR values at the start of therapy than those with wild-type genotypes [12].
Figure 3.
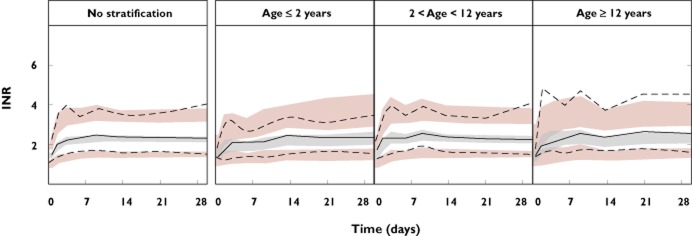
Prediction corrected VPCs for the optimized model applied on data from warfarin treated children. The panel to the left represents data from all children and the three panels to the right after stratification into three age groups; ≤2 years, >2 and <12 years, and ≥12 years. Solid lines denote the medians of observed data and dashed lines denote the 10th and 90th percentiles of observed data. Shaded areas represent 95% confidence intervals of simulated medians (grey) and 80% prediction intervals (pink). A model is considered to have good predictive properties if the lines representing the observed data mainly fall inside the simulated confidence intervals
Figure 4.
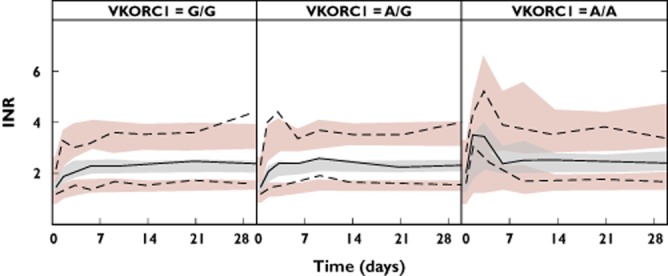
Prediction corrected VPCs but with data stratified on VKORC1 genotype, with from left to right VKORC1 G/G (decreased sensitivity to warfarin), VKORC1 A/G (intermediate sensitivity to warfarin) and VKORC1 A/A (increased sensitivity to warfarin)
Predictions of maintenance dose
Predicted maintenance doses for 54 typical children aged 2, 8 and 14 years old and with all possible combinations of CYP2C9 and VKORC1 genotypes, a baseline INR of 1 and a target INR of 2.5 are presented in Table 3. For example, the model predicts a three-fold difference in maintenance dose between a typical 2-year-old with a bodyweight of 13 kg and a 14-year-old with a bodyweight of 52 kg, conditioned on the same genotype combinations, despite a four-fold difference in bodyweight. For two children with the same age, bodyweight and CYP2C9 genotype, the model predicts approximately a two-fold difference in dose between a child carrying the VKORC1 −1639 G/G genotype and a child homozygous for the variant A allele. For two children who differ only in CYP2C9 genotype, the model predicts approximately a four-fold difference in dose between subjects having the wild-type genotype (CYP2C9*1/*1) and those being homozygous for the *3 allele (CYP2C9*3/*3). Predicted maintenance doses were 10–12% lower if the baseline INR was 1.2 instead of 1, and 25–30% higher if the target INR was set to 3 instead of 2.5.
Table 3.
