

Abstract
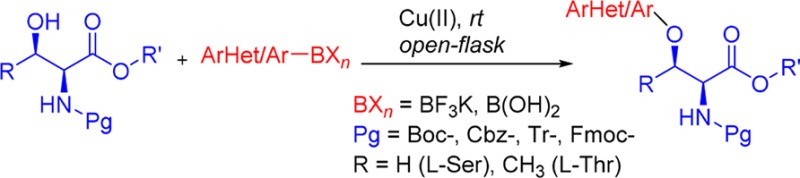
An effective protocol toward the O-arylation of β-hydroxy-α-amino acid substrates serine and threonine has been developed via Chan–Lam cross-coupling. This Cu(II)-catalyzed transformation involves benign open-flask conditions that are well-tolerated with a variety of protected (Boc-, Cbz-, Tr-, and Fmoc-) serine and threonine derivatives and various potassium organotrifluoroborates and boronic acids.
In nature, structural diversity in protein synthesis is achieved with a small number of naturally occurring amino acids. In the laboratory setting, the continuing development of novel reactions provides a much larger palette for exploring structural space, and indeed, on this basis, peptide synthesis has gone through major advances since the first report of peptide composition by Emil Fisher. In fact, unnatural peptides have now become a significant part of drug discovery efforts.1 Among other chemical modification methods, cross-coupling reactions on peptides and proteins have received much attention, leading to selective formation of nonlabile linkages at residues functionally orthogonal to natural amino acids. Various unnatural amino acids synthesized via cross-coupling protocols can be genetically incorporated into peptides and proteins, leading to valuable protein modifications.2 For example, the cross-coupling of 4-halophenylalanine, 4-boronophenylalanine, and l-tyrosine have been successfully accomplished.3 In the same vein, β-hydroxy-α-amino acid derivatives have been used as constructs of drugs for the treatment of Alzheimer’s disease (Figure 1, I) or inhibition of β-amyloid peptide release.4 Benzoxazepinone II (Figure 1) was recently discovered as a potential target for cancer therapy.5 It is noteworthy that compounds I and II are arylated at the O-site of serine.
Figure 1.
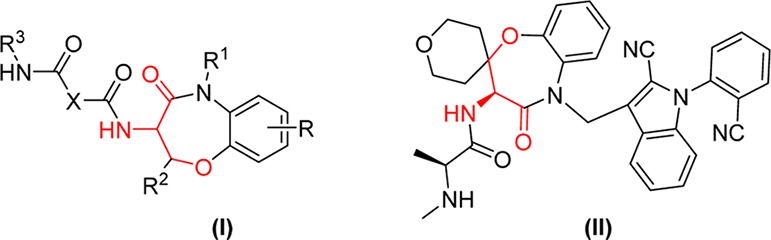
Serine-containing biologically active compounds.
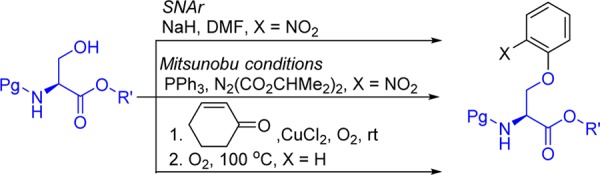
To date, there are few general methods described for the O-arylation of l-serine. This transformation has been achieved by nucleophilic aromatic substitution, but this protocol is limited by the use of strong bases (NaH and KHMDS) and the need for 1-fluoro-2-nitrobenzene substrates.4,5 Alternatively, the Mitsunobu reaction affords the arylated product but with lowered yields. This protocol also requires triphenylphosphine as a reagent,4b resulting in tedious workup procedures. A new approach to the O-arylation of serine involves the copper-mediated reaction of nonaromatic precursors, α,β-unsaturated ketones, under aerobic conditions (Scheme 1), affording aromatized O-arylated products. However, this protocol is substrate specific and requires higher temperatures, affording the O-arylated l-Ser in only 33% yield.6 Thus, development of an efficient, mild and general method to arylate the hydroxyl position of l-serine to access structurally complex aryl alkyl ethers in natural products and pharmaceuticals remains a challenging goal.
