

Abstract
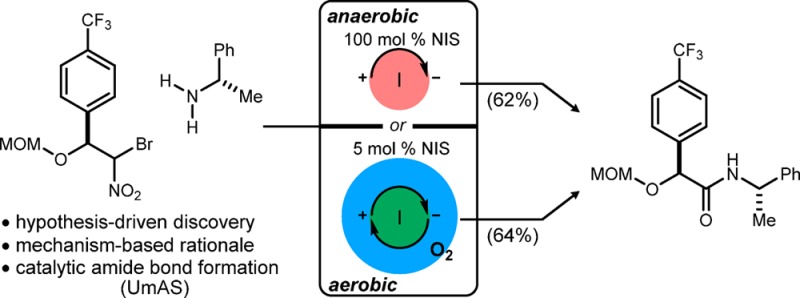
Umpolung Amide Synthesis (UmAS) provides direct access to amides from an α-bromo nitroalkane and an amine. Based on its mechanistic bifurcation after convergent C–N bond formation, depending on the absence or presence of oxygen, UmAS using substoichiometric N-iodosuccinimide (NIS) under aerobic conditions has been developed. In combination with the enantioselective preparation of α-bromo nitroalkane donors, this protocol realizes the goal of enantioselective α-amino amide and peptide synthesis based solely on catalytic methods.
“Amide formation avoiding poor atom economy reagents”1 has been hailed as a high level challenge and in need of critical importance in organic chemistry due to the ubiquity of amide bonds in biologically active molecules.2 Currently, solid-phase peptide synthesis3 and native chemical ligation4 are the most used amide forming transformations involving stereochemically complex products.5−8 Solid-phase peptide synthesis, however, uses superstoichiometric amounts of coupling reagents and is thus not economical. Catalytic methods9 based on boronic acids,10N-heterocyclic carbenes,11 and transfer hydrogenation12−14 have limited or no adaptation to more complex targets that present Lewis basic functionality and stereochemical complexity; in cases where turnover is documented, reagent loadings increase substantially as substrate complexity increases. Hence, it remains a challenge to prepare complex α-amino amides relying solely on enantioselective catalysis and amide formation using substoichiometric promoters.2,5,15
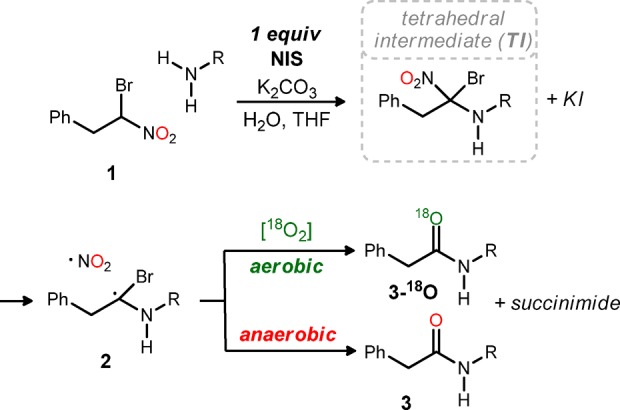
We recently reported a unique method to prepare amides from an α-bromo nitroalkane and amine using N-iodosuccinimide (NIS) under semiaqueous conditions, typically in THF with 5 equiv of H2O.16 The fact that the α-bromonitroalkane in these transformations is a functional equivalent of an acyl anion led us to suggest the term Umpolung Amide Synthesis (UmAS).17 When the chiral proton catalyzed aza-Henry reaction is used to prepare the α-bromonitroalkane components, chiral α-amino acids, specifically aryl glycines,18 can be accessed in high enantiomeric excess.16 The Henry reaction was deployed using a similar strategy to develop an enantioselective α-oxy amide synthesis.19 Concomitant with the development of these new reactions, we sought knowledge related to the mechanism of UmAS. Formation of a tetrahedral intermediate was posited on evidence for the formation of nucleophilic nitronate and electrophilic N-halamine. More recently, two pathways from the putative tetrahedral intermediate to an amide product were outlined, supported by isotopic labeling studies (Scheme 1).20 These findings served as the basis for the studies described here which document the feasibility of UmAS using substoichiometric NIS. Minimization of the cost and waste generation associated with amide and peptide synthesis based solely on enantioselective methods is a widely regarded goal.1
