

Abstract

A new method for the synthesis of 2-aminoimidazole products is described. The heterocyclic products are generated in good yields via Pd-catalyzed carboamination reactions of N-propargyl guanidines and aryl triflates. This methodology generates both a C–N and C–C bond during the annulation step and facilitates the rapid construction of 2-aminoimidazole products with different aryl groups. The utility of this methodology was demonstrated in the total synthesis of preclathridine A, preclathridine B, and dorimidazole B from a single intermediate.
Cyclic guanidine natural products have garnered widespread attention due to their wide-ranging biological properties.1 One interesting subclass of cyclic guanidines are substituted 2-aminoimidazole derivatives (1–5) (Figure 1). These compounds display a myriad of potentially useful biological activities. Members of this family of alkaloids have been shown to be human β-secretase (BACE1) inhibitors,2 tubulin-binding agents,3 and epidermal growth factor receptor inhibitors,4 and also exhibit interesting antimicrobial5 and antibiotic activity.6 Classical methods for the construction of 2-aminoimidazoles generally involve either condensation reactions of α-amino- or α-haloketones or functionalization of imidazole derivatives.7 More recently, the metal-catalyzed hydroamination of acyclic terminally substituted N-propargyl guanidines was reported as a useful means for generating substituted 2-aminoimidazoles.8
Figure 1.

2-Aminoimidazole natural products.
Although these strategies have proven to be highly valuable for the construction of 2-aminoimidazole-containing alkaloids, none involve C–C bond formation during the ring-closing step, and as a result, the synthesis of closely related alkaloids such as 1–3 cannot currently be accomplished from a single intermediate. Rather, different substrates are required to access compounds that differ in the nature of the 4-(arylmethyl) group. A more attractive synthetic approach to these compounds would be one that facilitates the conversion of a single, simple, starting material to any of these derivatives via installation of an appropriate aryl group, which could be obtained from a readily available compound such as an aryl halide or triflate.
We recently reported a method for the construction of saturated cyclic guanidines via Pd-catalyzed carboamination reactions between N-allyl guanidines 6 and aryl or alkenyl halides (Scheme 1a).9 These cross-coupling reactions led to the formation of both a C–N bond and a C–C bond and afforded the products 7 in good yields.10,11 We envisioned that this methodology could be employed for the synthesis of 2-aminoimidazole-containing products by simply using N-propargyl guanidine substrates 8 in place of the N-allyl substrates 6 (Scheme 1b). These transformations should generate products 9 bearing an exocyclic alkene, which would then isomerize either under the reaction conditions or during a workup step to afford the desired 2-aminoimidazole products 10.8a,8c Importantly, these transformations would effect installation of a C4′ aryl group during the ring-closing step. This would allow for straightforward introduction of different groups at the C4′ position, which cannot currently be accomplished using other existing methods. Herein we describe our initial findings on this new approach to the construction of 2-aminoimidazoles and the application of this method to the synthesis of three different alkaloid natural products (1–3) from a single intermediate.
Scheme 1. Synthesis of Cyclic Guanidines via Pd-Catalyzed Carboamination Reactions.

In our original studies on the synthesis of saturated guanidines, we utilized substrates 6 bearing two PMP protecting groups. Despite the utility of these transformations, we were not able to develop conditions to remove both PMP aryl groups from the carboamination products. As such, in the present studies we elected to examine substrates bearing N-sulfonyl protecting groups, as these compounds can be prepared in a straightforward manner, and cleavage of N-sulfonyl groups from 2-aminoimidazoles has previously been demonstrated.12
At the outset of this project we decided to explore the reactivity of N-tosyl-N-propargyl guanidine substrate 8a, which was prepared in four steps (63% overall yield) from commercially available materials (N-methylallylamine, trimethylsilylacetylene, and formaldehyde). Unfortunately efforts to couple 8a with aryl bromide 3-bromophenyl-1,3-dioxolane using conditions previously employed for reactions of 6 were unsuccessful (eq 1). The desired 2-aminoimidazole was not obtained, and instead a mixture of hydroamination products 11a–b was generated.