Scheme 1. Methods for Preparation of O-Arylated l-Serine.
This need has led to alternative methods for the synthesis of such structural motifs.7 The direct formation of the C–O bond in alkyl aryl ethers by metal-catalyzed arylation of aliphatic alcohols with aryl halides is a formidable task compared to the classic Ullman-type synthesis of diaryl ethers because of the low reactivity of aliphatic alcohols.7,8 Investigations by Buchwald and co-workers have shown that a reaction with Cu(I) salts in the presence of strongly basic alkoxides, performed under refluxing conditions, can efficiently promote the cross-coupling of aryl halides with phenols and aliphatic alcohols,9 an alternative to the analogous Pd-catalyzed reactions developed by Buchwald and Hartwig.10 It is noteworthy that Buchwald’s conditions have been used on hydroxyproline methyl esters but failed to afford the desired O-arylated products because of a lack of reactivity or elimination after the resulting arylation.10g Another protocol involves less common arylating agents such as organobismuths, but this approach requires in situ generation of the reagents by oxidation of the triarylbismuthine.7,11
In 1998, Chan,12 Evans,3a and Lam13 independently reported heteroatom arylation reactions that modernized alkyl-aryl ether synthesis. The development of this coupling reaction led to mild transformations that took place at room temperature under weakly basic conditions and could be carried out in an open reaction flask. For the C–O bond-forming reactions, this was limited primarily to phenols3b,3c,13,14 with the first examples utilizing catalytic amounts of Cu(OAc)2 reported in 2001 by Lam.8a,13 In addition to phenols, the development of copper-mediated C–O bond formation has flourished with the discovery that a broad range of oxygen nucleophiles, including carboxylic acids,15 aliphatic alcohols,8d aryloximes,16 silanols,17N-hydroxyphthalimides,18 and water,2a,19 have been coupled successfully from boron reagents using Cu(OAc)2 as the catalyst of choice.
Herein, we report the first Chan–Lam cross-coupling of β-hydroxy-α-amino acid derivatives. The results reveal an effective and practical protocol for the formation of C–O alkyl aryl ethers from β-hydroxy-α-amino acid substrates serine and threonine, using benign conditions with both arylboronic acids and aryltrifluoroborates.
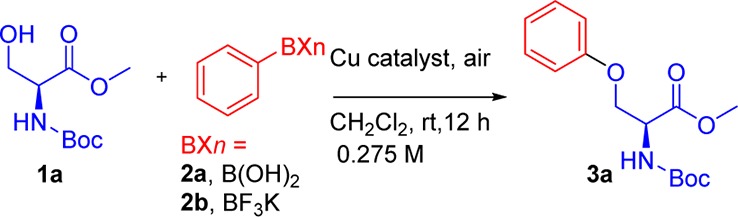
In an initial attempt at arylation, the reaction of Boc-l-Ser-OMe (1a) with both phenylboronic acid (2a) and potassium phenyltrifluoroborate (2b) in the presence of various copper catalysts, bases, and solvents in an open flask at room temperature was examined (Table 1). Comprehensive results and all screenings employed are shown in Table S1 of the Supporting Information). The use of CuI and CuSO4·5H2O with different catalyst loadings in the presence of Cs2CO3 and 1,10-phenanthroline using MeCN, CH2Cl2, or toluene as solvent was deemed ineffective (entries 1–6, Table 1). Anhydrous Cu(OAc)2 (10 mol %) in the presence of DMAP (20 mol %) afforded the desired product in 75% yield when 2a was utilized (entry 7, Table 1), but not in the case of 2b (entry 7, Table 1). The use of Cu(OAc)2 in pyridine and DMF as solvent was ineffective with both types of boron species (entry 9, Table 1). Fortunately, the reaction with Cu(OAc)2·H2O in the presence of DMAP afforded the desired product with both 2a and 2b (entry 10, Table 1). We set out to examine the reaction conditions further in the presence of Cu(OAc)2·H2O. The use of excess 2b (2 equiv, entry 11, Table 1) was deemed ineffective, while the most effective catalyst loading proved to be 10 mol % of Cu(OAc)2·H2O (entries 10 and 12–13, Table 1). DMAP was found to be superior to tetramethylguanidine, DBU, and DIPEA, and its absence gave zero yield (entries 10, 14–16, 23, Table 1). The addition of water (10 mol %) has no apparent effect on the boronic acid reaction, but is believed to assist the trifluoroborate reaction by facilitating the hydrolysis of PhBF3K. The use of ClCH2CH2Cl at higher temperatures (60 °C) gave homocoupling and ether formation of 2a or 2b as major side reactions, and thus CH2Cl2 was the solvent of choice for such transformations (entries 17–22, Table 1).