Model predicted warfarin maintenance doses in mg (mg kg−1) for typical children 2, 8 or 14 years old, and with different combinations of CYP2C9 and VKORC1 genotypes, all with a baseline INR of 1 and a target INR of 2.5. The following bodyweights were assumed for a 2, 8 and 14-year-old: 13 kg, 26 kg and 52 kg, respectively
| VKORC1 G/G | VKORC1 A/G | VKORC1 A/A | |||||||
|---|---|---|---|---|---|---|---|---|---|
| CYP2C9 | 2 years | 8 years | 14 years | 2 years | 8 years | 14 years | 2 years | 8 years | 14 years |
| (13 kg) | (26 kg) | (52 kg) | (13 kg) | (26 kg) | (52 kg) | (13 kg) | (26 kg) | (52 kg) | |
| *1/*1 | 2.74 | 5.00 | 8.43 | 2.05 | 3.73 | 6.29 | 1.35 | 2.47 | 4.16 |
| (0.211) | (0.192) | (0.162) | (0.158) | (0.144) | (0.121) | (0.104) | (0.095) | (0.080) | |
| *1/*2 | 2.11 | 3.78 | 6.35 | 1.57 | 2.82 | 4.74 | 1.04 | 1.86 | 3.13 |
| (0.162) | (0.145) | (0.122) | (0.121) | (0.108) | (0.091) | (0.080) | (0.072) | (0.060) | |
| *1/*3 | 1.76 | 3.11 | 5.24 | 1.32 | 2.32 | 3.91 | 0.87 | 1.53 | 2.58 |
| (0.135) | (0.120) | (0.101) | (0.101) | (0.089) | (0.075) | (0.067) | (0.059) | (0.050) | |
| *2/*2 | 1.45 | 2.54 | 4.27 | 1.08 | 1.89 | 3.19 | 0.72 | 1.25 | 2.11 |
| (0.112) | (0.098) | (0.082) | (0.083) | (0.073) | (0.061) | (0.055) | (0.048) | (0.040) | |
| *2/*3 | 1.09 | 1.87 | 3.15 | 0.81 | 1.40 | 2.35 | 0.54 | 0.92 | 1.56 |
| (0.084) | (0.072) | (0.061) | (0.062) | (0.054) | (0.045) | (0.041) | (0.036) | (0.030) | |
| *3/*3 | 0.71 | 1.21 | 2.04 | 0.53 | 0.90 | 1.52 | 0.35 | 0.60 | 1.01 |
| (0.055) | (0.047) | (0.039) | (0.041) | (0.035) | (0.029) | (0.027) | (0.023) | (0.019) | |
Discussion
This is the largest study to date, which has evaluated the effect of genetic, clinical and demographic factors on warfarin dose variability in children. Despite warfarin being increasingly used to treat thromboembolic disorders in children, the number of children treated and eligible for pharmacogenetic studies is still limited. Novel approaches, such as modelling and simulation that can incorporate prior knowledge and pool data across studies, have been promoted as one way to improve the quality and robustness of analysis in small patient populations. To our knowledge this is the first time that data from warfarin treated children have been analyzed using modelling and simulation.
We have previously developed a PKPD-based warfarin model from large adult patient populations using population modelling. This model has now been updated and optimized for use in children. A major advantage of analyzing data using population modelling is that all available observations can be used in the analyses, irrespective of when during treatment data were collected (at treatment start, during stable anticoagulation or following dose revisions). To get accurate and reliable results it is imperative that the dose history is correctly reproduced, but this holds true regardless of the method of analysis. In the majority of published warfarin studies in both adults and children, only treatment data from a single time point at a period of stable anticoagulation are included in the analysis. This is likely to introduce a selection bias by excluding the patients who are the most difficult to treat, and also leads to a poor utilization of available treatment data. The definition of stable anticoagulation also varies between studies [2], which further complicates interpretation and comparability of study results. In the current analysis, treatment data from 163 warfarin treated children have been used, including data from all phases of therapy, and representing over 183 treatment years, i.e. on average >1 treatment year/child. A limitation might on the other hand be that our study was restricted to mainly Caucasian children (77%), all with congenital or acquired heart disease. However, the homogeneity of our study population enabled us to detect an increased sensitivity to warfarin at the start of therapy (as a time-effect on EC50). This is in line with results reported by Moffett et al. who discovered that cardiac surgery was a risk factor for elevated INR values in children starting warfarin therapy, with an odds ratio of 4.1 (95% CI 1.5, 11.6) [31]. Rahman et al. also reported an increased sensitivity to warfarin at the start of therapy in adults undergoing heart valve replacement. The mean maintenance dose (1–3 months after surgery) was 43% higher than the mean initiation dose (5.09 vs. 3.55 mg; P < 0.001) [32]. It can therefore be speculated that both children and adults undergoing major surgery are more sensitive to warfarin. Reasons for this could be temporary physiological changes after major surgery that may influence the PK and/or PD of warfarin, combined with a decreased appetite and a lower intake of vitamin K postoperatively. Lack of physical activity postoperatively may also influence dose requirements, since it has been shown that increased physical activity may lead to increased warfarin dose requirements in adults [33].