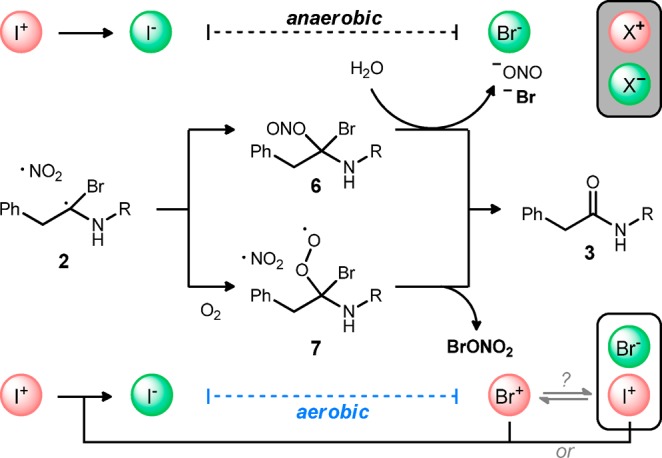
Scheme 1. Umpolung Amide Synthesis (UmAS): Competing Aerobic and Anaerobic Pathways to Amide from the Putative Tetrahedral Intermediate.
Our initial studies of amide formation from α-bromonitroalkanes uniformly required a full equivalent of N-iodosuccinimide, or an alternative electrophilic halonium source, in order to achieve complete conversion and the highest yields. This finding was more rigorously affirmed following the discovery of the mechanistic bifurcation outlined in Scheme 1.21 An experimental protocol was designed to track substrate conversion (1H NMR analysis of crude reaction mixtures) and the isolated yield (to corroborate conversion estimates), and these two measures tracked closely with one another at high levels of NIS (Table 1).22 When the reaction setup rigorously excluded oxygen (i.e., three freeze–pump–thaw cycles ending with an argon backflush) and maintained an identical overall reaction time, increasing NIS loadings led to increasing isolated yields of amide, reflecting the trend normally observed (Table 1, entries 1–5). The crude reaction mixtures of an experiment series with low NIS loadings (≤20 mol %, Table 1, entries 5–7) were suggestive of the sacrificial role that the substrate might play as an electrophilic halogen (bromonium ion) source, since debrominated nitroalkane (5) was observed in these experiments. In these cases, the substrate acts competitively as a bromonium source, leading to higher than expected yields for the 0 and 10 mol % NIS experiments as well as the nonlinear trend observed below 20 mol % NIS (Figure 1). At higher NIS loadings (≥30 mol %), 5 was not observed, signaling that the substrate contributes negligibly as a bromonium source; in this regime, a linear relationship is observed between the yield and NIS loading. This behavior suggests that the substrate only acts as the bromonium source when the reaction is starved of NIS leading to two different pathways (iodonium- vs bromonium-mediated) to form the halamine.
Table 1. Investigation of NIS Reagent Loading Under Anaerobic and Aerobic Conditions: Catalyzed Umpolung Amide Synthesis.
| entrya | O2b | NIS amount (mol %) | yield (%)c |
|---|---|---|---|
| 1 | no | 100 | 69 |
| 2 | no | 75 | 52 |
| 3 | no | 50 | 36 |
| 4 | no | 30 | 25 |
| 5 | no | 20 | 20 |
| 6 | no | 10 | 20 |
| 7 | no | 0 | 19 |
| 8 | yes | 20 | 76 |
| 9 | yes | 10 | 73 |
| 10 | yes | 5 | 76 |
| 11 | yes | 1 | 55 |
| 12e | yes | 0 | 51 |
| 13e | yesd | 5 | 69 |
Reactions are 0.2 M in nitroalkane and employ 1.2 equiv of amine and 5 equiv of H2O and run for 24 h. Under anaerobic conditions (entries 1–7), NIS was added once the nitroalkane was no longer visible by TLC, indicating that the nitronate predominated (see Supporting Information).
All solutions were first degassed (freeze–pump–thaw cycles), and entries not using oxygen were performed under an argon atmosphere (balloon), whereas entries using oxygen were performed under an oxygen atmosphere (balloon).
Isolated yield.
Open to air.