Table 1. Optimization of Reaction Conditions with Phenylboronic Acid and Potassium Phenyltrifluoroborate.
| yields of 3a (%) |
||||
|---|---|---|---|---|
| entry | catalyst (10 mol %) | base/ligand (equiv) | 2a, B(OH)2 | 2b, BF3K |
| 1a | CuI | Cs2CO3 (3.0), 1,10-phen (0.2) | n.r. | n.r. |
| 2b,20 | CuI | Cs2CO3 (3.0), 1,10-phen (0.1) | n.r. | n.r. |
| 3b | CuI | 1,10-phen (0.2) | n.r. | n.r. |
| 4a | CuSO4·5H2O | Cs2CO3 (3.0) | n.r. | n.r. |
| 5c | CuSO4·5H2O | Cs2CO3 (3.0) | n.r. | n.r. |
| 6a | CuSO4·5H2O | n.r. | n.r. | |
| 7 | Cu(OAc)2 | DMAP (0.2) | 75% | n.r. |
| 8 | Cu(OAc)2 | DMAP (1.0) | traces | n.r. |
| 9d,21 | Cu(OAc)2 | py (3.0) | n.r. | n.r. |
| 10 | Cu(OAc)2·H2O | DMAP (0.2) | 85% | 80% |
| 11e,8d | Cu(OAc)2·H2O | DMAP (0.2) | tracese | |
| 12f | Cu(OAc)2·H2O | DMAP (0.2) | 65% | |
| 13g | Cu(OAc)2·H2O | DMAP (0.2) | 81% | |
| 14 | Cu(OAc)2·H2O | tetramethylguanidine (0.2) | hc | hc |
| 15 | Cu(OAc)2·H2O | DBU (0.2) | hc | hc |
| 16 | Cu(OAc)2·H2O | DIPEA (0.2) | 79% | 70% |
| 17f,h | Cu(OAc)2·H2O | DMAP (0.2) | hc | hc |
| 18h | Cu(OAc)2·H2O | n.r. | n.r. | |
| 19a | Cu(OAc)2·H2O | DMAP (0.2) | n.r. | n.r. |
| 20i | Cu(OAc)2·H2O | DMAP (0.2) | 60% | 55% |
| 21j | Cu(OAc)2·H2O | DMAP (0.2) | 84% | 81% |
| 22k | Cu(OAc)2·H2O | DMAP (0.2) | 85% | 87% |
| 23 | Cu(OAc)2·H2O | n.r. | n.r. | |
Under the conditions developed, the scope and limitations of this coupling were evaluated (Schemes 2 and 3). Various aryl and heteroaryltrifluoroborates and boronic acids as well as a series of serine derivatives with different protecting groups and ester derivatives of l-Ser (Scheme 2) were employed. Several protecting groups, Boc- (3a–e), Cbz- (3f–3p, 3s), Tr-(3q), and the base-sensitive Fmoc- (3r) were well-tolerated. In general, the reaction was unaffected by steric factors but was more sensitive to the electronic nature of the boron substrate as demonstrated by the yields.
Scheme 2. Substrate Scope of O-Arylation of l-Serine Derivatives.