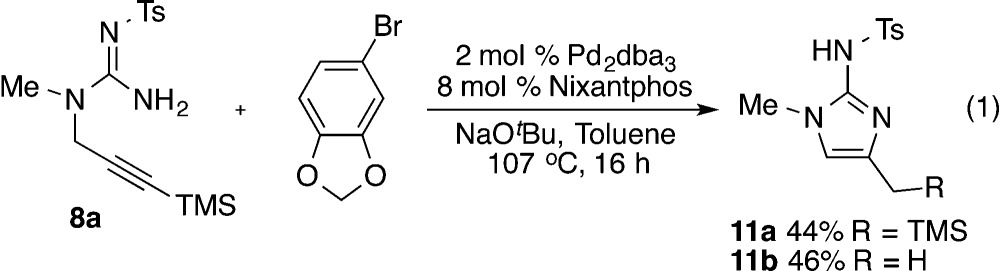 |
1 |
It seemed likely that the failure of 8a to undergo successful carboamination was due to the decreased nucleophilicity of the N-tosyl guanidine as compared to the bis-PMP-protected guanidine present in 6. This decreased nucleophilicity likely disfavors the key syn-aminopalladation step in the catalytic cycle;13 previously reported cases of reactions that proceed via alkyne syn-aminopalladation involve relatively nucleophilic amines.14 Our group has recently discovered that Pd-catalyzed carboamination reactions of relatively weak nitrogen nucleophiles (N-allylsulfamides) can be induced to proceed via anti-aminopalladation pathways under appropriate conditions, which leads to significantly improved chemical yields for transformations of these types of substrates.15,16 Thus, we reasoned that altering the reaction conditions to favor an anti-aminopalladation mechanistic pathway might promote the desired transformations of 8a to 2-aminoimidazoles 10.
To probe the hypothesis outlined above, N-propargyl guanidine substrate 8a was subjected to the reaction conditions that had provided optimal results for Pd-catalyzed carboamination reactions of N-allyl sulfamides.15a As shown in Table 1, the coupling of 8a with aryl triflate 12 to afford 2-aminoimidazole 10a was successfully achieved when Pd(OAc)2 was employed as the palladium source along with the base LiOtBu and the solvent PhCF3. Significant amounts of desired product were obtained with several different phosphine ligands, including Nixantphos17 and a variety of different Buchwald-type biarylphosphines (entries 1–5).18 The primary side products generated in these reactions resulted from competing hydroamination of the alkyne (11a and 11b), but this side reaction was less problematic than with the conditions shown in eq 1. Ultimately the RuPhos ligand was found to provide the best results (entry 5), affording the desired product 10a in 80% isolated yield while only trace amounts of hydroamination side products were generated.