The most common way to report the contribution of individual factors to warfarin dose requirement is by estimating the percentage in dose variability explained by a given factor [3–7]. However, the clinical utility of this measure, when evaluating predictors important for individualized dosing, may be questioned. The percentage of variability explained by a given factor is a measure of what is most important for the overall variability in the population, and takes into account the effect size of a given factor and also how common this factor is in the study population. For dose prediction on an individual basis, a more relevant measure would be how much a given factor can influence the dose in an individual patient. VKORC1 is usually ascribed a more important role on a population level due to the fact that VKORC1 polymorphisms are more common than CYP2C9 polymorphisms in most populations [8]. However, we have previously shown that polymorphisms in the VKORC1 gene typically explain up to a two-fold difference in warfarin dose in adult patients, while the difference in dose between typical individuals with different CYP2C9 genotypes can be up to 4.2-fold. With the optimized pharmacometric model for children we demonstrate that CYP2C9 and VKORC1 genotypes also have comparable quantitative effects on dose variability in children.
Evidence that the CYP4F2 genotype can influence warfarin dose requirement was first presented in 2008 [28] and later confirmed by others [29,30]. However, it has been questioned whether the contribution of CYP4F2 is sufficient for inclusion in pharmacogenetic dosing algorithms [34]. In our study we found a 9% increase in warfarin dose per CYP4F2 T (*3) allele, but with the 95% CI ranging from a 3% reduction to a 21% increase in dose per T allele. The relatively small effect, together with poor precision in the estimate that also included zero, did not support retention of the CYP4F2 genotype in the final model. This is also in agreement with results reported by Moreau et al. [6] and Biss et al. [7], who found no association between warfarin maintenance dose and CYP4F2 genotype in children.
A general dosing recommendation in children is to adjust the warfarin dose according to bodyweight [1,4,5]. The final model also supports that individual dose requirements are dependent on bodyweight, but that the relationship is non-linear. For example, the model only predicts a three-fold difference in dose (in mg) for a four-fold difference in bodyweight, which means that if adult doses are linearly scaled to children this will lead to under-dosing. This is in line with findings from other warfarin studies in children, which have concluded that a higher weight-normalized dose is required, especially in the younger age groups [35,36].
The model-predicted maintenance dose for typical children 2, 8 and 14 years old ranged from 0.019 mg kg−1 to 0.21 mg kg−1 (Table 3). This should be compared with observed maintenance doses of warfarin in children, which are reported to range from 0.03 to 0.39 mg kg−1 by Nowak-Göttl et al. [4], from 0.04 to 0.32 mg kg−1 by Nguyen et al. [5], from 0.03 to 0.50 mg kg−1 by Moreau et al. [6], from 0.02 to 0.43 mg kg−1 by Biss et al. [7], and from 0.04 to 0.29 mg kg−1 by Hamberg et al. [21]. The upper range of model-predicted warfarin doses was hence lower (0.21 mg kg−1) than reported in these five studies (0.50 mg kg−1). However, this is not unexpected since the model-predicted doses are conditioned on a target INR of 2.5 and limited to children 2–14 years old, whereas observed doses also included children with a higher target INR and a broader age range (3 months–18.6 years). In addition, the model-predicted doses presented here only represent typical doses and do not take the unexplained between-subject variability into account. Overall the agreement between model-predicted and observed maintenance doses appeared to be good.
In conclusion, we have developed the first PKPD-based pharmacometric model that provides quantitative estimates of genetic, clinical and demographic sources of warfarin dose variability in children. The impact of genotype on dose variability in children was found to be comparable with what has been reported for adults, with CYP2C9 explaining up to a four-fold difference in dose (CYP2C9 *1/*1 vs. *3/*3) and VKORC1 explaining up to a two-fold difference in dose (VKORC1 G/G vs. A/A), respectively. Bodyweight, age, baseline and target INR, and time since initiation of therapy, but not CYP4F2 genotype, were also found to influence significantly typical warfarin dose requirements in children. The dependence of warfarin dose requirements on bodyweight is non-linear, and an increased sensitivity to warfarin at the start of therapy can be speculated to only be present after major surgery.