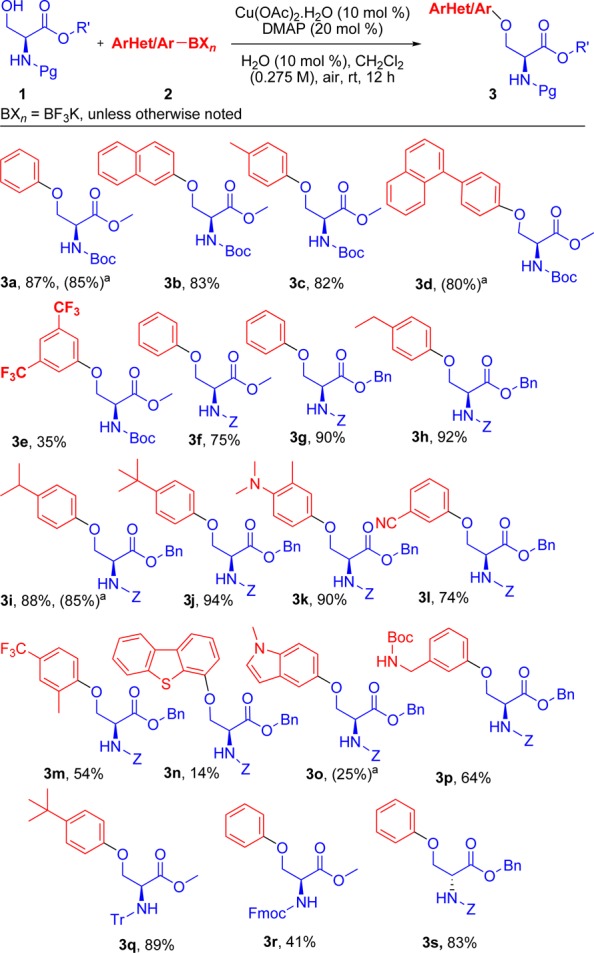
Ar/HetAr-BXn = B(OH)2 was utilized, and no water was added to the reaction.
Scheme 3. Substrate Scope of O-Arylation of l-Threonine Derivatives.
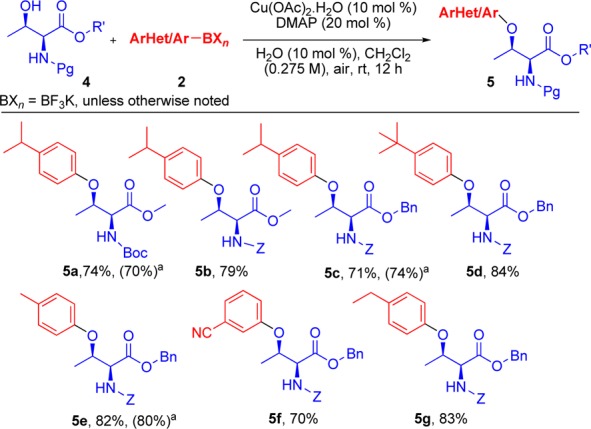
Ar/HetAr-BXn = B(OH)2 was utilized, and no water was added to the reaction.
Strongly electron-donating substituents with p-methyl (3c), p-ethyl (3h), p-isopropyl (3i), p-tert-butyl (3j), and p-N,N-dimethylamino (3k) afforded O-arylated l-serines in high yields. Electron-withdrawing substituents were also tolerated under the reaction conditions but gave lower yields, as in 3,5-trifluoromethyl (3e), p-trifluoromethyl (3m), and the m-nitrile (3l) substituted products. Both methyl and benzyl esters were examined, revealing that benzyl esters afford better yields (3f vs 3g). Using bulky protecting groups, such as the trityl group in 3q, as well as o-methyl (3m) and m-methyl (3k) substituents seemed to have no effect on the yield of the reaction. Although resulting in very low yields, we were pleased to observe that the reaction works with some heterocycles, including N-methylindole (3o) and dibenzothiophene (3n). However, the use of thiophenes and furans was detrimental, and no cross-coupling was observed when using organoboron reagents incorporating these ring systems. Instead, such substrates afforded unreacted starting materials and homocoupling as major side-products, especially at higher reaction temperatures.
We next turned our attention to applying this method to l-threonine (Scheme 3). With these substrates as well, the reaction conditions developed proved highly suitable and, most importantly, afforded single diastereomers with no epimerization observed. Both Boc- (5a) and Cbz- (5b–5g) protecting groups were employed and also proved to be well-tolerated.