Table 1. Ligand Optimizationa.
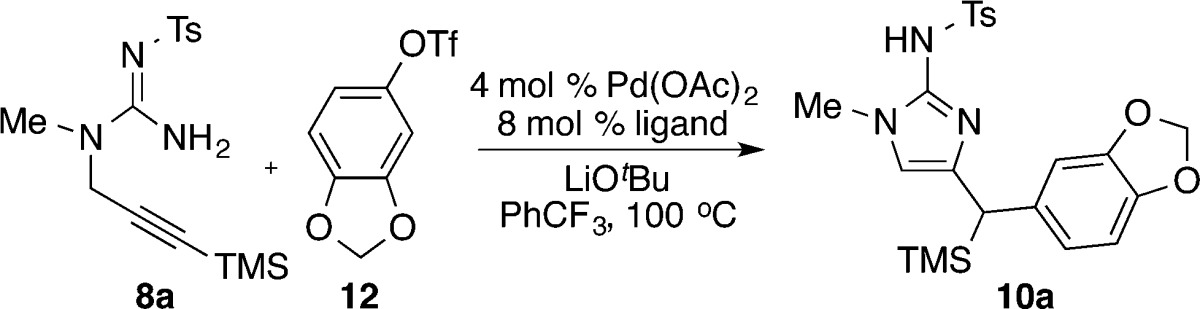
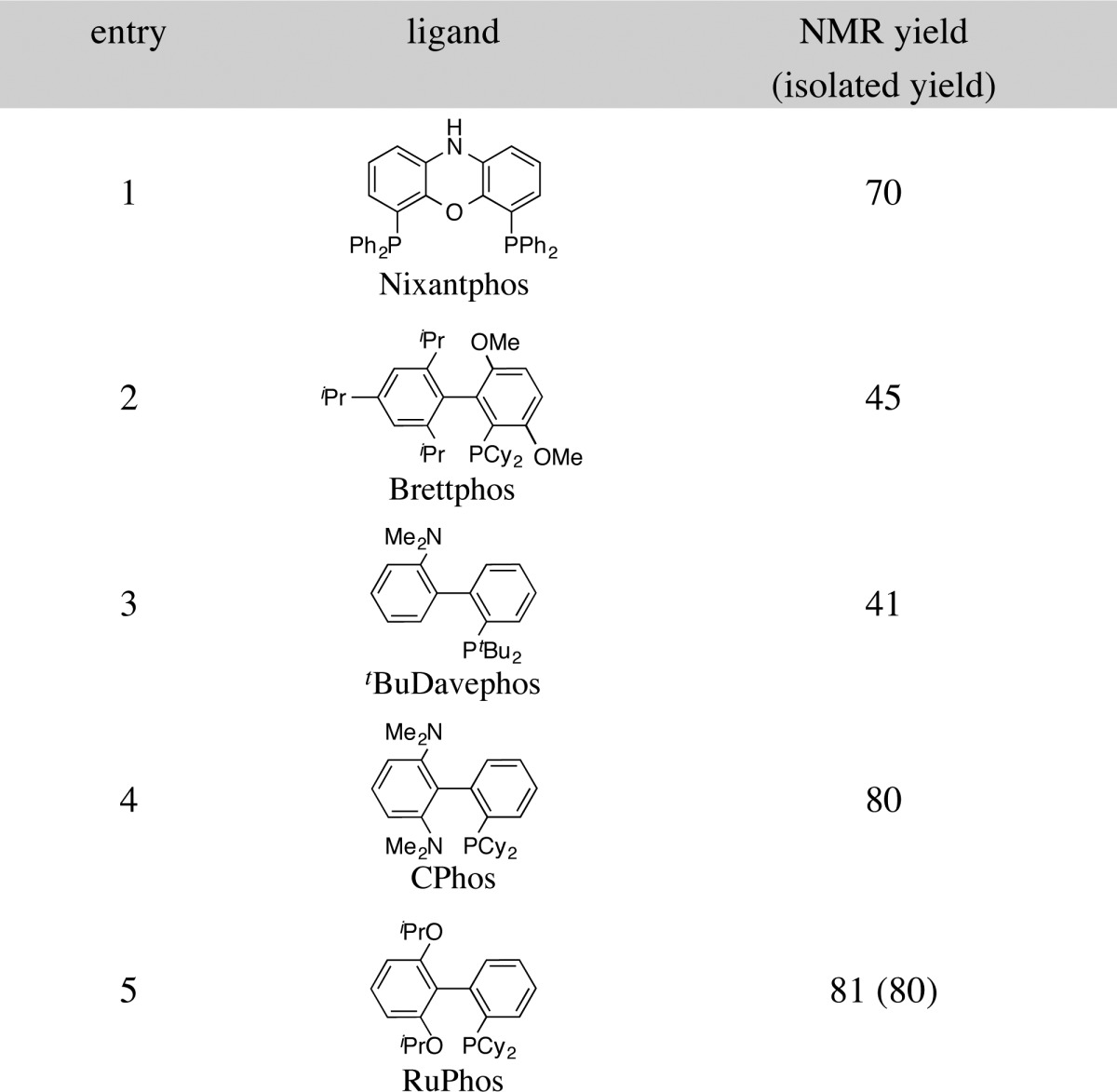
Reaction conditions: 1.0 equiv of 8a, 1.2 equiv of 12, 2.4 equiv of LiOtBu, 4 mol % Pd(OAc)2, 8 mol % ligand, PhCF3 (0.1 M), 100 °C, 16 h.
With optimized conditions in hand, the scope of these transformations was examined by coupling 8a with a variety of aryl triflates (Table 2). Gratifyingly, aryl triflates bearing electron-donating, electron-neutral, and electron-withdrawing groups (entries 1–5) afforded the corresponding 2-aminoimidazole derivatives in good yields. Moreover, an ortho-methyl substituted aryl triflate also underwent smooth coupling with 8a to generate product 10f in high yield (entry 4). However, it was necessary to employ 2 equiv of most aryl triflates due to competing base-mediated cleavage of the triflate to the corresponding phenol. In most instances products bearing TMS-groups were stable under these conditions, and it was not necessary to rigorously monitor reactions to determine the exact time of completion; reactions could be conducted for up to 16 h with no product degradation (although efforts to optimize reaction times indicated most were complete in ca. 3 h). However, a relatively short reaction time (3 h) and somewhat careful monitoring of reaction progress were required for the coupling of substrate 8a with 4-benzoylphenyl trifluoromethanesulfonate in order to prevent conversion of product 10e to the corresponding desilylated compound (entry 5). Related transformations of substrate 8b, which contains a phenyl-substituted alkyne, also provided the desired 2-aminoimidazole products. However, yields were lower than those in analogous transformations of 8a due to competing hydroamination of the substrate.