The model provides the basis for development of a computerized tool for a priori and a posteriori dose-individualization in the pursuit of improving efficacy and safety of warfarin therapy in children. A prototype for a dose individualization tool has already been developed for adults [37]. Our dosing model should now be validated and prospectively evaluated to determine whether it will aid effective and safe warfarin therapy in children. A randomized, controlled clinical trial against standard-of-care would be the ultimate way to investigate if the model could improve INR control in warfarin treated children. However, this is a huge task and is probably best done through an international collaboration that includes children with a diverse clinical and ethnic background.
Acknowledgments
We thank all the children and young adults and their parents/caregivers who participated in the studies. We also want to acknowledge the following persons for involvement in patient recruitment: B-M Ekman-Joelsson and J Sunnegårdh, The Queen Silvia Children's Hospital, Gothenburg; K Hanséus, Children's Heart Centre, Lund; B Lundell, Astrid Lindgren Children's Hospital, Stockholm; A Jonzon, Uppsala University Children's Hospital, Uppsala; L.R. Brandao, The Hospital for Sick Children, Toronto; E.A. Chalmers, Royal Hospital for Sick Children, Glasgow; M.D. Williams, Birmingham Children's Hospital, Birmingham; J.D. Grainger, Royal Manchester Children's Hospital, Manchester; P Walsh, The Newcastle upon Tyne Hospitals National Health Services Trust, Newcastle upon Tyne.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf and declare: A-K.H. had support from the Ränk family via the Swedish Heart and Lung Foundation and the Swedish Academy of Pharmaceutical Sciences, M.W. had support from the Swedish Research Council (Medicine 523–2008-5568 and 521–2011-2440), the Swedish Heart and Lung Foundation and the Clinical Research Support (ALF) at Uppsala University and T.T.B. had support from Baxter and the Royal College of Pathologists (research training fellowship) for the submitted work. There was no financial relationship with any other organization that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Prediction corrected VPCs for the optimized model applied on data from 163 children but stratified on (A) CYP2C9 genotype or (B) CYP4F2 genotype. Figure 1A includes from left to right data from children with CYP2C9 *1/*1 (high enzyme activity), CYP2C9 *1/*2 or *1/*3 (intermediate enzyme activity) and CYP2C9 *2/*2, *2/*3 or *3/*3 (low enzyme activity). The right panel, representing children with rare CYP2C9 genotypes, includes data from only four children and results in wide prediction intervals that overlap. Figure 1B includes from left to right data from children with CYP4F2 *1/*1 (wild-type), CYP4F2 *1/*3 (one variant allele) and CYP4F2 *3/*3 (two variant alleles). There is no systematic bias in the VPCs suggesting that a child with the CYP4F2 variant allele requires a higher warfarin dose
Figure S2 Prediction corrected VPCs for the optimized model applied on data from 163 warfarin treated children but using body weight (A) or age (B) as the independent variable (x-axis) instead of time. Solid lines denote the medians of observed data and dashed lines denote the 10th and 90th percentiles of observed data. Shaded areas represent 95% confidence intervals of simulated medians (grey) and 80% prediction intervals (pink). A model is considered to have good predictive properties if the lines representing the observed data mainly fall inside the simulated prediction intervals
References