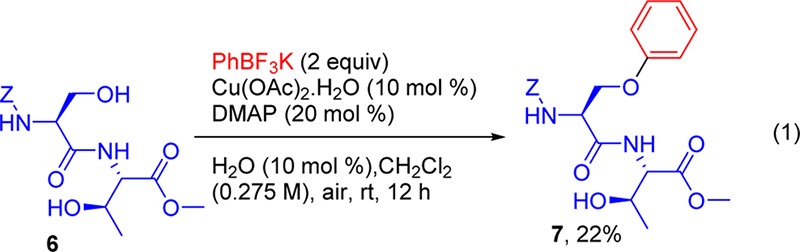
Surprisingly, the reaction of Cbz-l-Ser-l-Thr-OMe 6 with 2 equiv of the phenyltrifluoroborate gave a single mono-O-arylated product 7 in 22% yield (eq 1). The mass spectral fragmentation pattern of the product indicated that arylation took place selectively at the l-serine site (see Supporting Information). The use of dichloroethane at 60 °C led to more homocoupling and ether formation of the corresponding phenyltrifluoroborate, with no significant change in product formation.
 |
1 |
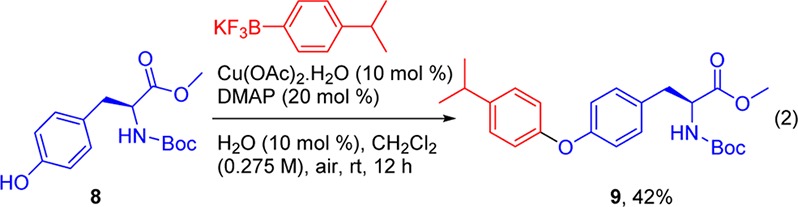
In addition, we examined the reaction conditions using the phenolic OH of l-tyrosine as the nucleophile, and indeed, the reaction of Boc-l-Tyr-OMe 8 afforded the desired product 9 in 42% yield (eq 2).
 |
2 |
We also studied the side-products of this transformation. Interestingly, the reaction affords ethers of the corresponding boron reagents (Scheme 4), particularly with organoborons possessing electron-donating groups. The side-product ether 10 was obtained when the reaction was run under the same conditions but in the absence of the amino acid. We believe that this is the result of copper(II)-promoted oxidation of ArBF3K23 with subsequent participation of the resulting phenol in the cross-coupling.
Scheme 4. Ether Formation from the Corresponding Boron Reagents.
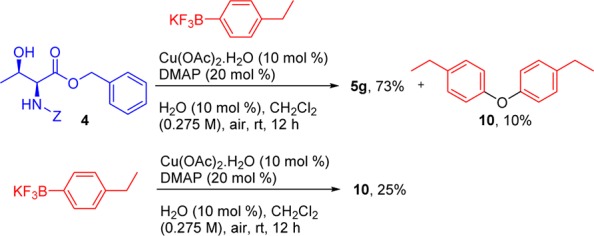
Finally, the reaction performed under argon, in the absence of air/oxygen, with (4-isopropylphenyl)boronic acid did not progress and afforded no product after 48 h.
In conclusion, this is the first report for the Chan–Lam cross-coupling of serine and threonine derivatives, allowing access to novel unnatural β-aryloxy-α-amino acid derivatives. This new Cu(II)-catalyzed transformation involves mild conditions (rt, open flask) and is well-tolerated with a variety of protected (Boc-, Cbz-, Tr-, and Fmoc-) serine and threonine derivatives and various potassium organotrifluoroborates and boronic acids.
Acknowledgments
This research was supported by the NIGMS (R01 GM-081376). Frontier Scientific is acknowledged for their generous donation of potassium organotrifluoroborates and boronic acids. Dr. Rakesh Kohli (University of Pennsylvania) is acknowledged for acquisition of HRMS spectra.