Table 2. Scope of Carboamination Reactionsa.
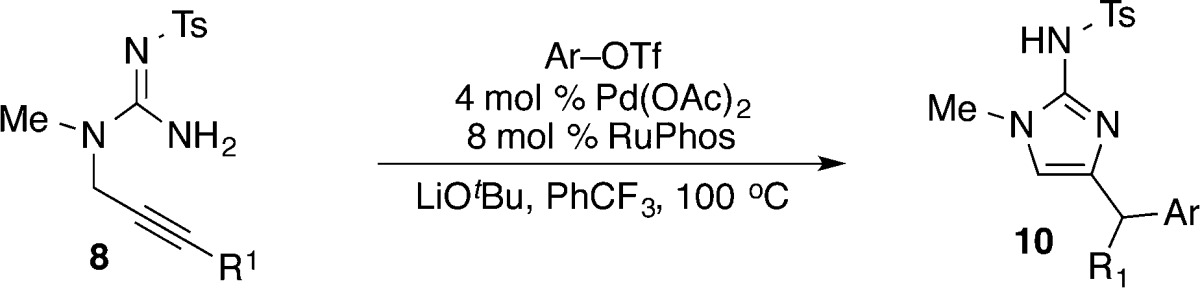
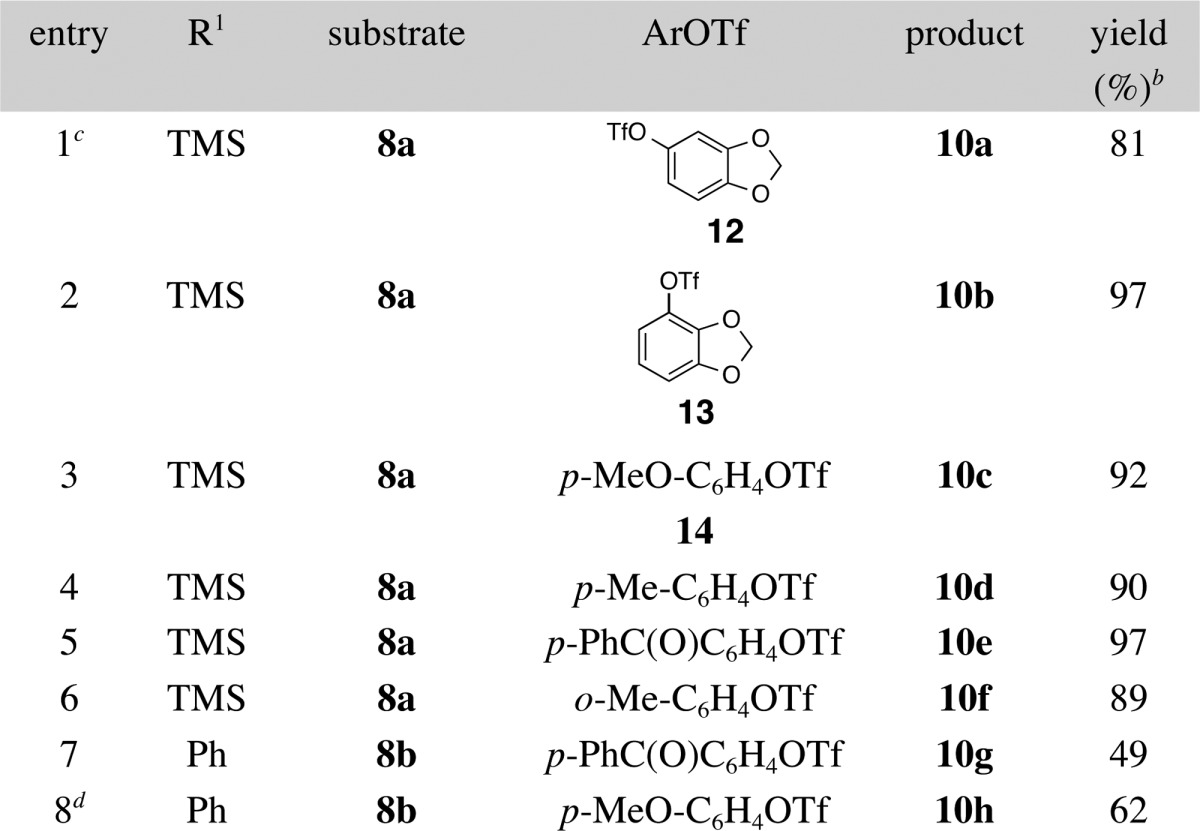
Reaction conditions: 1.0 equiv of 8a or 8b, 2.0 equiv of Ar–OTf, 2.4 equiv of LiOtBu, 4 mol % Pd(OAc)2, 8 mol % RuPhos, PhCF3 (0.1 M), 100 °C, 3 h.