- 1.Monagle P, Chan AKC, Goldenberg NA, Ichord RN, Journeycake JM, Nowak-Gottl U, Vesely SK. Antithrombotic Therapy in Neonates and Children: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 Suppl):e737S–e801S. doi: 10.1378/chest.11-2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jorgensen AL, FitzGerald RJ, Oyee J, Pirmohamed M, Williamson PR. Influence of CYP2C9 and VKORC1 on patient response to warfarin: a systematic review and meta-analysis. Novelli G, editor. PLoS ONE. 2012;7:e44064. doi: 10.1371/journal.pone.0044064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wadelius M, Chen LY, Lindh JD, Eriksson N, Ghori MJR, Bumpstead S, Holm L, McGinnis R, Rane A, Deloukas P. The largest prospective warfarin-treated cohort supports genetic forecasting. Blood. 2009;113:784–792. doi: 10.1182/blood-2008-04-149070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nowak-Göttl U, Dietrich K, Schaffranek D, Eldin NS, Yasui Y, Geisen C, Mitchell LG. In pediatric patients, age has more impact on dosing of vitamin K antagonists than VKORC1 or CYP2C9 genotypes. Blood. 2010;116:6101–6105. doi: 10.1182/blood-2010-05-283861. [DOI] [PubMed] [Google Scholar]
- 5.Nguyen N, Anley P, Yu MY, Zhang G, Thompson AA, Jennings LJ. Genetic and clinical determinants influencing warfarin dosing in children with heart disease. Pediatr Cardiol. 2012;34:984–990. doi: 10.1007/s00246-012-0592-1. [DOI] [PubMed] [Google Scholar]
- 6.Moreau C, Bajolle F, Siguret V, Lasne D, Golmard JL, Elie C, Beaune P, Cheurfi R, Bonnet D, Loriot MA. Vitamin K antagonists in children with heart disease: height and VKORC1 genotype are the main determinants of the warfarin dose requirement. Blood. 2012;119:861–867. doi: 10.1182/blood-2011-07-365502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biss TT, Avery PJ, Brandao LR, Chalmers EA, Williams MD, Grainger JD, Leathart JBS, Hanley JP, Daly AK, Kamali F. VKORC1 and CYP2C9 genotype and patient characteristics explain a large proportion of the variability in warfarin dose requirement among children. Blood. 2012;119:868–873. doi: 10.1182/blood-2011-08-372722. [DOI] [PubMed] [Google Scholar]
- 8.Limdi NA, Wadelius M, Cavallari L, Eriksson N, Crawford DC, Lee MTM, Chen CH, Motsinger-Reif A, Sagreiya H, Liu N, Wu AHB, Gage BF, Jorgensen A, Pirmohamed M, Shin JG, Suarez-Kurtz G, Kimmel SE, Johnson JA, Klein TE, Wagner MJ. Warfarin pharmacogenetics: a single VKORC1 polymorphism is predictive of dose across 3 racial groups. Blood. 2010;115:3827–3834. doi: 10.1182/blood-2009-12-255992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leeder JS, Kearns GL. Interpreting pharmacogenetic data in the developing neonate: the challenge of hitting a moving target. Clin Pharmacol Ther. 2012;92:434–436. doi: 10.1038/clpt.2012.130. [DOI] [PubMed] [Google Scholar]
- 10.Johnson TN, Rostami-Hodjegan A, Tucker GT. Prediction of the clearance of eleven drugs and associated variability in neonates, infants and children. Clin Pharmacokinet. 2006;45:931–956. doi: 10.2165/00003088-200645090-00005. [DOI] [PubMed] [Google Scholar]
- 11.Meckley LM, Wittkowsky AK, Rieder MJ, Rettie AE, Veenstra DL. An analysis of the relative effects of VKORC1 and CYP2C9 variants on anticoagulation related outcomes in warfarin-treated patients. Thromb Haemost. 2008;100:229–239. [PubMed] [Google Scholar]
- 12.Biss TT, Avery P, Williams MD, Brandao LR, Grainger JD, Kamali F. The VKORC1 and CYP2C9 genotypes are associated with over-anticoagulation during initiation of warfarin therapy in children. J Thromb Haemost. 2013;11:373–375. doi: 10.1111/jth.12072. [DOI] [PubMed] [Google Scholar]
- 13.Edginton AN. Knowledge-driven approaches for the guidance of first-in-children dosing. Pediatr Anesth. 2010;21:206–213. doi: 10.1111/j.1460-9592.2010.03473.x. [DOI] [PubMed] [Google Scholar]
- 14.Cella M, Gorter de Vries F, Burger D, Danhof M, Della Pasqua O. A model-based approach to dose selection in early pediatric development. Clin Pharmacol Ther. 2010;87:294–302. doi: 10.1038/clpt.2009.234. [DOI] [PubMed] [Google Scholar]