Supporting Information Available
Experimental procedures and product characterizations, 1H and 13C NMR spectra, mass spectral fragmentation for 7, and Table S1. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Fischer E.; Fourneau E. Ber. Dtsch. Chem. Ges. 1901, 34, 2868. [Google Scholar]; b Shen B.; Makley D. M.; Johnston J. N. Nature 2010, 465, 1027. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hanessian S. J. Org. Chem. 2012, 77, 6657. [DOI] [PubMed] [Google Scholar]; d Merrifield R. B. J. Am. Chem. Soc. 1963, 85, 2149. [Google Scholar]; e Masurier N.; Zajdel P.; Verdie P.; Pawlowski M.; Amblard M.; Martinez J.; Subra G. Chem.—Eur. J. 2012, 18, 11536. [DOI] [PubMed] [Google Scholar]; f Vlieghe P.; Lisowski V.; Martinez J.; Khrestchatisky M. Drug Discovery Today 2010, 15, 40. [DOI] [PubMed] [Google Scholar]; g El Khatib M.; Elagawany M.; Caliskan E.; Davis E. F.; Faidallah H. M.; El-Feky S. A.; Katritzky A. R. Chem. Commun. 2013, 49, 2631. [DOI] [PubMed] [Google Scholar]
- a Inamoto K.; Nozawa K.; Yonemoto M.; Kondo Y. Chem. Commun. 2011, 47, 11775. [DOI] [PubMed] [Google Scholar]; b Dibowski H.; Schmidtchen F. P. Angew. Chem., Int. Ed. 1998, 37, 476. [DOI] [PubMed] [Google Scholar]; c Bong D. T.; Ghadiri M. R. Org. Lett. 2001, 3, 2509. [DOI] [PubMed] [Google Scholar]; d Vilaro M.; Arsequell G.; Valencia G.; Ballesteros A.; Barluenga J. Org. Lett. 2008, 10, 3243. [DOI] [PubMed] [Google Scholar]; e Ojida A.; Tsutsumi H.; Kasagi N.; Hamachi I. Tetrahedron Lett. 2005, 46, 3301. [Google Scholar]; f Kodama K.; Fukuzawa S.; Nakayama H.; Kigawa T.; Sakamoto K.; Yabuki T.; Matsuda N.; Shirouzu M.; Takio K.; Tachibana K.; Yokoyama S. ChemBioChem 2006, 7, 134. [DOI] [PubMed] [Google Scholar]; g Kodama K.; Fukuzawa S.; Nakayama H.; Sakamoto K.; Kigawa T.; Yabuki T.; Matsuda N.; Shirouzu M.; Takio K.; Yokoyama S.; Tachibana K. ChemBioChem 2007, 8, 232. [DOI] [PubMed] [Google Scholar]; h Brustad E.; Bushey M. L.; Lee J. W.; Groff D.; Liu W.; Schultz P. G. Angew. Chem., Int. Ed. 2008, 47, 8220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Evans D. A.; Katz J. L.; West T. R. Tetrahedron Lett. 1998, 39, 2937. [Google Scholar]; b Hitotsuyanagi Y.; Ishikawa H.; Naito S.; Takeya K. Tetrahedron Lett. 2003, 44, 5901. [Google Scholar]; c Deng H.; Jung J.-K.; Liu T.; Kuntz K. W.; Snapper M. L.; Hoveyda A. H. J. Am. Chem. Soc. 2003, 125, 9032. [DOI] [PubMed] [Google Scholar]; d Chalker J. M.; Wood C. S. C.; Davis B. G. J. Am. Chem. Soc. 2009, 131, 16346. [DOI] [PubMed] [Google Scholar]; e Jung M. E.; Lazarova T. I. J. Org. Chem. 1999, 64, 2976. [DOI] [PubMed] [Google Scholar]; f Gao Z.; Gouverneur V.; Davis B. G. J. Am. Chem. Soc. 2013, 135, 13612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Thorsett E. D.; Porter W. J.; Nissen J. S.; Latimer L. H.; Audia J. E.; Droste J. Athena Neurosciences, Inc., Eli Lilly and Company. US, 506,782BI, 2003. [Google Scholar]; b Flohr A.; Jakob-Roetne R.; Wostl W. Hoffmann-La Roche Inc. US WO2006061136, 2006.