Isolated yield.
The reaction was conducted using 1.2 equiv of Ar–OTf.
The reaction was conducted using 5 equiv of Ar–OTf.
To demonstrate the utility of this methodology we undertook the total synthesis of 2-aminoimidazole alkaloids 1–3. As shown in Scheme 2, N-tosyl protected substrate 8a was coupled with three different aryl triflates using the optimized reaction conditions from above. However, in the synthesis of 1–3 the TMS-substituted products of these reactions were not isolated but, instead, were treated with 4 M HCl in dioxane to effect protodesilyation.19 The N-tosyl groups were then cleaved from the resulting intermediates using Li/naphthalene, thereby providing alkaloids preclathridine A, preclathridine B, and dorimidazole B.20,21 Overall, the three 2-aminoimidizole natural products 1–3 were synthesized in just six steps and good overall yields (40–48%) from commercially available materials. Importantly, this work highlights the power of Pd-catalyzed carboamination reactions as a synthetic strategy for the construction of 2-aminoimidazole products and illustrates the potential to generate several different alkaloid derivatives from one intermediate (8a).
Scheme 2. Synthesis of 2-Aminoimidazole Natural Products.

The rapid isomerization of putative intermediate 9 prevents us from making definitive statements about the mechanism of these reactions, as the stereochemistry of addition to the alkyne cannot be unambiguously established. However, given our prior observations that reaction conditions such as those employed in these transformations tend to favor anti-aminopalladation pathways, we suggest the mechanism illustrated in Scheme 3 may be in operation.15 Oxidative addition of the aryl triflate to palladium(0) generates cationic palladium complex 15.22 Coordination of the Pd-catalyst to the alkyne produces 16 and facilitates outer-sphere attack of the guanidine nucleophile onto the alkyne (anti-aminopalladation). Reductive elimination from Pd-alkenyl intermediate 17 affords the exocyclic product 9, which then undergoes double-bond isomerization to give the desired 2-aminoimidazole product 10.
Scheme 3. Catalytic Cycle.

In summary, we have developed a new strategy for the construction of 2-aminoimidazoles via the Pd-catalyzed alkyne carboamination of tosyl-protected N-propargyl guanidines and aryl triflates. The N-tosyl-2-aminoimidazole products are generated in good yields, and the N-tosyl group can be reductively cleaved in a single step. Notably, this strategy generates a C–C bond during the annulation event, thus facilitating late-stage derivitization as illustrated through the total synthesis of preclathridine A, preclathridine B, and dorimidazole B in just two steps from a common intermediate. Continued studies on the use of Pd-catalyzed carboamination reactions of alkynes and alkenes in the synthesis of guanidine alkaloids are currently underway.
Acknowledgments
We acknowledge the NIH-NIGMS (GM-071650) and the University of Michigan Associate Professor Support Fund for financial support of this work. B.Z. acknowledges the University of Michigan Department of Chemistry for a Summer Undergraduate Research Fellowship.