48 h reaction time.
Figure 1.
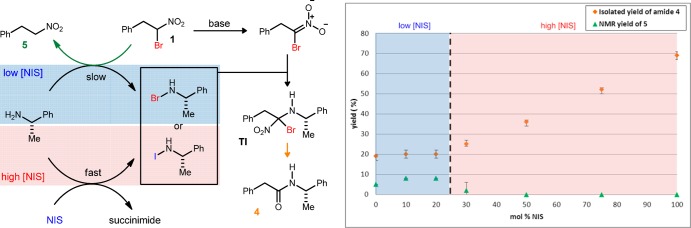
Correlation of electrophilic halogen loading and amide yield under anaerobic conditions.
A separate investigational line was aimed at the effect of an oxygen atmosphere under carefully controlled conditions, vis-à-vis, reaction degassing followed by an oxygen atmosphere (balloon). The use of a 20 mol % NIS loading in an otherwise identical reaction (cf. Table 1, entry 5) to which a balloon of oxygen was attached led to complete substrate conversion and isolation of the amide in 76% yield (Table 1, entry 8). It was further discovered that the magnitude of turnover under these conditions can be quite high, as the amide was isolated in 55% yield when 1 mol % NIS was used (Table 1, entry 11). We therefore examined, under oxygen-rich conditions, the possibility that the substrate might substitute for NIS to form the necessary halamine intermediate to initiate the reaction. Indeed, amide was isolated in 51% yield in the absence of a halonium source (Table 1, entry 12). The rate of amide production in this reaction was noticeably slower. The role of oxygen is critical for turnover, as the corresponding experiment under anaerobic conditions (no NIS) provided a 19% yield of the amide product (Table 1, entry 7). The reaction also proceeds with a less oxygen-rich atmosphere, although at a slower rate (Table 1, entry 13).
The behavior outlined in Table 1 is consistent with the intermediates and pathways depicted in Scheme 2. Recombination of the two radicals under anaerobic conditions leads to an alkyl nitrite that can hydrolyze to amide. The coproducts of this step are formally nitrite and bromide. Aerobic conditions, however, provide for formation of an alkyl peroxy intermediate competitive with nitrite formation.23 A straightforward balance of 7 and ·NO2 with amide products leads to the elements of bromonium nitrate. Hence, aerobic conditions and NIS lead to the production of an equivalent of electrophilic bromine (or iodine), in contrast to anaerobic conditions that lead to an equivalent each of bromide and iodide. The finer details remain to be clarified, as several pathways can be drawn from 7 to amide, consistent with oxidation of bromine, or oxidation of iodide to iodonium (e.g., I–Br) to form the key electrophilic halamine in a subsequent cycle.24 Nitrite radicals have been shown in the literature to react with superoxide,23 and XONO2 (X = Cl, Br, I) compounds are known to form via radical processes.25 The proposed formation of BrONO2 is of particular interest, as this species can oxidize iodide to iodonium. The uncertainty surrounding the actual redox pathway leading from 7 to amide may be of more academic value, and it may be difficult to discern whether iodonium or bromonium is the actual propagating species (or whether they are mutually exclusive). However, we have noted that reactions using NIS exhibit both faster conversion and less complex crude reaction mixtures than alternatives.26
Scheme 2. Umpolung Amide Synthesis (UmAS): Competing Aerobic and Anaerobic Pathways to Amide from the Putative Tetrahedral Intermediate.
Notwithstanding this uncertainty, analysis of the aerobic UmAS protocol substoichiometrically in NIS was evaluated in several existing UmAS contexts. A direct comparison of UmAS couplings of α-bromonitroalkanes and α-methyl benzyl amine led to comparable yields in most cases (Table 2). Several examples provided a higher yield of amide using aerobic conditions and a 5 mol % NIS loading. The yield was significantly lower (e.g., 66% vs 51% yield) in only two cases when using substoichiometric NIS (Table 2, entries 5–6). A similar trend was observed when a range of functionalized amines were compared using the two protocols (Table 3).
Table 2. NIS-Catalyzed Umpolung Amide Synthesis: Scope of Bromonitroalkane.