- 15.Krekels EHJ, van den Anker JN, Baiardi P, Cella M, Cheng KY, Gibb DM, Green H, Iolascon A, Jacqz-Aigrain EM, Knibbe CA, Santen GW, van Schaik RH, Tibboel D, Della Pasqua OE. Pharmacogenetics and paediatric drug development: issues and consequences to labelling and dosing recommendations. Expert Opin Pharmacother. 2007;8:1787–1799. doi: 10.1517/14656566.8.12.1787. [DOI] [PubMed] [Google Scholar]
- 16.Läer S, Barrett JS, Meibohm B. The in silico child: using simulation to guide pediatric drug development and manage pediatric pharmacotherapy. J Clin Pharmacol. 2009;49:889–904. doi: 10.1177/0091270009337513. [DOI] [PubMed] [Google Scholar]
- 17.Manolis E, Pons G. Proposals for model-based paediatric medicinal development within the current European Union regulatory framework. Br J Clin Pharmacol. 2009;68:493–501. doi: 10.1111/j.1365-2125.2009.03484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manolis E, Osman TE, Herold R, Koenig F, Tomasi P, Vamvakas S, Saint Raymond A. Role of modeling and simulation in pediatric investigation plans. Paediatr Anaesth. 2011;21:214–221. doi: 10.1111/j.1460-9592.2011.03523.x. [DOI] [PubMed] [Google Scholar]
- 19.Hamberg A-K, Dahl ML, Barban M, Scordo MG, Wadelius M, Pengo V, Padrini R, Jonsson EN. A PK-PD model for predicting the impact of age, CYP2C9, and VKORC1 genotype on individualization of warfarin therapy. Clin Pharmacol Ther. 2007;81:529–538. doi: 10.1038/sj.clpt.6100084. [DOI] [PubMed] [Google Scholar]
- 20.Hamberg A-K, Wadelius M, Lindh JD, Dahl ML, Padrini R, Deloukas P, Rane A, Jonsson EN. A pharmacometric model describing the relationship between warfarin dose and INR response with respect to variations in CYP2C9, VKORC1, and age. Clin Pharmacol Ther. 2010;87:727–734. doi: 10.1038/clpt.2010.37. [DOI] [PubMed] [Google Scholar]
- 21.Hamberg A-K, Friberg LE, Hanséus K, Ekman-Joelsson B-M, Sunnegårdh J, Jonzon A, Lundell B, Jonsson EN, Wadelius M. Warfarin dose prediction in children using pharmacometric bridging – comparison with published pharmacogenetic dosing algorithms. Eur J Clin Pharmacol. 2013;69:1275–1283. doi: 10.1007/s00228-012-1466-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anderson BJ, Holford NHG. Mechanism-based concepts of size and maturity in pharmacokinetics. Annu Rev Pharmacol Toxicol. 2008;48:303–332. doi: 10.1146/annurev.pharmtox.48.113006.094708. [DOI] [PubMed] [Google Scholar]
- 23.Bauer RJ. NONMEM User's Guide. Ellicott City, MD: Icon Development Solutions; 2011. pp. 1–128. [Google Scholar]
- 24.Jonsson EN, Karlsson MO. Xpose–an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed. 1999;58:51–64. doi: 10.1016/s0169-2607(98)00067-4. [DOI] [PubMed] [Google Scholar]
- 25.Jacqmin P, Snoeck E, Schaick EA, Gieschke R, Pillai P, Steimer JL, Girard P. Modelling response time profiles in the absence of drug concentrations: definition and performance evaluation of the K–PD model. J Pharmacokinet Pharmacodyn. 2006;34:57–85. doi: 10.1007/s10928-006-9035-z. [DOI] [PubMed] [Google Scholar]
- 26.Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011;13:143–151. doi: 10.1208/s12248-011-9255-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jönsson S, Karlsson MO. Estimation of dosing strategies aiming at maximizing utility or responder probability, using oxybutynin as an example drug. Eur J Pharm Sci. 2005;25:123–132. doi: 10.1016/j.ejps.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 28.Caldwell MD, Awad T, Johnson JA, Gage BF, Falkowski M, Gardina P, Hubbard J, Turpaz Y, Langaee TY, Eby C, King CR, Brower A, Schmelzer JR, Glurich I, Vidaillet HJ, Yale SH, Zhang KQ, Berg RL, Burmester JK. CYP4F2 genetic variant alters required warfarin dose. Blood. 