Supporting Information Available
Experimental procedures, characterization data for all new compounds, descriptions of stereochemical assignments, and copies of 1H and 13C NMR spectra for all new compounds reported in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Berlinck R. G. S.; Trindade-Silva A. E.; Santos M. F. C. Nat. Prod. Rep. 2012, 29, 1382–1406. [DOI] [PubMed] [Google Scholar]; b Berlinck R. G. S.; Burtoloso A. C. B.; Trindade-Silva A. E.; Romminger S.; Morais R. P.; Bandeira K.; Mizuno C. M. Nat. Prod. Rep. 2010, 27, 1871–1907. [DOI] [PubMed] [Google Scholar]; c Berlinck R. G. S.; Burtoloso A. C. B.; Kossuga M. H. Nat. Prod. Rep. 2008, 25, 919–954. [DOI] [PubMed] [Google Scholar]; d Berlinck R. G. S.; Kossuga M. H. Nat. Prod. Rep. 2005, 22, 516–550. [DOI] [PubMed] [Google Scholar]
- a Malamas M. S.; Erdei J.; Gunawan I.; Barnes K.; Johnson M.; Hui Y.; Turner J.; Hu Y.; Wagner E.; Fan K.; Olland A.; Bard J.; Robichaud A. J. J. Med. Chem. 2009, 52, 6314–6323. [DOI] [PubMed] [Google Scholar]; b Malamas M. S.; Erdei J.; Gunawan I.; Turner J.; Hu Y.; Wagner E.; Fan K.; Chopra R.; Olland A.; Bard J.; Jacobsen S.; Magolda R. L.; Pangalos M.; Robichaud A. J. J. Med. Chem. 2010, 53, 1146–1158. [DOI] [PubMed] [Google Scholar]
- a Nodwell M.; Pereira A.; Riffell J. L.; Zimmerman C.; Patrick B. O.; Roberge M.; Andersen R. J. J. Org. Chem. 2009, 74, 995–1006. [DOI] [PubMed] [Google Scholar]; b Coleman R. S.; Campbell E. L.; Carper D. J. Org. Lett. 2009, 11, 2133–2136. [DOI] [PubMed] [Google Scholar]
- Copp B. R.; Fairchild C. R.; Cornell L.; Casazza A. M.; Robinson S.; Ireland C. M. J. Med. Chem. 1998, 41, 3909–3911. [DOI] [PubMed] [Google Scholar]
- a Carmely S.; Kashman Y. Tetrahedron Lett. 1987, 28, 3003–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Carmely S.; Ilan M.; Kashman Y. Tetrahedron 1989, 45, 2193–2200. [Google Scholar]; c Mancini I.; Guella G.; Debitus C.; Pietra F. Helv. Chem. Acta 1995, 78, 1178–1184. [Google Scholar]
- Dunbar D. C.; Rimoldi J. M.; Clark A. M.; Kelly M.; Hamann M. T. Tetrahedron 2000, 56, 8795–8798. [Google Scholar]
- For reviews of 2-aminoimidazoles, see:; a Sullivan J. D.; Giles R. L.; Looper R. E. Curr. Bioact. Compd. 2009, 5, 39–78. [Google Scholar]; b Weinreb S. M. Nat. Prod. Rep. 2007, 24, 931–948. [DOI] [PubMed] [Google Scholar]; c Hoffman H.; Lindel T. Synthesis 2003, 12, 1753–1783. [Google Scholar]
- a Giles R. L.; Sullivan J. D.; Steiner A. M.; Looper R. E. Angew. Chem., Int. Ed. 2009, 48, 3116–3120. [DOI] [PubMed] [Google Scholar]; b Gibbons J. B.; Gligorich K. M.; Welm B. E.; Looper R. E. Org. Lett. 2012, 14, 4734–4737. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ermolat’ev D. S.; Bariwal J. B.; Steenackers H. P. L.; De Keersmaecker S. C. J.; Van der Eycken E. V. Angew. Chem., Int. Ed. 2010, 49, 9655–9658. [DOI] [PubMed] [Google Scholar]
- Zavesky B. P.; Babij N. R.; Fritz J. A.; Wolfe J. P. Org. Lett. 2013, 15, 5420–5423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For reviews on Pd-catalyzed carboamination reactions, see:; a Wolfe J. P. Eur. J. Org. Chem. 2007, 571–582. [PMC free article] [PubMed] [Google Scholar]; b Wolfe J. P. Synlett 2008, 2913–2937. [Google Scholar]; c Schultz D. M.; Wolfe J. P. Synthesis 2012, 44, 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Wolfe J. P. Top. Heterocycl. Chem. 2013, 32, 1–38. [Google Scholar]