- Donnell A. F.; Michoud C.; Rupert K. C.; Han X.; Aguilar D.; Frank K. B.; Fretland A. J.; Gao L.; Goggin B.; Hogg J. H.; Hong K.; Janson C. A.; Kester R. F.; Kong N.; Le K.; Li S.; Liang W.; Lombardo L. J.; Lou Y.; Lukacs C. M.; Mischke S.; Moliterni J. A.; Polonskaia A.; Schutt A. D.; Solis D. S.; Specian A.; Taylor R. T.; Weisel M.; Remiszewski S. W. J. Med. Chem. 2013, 56, 7772. [DOI] [PubMed] [Google Scholar]
- Simon M.-O.; Girard S. A.; Li C.-J. Angew. Chem., Int. Ed. 2012, 51, 7537. [DOI] [PubMed] [Google Scholar]
- Evano G.; Blanchard N.; Toumi M. Chem. Rev. 2008, 108, 3054. [DOI] [PubMed] [Google Scholar]
- a Allen S. E.; Walvoord R. R.; Padilla-Salinas R.; Kozlowski M. C. Chem. Rev. 2013, 113, 6234. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Qiao J. X.; Lam P. Y. S. Synthesis 2011, 829. [Google Scholar]; c Ley S. V.; Thomas A. W. Angew. Chem., Int. Ed. 2003, 42, 5400. [DOI] [PubMed] [Google Scholar]; d Quach T. D.; Batey R. A. Org. Lett. 2003, 5, 1381. [DOI] [PubMed] [Google Scholar]; e Lindley J. Tetrahedron 1984, 40, 1433. [Google Scholar]; f Finet J.-P.; Fedorov A. Y.; Combes S.; Boyer G. Curr. Org. Chem. 2002, 6, 597. [Google Scholar]; g Hassan J.; Sevignon M.; Gozzi C.; Schulz E.; Lemaire M. Chem. Rev. 2002, 102, 1359. [DOI] [PubMed] [Google Scholar]
- a Wolter M.; Nordmann G.; Job G. E.; Buchwald S. L. Org. Lett. 2002, 4, 973. [DOI] [PubMed] [Google Scholar]; b Zhu J.; Price B. A.; Zhao S. X.; Skonezny P. M. Tetrahedron Lett. 2000, 41, 4011. [Google Scholar]; c Marcoux J.-F.; Doye S.; Buchwald S. L. J. Am. Chem. Soc. 1997, 119, 10539. [Google Scholar]
- a Parrish C. A.; Buchwald S. L. J. Org. Chem. 2001, 66, 2498. [DOI] [PubMed] [Google Scholar]; b Kuwabe S.-I.; Torraca K. E.; Buchwald S. L. J. Am. Chem. Soc. 2001, 123, 12202. [DOI] [PubMed] [Google Scholar]; c Torraca K. E.; Huang X.; Parrish C. A.; Buchwald S. L. J. Am. Chem. Soc. 2001, 123, 10770. [DOI] [PubMed] [Google Scholar]; d Shelby Q.; Kataoka N.; Mann G.; Hartwig J. J. Am. Chem. Soc. 2000, 122, 10718. [Google Scholar]; e Mann G.; Hartwig J. F. J. Am. Chem. Soc. 1996, 118, 13109. [Google Scholar]; f Palucki M.; Wolfe J. P.; Buchwald S. L. J. Am. Chem. Soc. 1997, 119, 3395. [Google Scholar]; g Toumi M.; Rincheval V.; Young A.; Gergeres D.; Turos E.; Couty F.; Mignotte B.; Evano G. Eur. J. Org. Chem. 2009, 3368. [Google Scholar]
- a Sinclair P. J.; Wong F.; Wyvratt M.; Staruch M. J.; Dumont F. Bioorg. Med. Chem. Lett. 1995, 5, 1035. [DOI] [PubMed] [Google Scholar]; b Sinclair P. J.; Wong F.; Staruch M. J.; Wiederrecht G.; Parsons W. H.; Dumont F.; Wyvratt M. Bioorg. Med. Chem. Lett. 1996, 6, 2193. [DOI] [PubMed] [Google Scholar]; c Brands K. M. J.; Dolling U.-H.; Jobson R. B.; Marchesini G.; Reamer R. A.; Williams J. M. J. Org. Chem. 1998, 63, 6721. [Google Scholar]; d Pietri S.; Liebgott T.; Finet J.-P.; Culcasi M.; Billottet L.; Bernard-Henriet C. Drug Dev. Res. 2001, 54, 191. [Google Scholar]