2008;111:4106–4112. doi: 10.1182/blood-2007-11-122010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takeuchi F, McGinnis R, Bourgeois S, Barnes C, Eriksson N, Soranzo N, Whittaker P, Ranganath V, Kumanduri V, McLaren W, Holm L, Lindh J, Rane A, Wadelius M, Deloukas P. A genome-wide association study confirms VKORC1, CYP2C9, and CYP4F2 as principal genetic determinants of warfarin dose. Visscher PM, editor. PLoS Genet. 2009;5:e1000433. doi: 10.1371/journal.pgen.1000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borgiani P, Ciccacci C, Forte V, Sirianni E, Novelli L, Bramanti P, Novelli G. CYP4F2 genetic variant (rs2108622) significantly contributes to warfarin dosing variability in the Italian population. Pharmacogenomics. 2009;10:261–266. doi: 10.2217/14622416.10.2.261. [DOI] [PubMed] [Google Scholar]
- 31.Moffett BS, Ung M, Bomgaars L. Risk factors for elevated INR values during warfarin therapy in hospitalized pediatric patients. Pediatr Blood Cancer. 2011;58:941–944. doi: 10.1002/pbc.23308. [DOI] [PubMed] [Google Scholar]
- 32.Rahman M, BinEsmael TM, Payne N, Butchart EG. Increased sensitivity to warfarin after heart valve replacement. Ann Pharmacother. 2006;40:397–401. doi: 10.1345/aph.1G407. [DOI] [PubMed] [Google Scholar]
- 33.Lenz TL, Lenz NJ, Faulkner MA. Potential interactions between exercise and drug therapy. Sports Med. 2004;34:293–306. doi: 10.2165/00007256-200434050-00002. [DOI] [PubMed] [Google Scholar]
- 34.Bejarano-Achache I, Levy L, Mlynarsky L, Bialer M, Muszkat M, Caraco Y. Effects of CYP4F2 polymorphism on response to warfarin during induction phase: a prospective, open-label, observational cohort study. Clin Ther. 2012;34:811–823. doi: 10.1016/j.clinthera.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 35.Takahashi H. Developmental changes in pharmacokinetics and pharmacodynamics of warfarin enantiomers in Japanese children. Clin Pharmacol Ther. 2000;68:541–555. doi: 10.1067/mcp.2000.110977. [DOI] [PubMed] [Google Scholar]
- 36.Kato Y, Ichida F, Saito K, Watanabe K, Hirono K, Miyawaki T, Yoshimura N, Horiuchi I, Taguchi M, Hashimoto Y. Effect of the VKORC1 genotype on warfarin dose requirements in Japanese pediatric patients. Drug Metab Pharmacokinet. 2011;26:295–299. doi: 10.2133/dmpk.DMPK-10-NT-082. [DOI] [PubMed] [Google Scholar]
- 37.Hamberg A-K. Pharmacometric Models for Individualisation of Warfarin in Adults and Children. 2013. Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine, Uppsala. Available at http://uu.diva-portal.org/smash/record.jsf?pid=diva2:615277 (last accessed 20 August 2013)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Prediction corrected VPCs for the optimized model applied on data from 163 children but stratified on (A) CYP2C9 genotype or (B) CYP4F2 genotype. Figure 1A includes from left to right data from children with CYP2C9 *1/*1 (high enzyme activity), CYP2C9 *1/*2 or *1/*3 (intermediate enzyme activity) and CYP2C9 *2/*2, *2/*3 or *3/*3 (low enzyme activity). The right panel, representing children with rare CYP2C9 genotypes, includes data from only four children and results in wide prediction intervals that overlap. Figure 1B includes from left to right data from children with CYP4F2 *1/*1 (wild-type), CYP4F2 *1/*3 (one variant allele) and CYP4F2 *3/*3 (two variant alleles). There is no systematic bias in the VPCs suggesting that a child with the CYP4F2 variant allele requires a higher warfarin dose
Figure S2 Prediction corrected VPCs for the optimized model applied on data from 163 warfarin treated children but using body weight (A) or age (B) as the independent variable (x-axis) instead of time. Solid lines denote the medians of observed data and dashed lines denote the 10th and 90th percentiles of observed data. Shaded areas represent 95% confidence intervals of simulated medians (grey) and 80% prediction intervals (pink). A model is considered to have good predictive properties if the lines representing the observed data mainly fall inside the simulated prediction intervals