- For Pd-catalyzed carboamination reactions utilized in the synthesis of polycyclic guanidine alkaloids, see:; Babij N. R.; Wolfe J. P. Angew. Chem., Int. Ed. 2012, 51, 4128–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Babij N. R.; Wolfe J. P. Angew. Chem., Int. Ed. 2013, 52, 9247–9250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina P.; Fresneda P. M.; Sanz M. A. J. Org. Chem. 1999, 64, 2540–2544. [Google Scholar]
- a Neukom J. D.; Perch N. S.; Wolfe J. P. J. Am. Chem. Soc. 2010, 132, 6276–6277. [DOI] [PubMed] [Google Scholar]; b Hanley P. S.; Marković D.; Hartwig J. F. J. Am. Chem. Soc. 2010, 132, 6302–6303. [DOI] [PubMed] [Google Scholar]; c Neukom J. D.; Perch N. S.; Wolfe J. P. Organometallics 2011, 30, 1269–1277. [Google Scholar]; d Hanley P. S.; Hartwig J. F. J. Am. Chem. Soc. 2011, 133, 15661–15673. [DOI] [PubMed] [Google Scholar]; e White P. B.; Stahl S. S. J. Am. Chem. Soc. 2011, 133, 18594–18597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected examples of reactions that involve alkyne syn-aminopalladation, see:; a Karstens W. F. J.; Stol M.; Rutjes F. P. J. T; Kooijman H.; Spek A. L.; Hiemstra H. J. Organomet. Chem. 2001, 624, 244–258. [Google Scholar]; b Jacobi P. A.; Liu H. Org. Lett. 1999, 1, 341–344. [DOI] [PubMed] [Google Scholar]; c Piou T.; Neuville L.; Zhu J. Tetrahedron 2013, 69, 4415–4420. [Google Scholar]; d Tang S.; Peng P.; Pi S.-F.; Liang Y.; Wang N.-X.; Li J.-H. Org. Lett. 2008, 10, 1179–1182. [DOI] [PubMed] [Google Scholar]; e Liu J.; Shen M.; Zhang Y.; Li G.; Khodabocus A.; Rodriguez S.; Qu B.; Farina V.; Senanayake C. H.; Lu B. Z. Org. Lett. 2006, 8, 3573–3575. [DOI] [PubMed] [Google Scholar]
- a Fornwald R. M.; Fritz J. A.; Wolfe J. P. Chem.—Eur. J. 2014, 20, 8782–8790. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Babij N. R.; McKenna G. M.; Fornwald R. M.; Wolfe J. P. Org. Lett. 2014, 16, 3412–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected recent examples of other transformations that proceed through alkyne anti-aminopalladation, see:; a Shen Z.; Lu X. Tetrahedron 2006, 62, 10896–10899. [Google Scholar]; b Han X.; Lu X. Org. Lett. 2010, 12, 3336–3339. [DOI] [PubMed] [Google Scholar]; c Liu Q.; Chen P.; Liu G. ACS Catal. 2013, 3, 178–181. [Google Scholar]; d Hu Z.; Wang J.; Liang D.; Zhu Q. Adv. Synth. Catal. 2013, 355, 3290–3294. [Google Scholar]; For reviews, see:; e Zeni G.; Larock R. C. Chem. Rev. 2006, 106, 4644–4680. [DOI] [PubMed] [Google Scholar]; f Zeni G.; Larock R. C. Chem. Rev. 2004, 104, 2285–2309. [DOI] [PubMed] [Google Scholar]
- van der Veen L. A.; Keeven P. H.; Schoemaker G. C.; Reek J. N. H.; Kamer P. C. J.; van Leeuwen P. W. N. M.; Lutz M.; Spek A. L. Organometallics 2000, 19, 872–883. [Google Scholar]
- a Surry D. S.; Buchwald S. L. Chem. Sci. 2011, 2, 27–50. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Han C.; Buchwald S. L. J. Am. Chem. Soc. 2009, 131, 7532–7533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steps 1a and 1b (overall) proceeded in 93%, 80%, and 94% yields in the synthesis of 1, 2, and 3, respectively. See the Supporting Information for full experimental details.
- The reductive detosylations (step 2) proceeded in 85%, 91%, and 68% yields in the synthesis of 1, 2, and 3, respectively.
- In prior syntheses of 1 and 3 reductive detosylation was accomplished using SmI2 rather than Li/naphthalene. See ref (12).
- Jutand A.; Mosleh A. Organometallics 1995, 14, 1810–1817. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.