- Chan D. M. T.; Monaco K. L.; Wang R.-P.; Winters M. P. Tetrahedron Lett. 1998, 39, 2933. [Google Scholar]
- Lam P. Y. S.; Vincent G.; Clark C. G.; Deudon S.; Jadhav P. K. Tetrahedron Lett. 2001, 42, 3415. [Google Scholar]
- a Simon J.; Salzbrunn S.; Prakash G. K. S.; Petasis N. A.; Olah G. A. J. Org. Chem. 2001, 66, 633. [DOI] [PubMed] [Google Scholar]; b Lam P. Y. S.; Vincent G.; Bonne D.; Clark C. G. Tetrahedron Lett. 2003, 44, 4927. [Google Scholar]; c Decicco C. P.; Song Y.; Evans D. A. Org. Lett. 2001, 3, 1029. [DOI] [PubMed] [Google Scholar]; d Evans D. A.; Katz J. L.; Peterson G. S.; Hintermann T. J. Am. Chem. Soc. 2001, 123, 12411. [DOI] [PubMed] [Google Scholar]; e Cherney R. J.; Duan J. J. W.; Voss M. E.; Chen L.; Wang L.; Meyer D. T.; Wasserman Z. R.; Hardman K. D.; Liu R.-Q.; Covington M. B.; Qian M.; Mandlekar S.; Christ D. D.; Trzaskos J. M.; Newton R. C.; Magolda R. L.; Wexler R. R.; Decicco C. P. J. Med. Chem. 2003, 46, 1811. [DOI] [PubMed] [Google Scholar]; f McKinley N. F.; O’Shea D. F. J. Org. Chem. 2004, 69, 5087. [DOI] [PubMed] [Google Scholar]; g Voisin A. S.; Bouillon A.; Lancelot J.-C.; Lesnard A.; Rault S. Tetrahedron 2006, 62, 6000. [Google Scholar]
- Zhang L.; Zhang G.; Zhang M.; Cheng J. J. Org. Chem. 2010, 75, 7472. [DOI] [PubMed] [Google Scholar]
- Villalobos J. M.; Srogl J.; Liebeskind L. S. J. Am. Chem. Soc. 2007, 129, 15734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan D. G.; Winternheimer D. J.; Merlic C. A. Org. Lett. 2011, 13, 2778. [DOI] [PubMed] [Google Scholar]
- Petrassi H. M.; Sharpless K. B.; Kelly J. W. Org. Lett. 2001, 3, 139. [DOI] [PubMed] [Google Scholar]
- Xu J.; Wang X.; Shao C.; Su D.; Cheng G.; Hu Y. Org. Lett. 2010, 12, 1964. [DOI] [PubMed] [Google Scholar]
- Bates C. G.; Saejueng P.; Doherty M. Q.; Venkataraman D. Org. Lett. 2004, 6, 5005. [DOI] [PubMed] [Google Scholar]
- Herradura P. S.; Pendola K. A.; Guy R. K. Org. Lett. 2000, 2, 2019. [DOI] [PubMed] [Google Scholar]
- Sun H.; Li X.; Sundermeyer J. J. Mol. Catal. A: Chem. 2005, 240, 119. [Google Scholar]
- a Xu X.; Wang X.; Shao C.; Su D.; Cheng G.; Hu Y. Org. Lett. 2010, 12, 1964. [DOI] [PubMed] [Google Scholar]; b Imamoto K.; Nozawa K.; Yonemoto M.; Kondo Y. Chem. Commun. 2011, 47, 11775. [DOI] [PubMed] [Google Scholar]; c Yang D.; An B.; Wei W.; Jiang M.; You J.; Wang H. Tetrahedron 2014, 70, 3630. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.