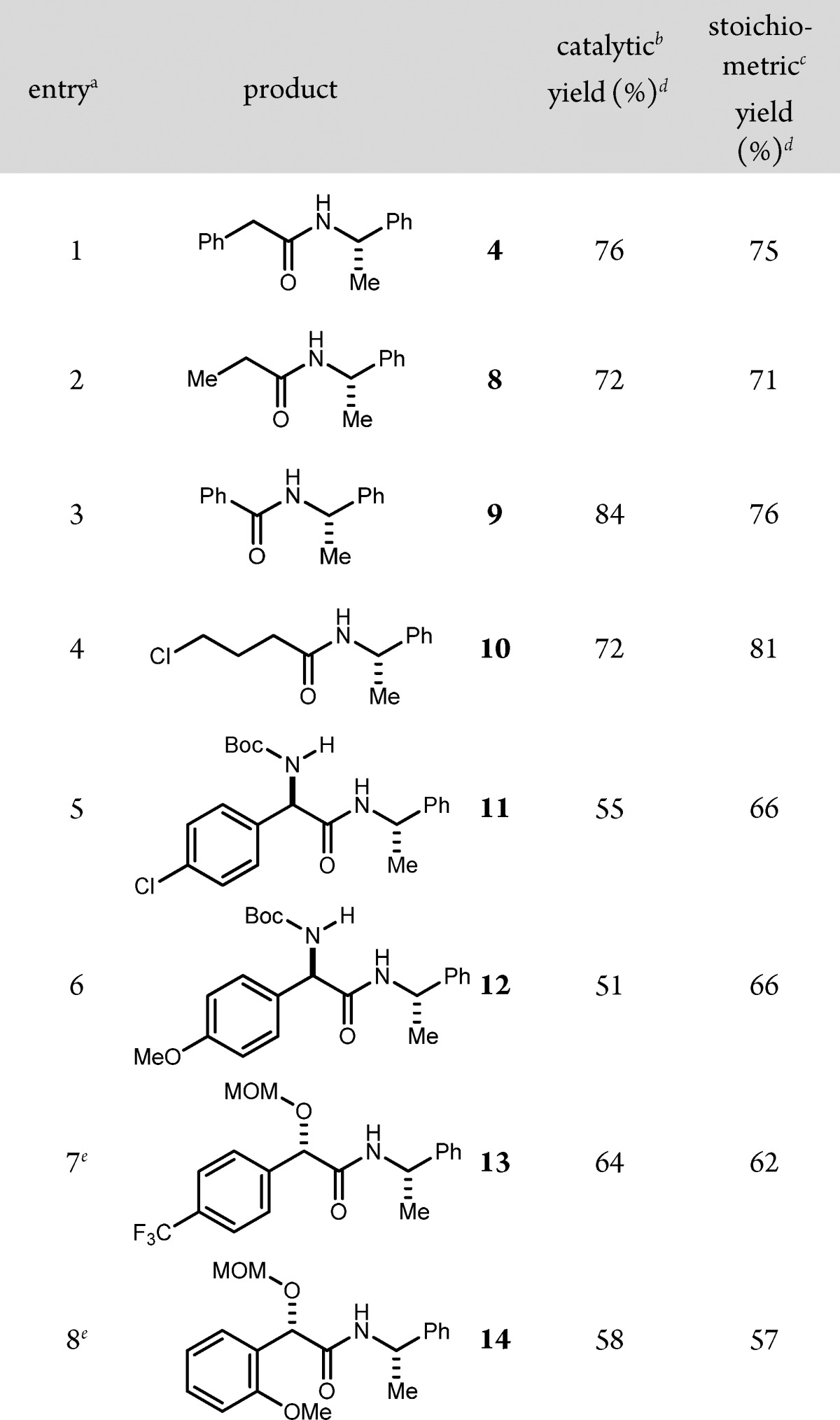
All reactions were conducted using bromonitroalkane (1 equiv, 0.2 M in DME), amine (1.2 equiv), K2CO3 (2 equiv), and H2O (5 equiv) at 0 °C. Entries 5–8 were >20:1 dr.
5 mol % NIS was used in the presence of an O2 balloon.
1 equiv of NIS was used.
Isolated yields.
10 mol % NIS was used.
Table 3. NIS-Catalyzed Umpolung Amide Synthesis: Scope of Amine.
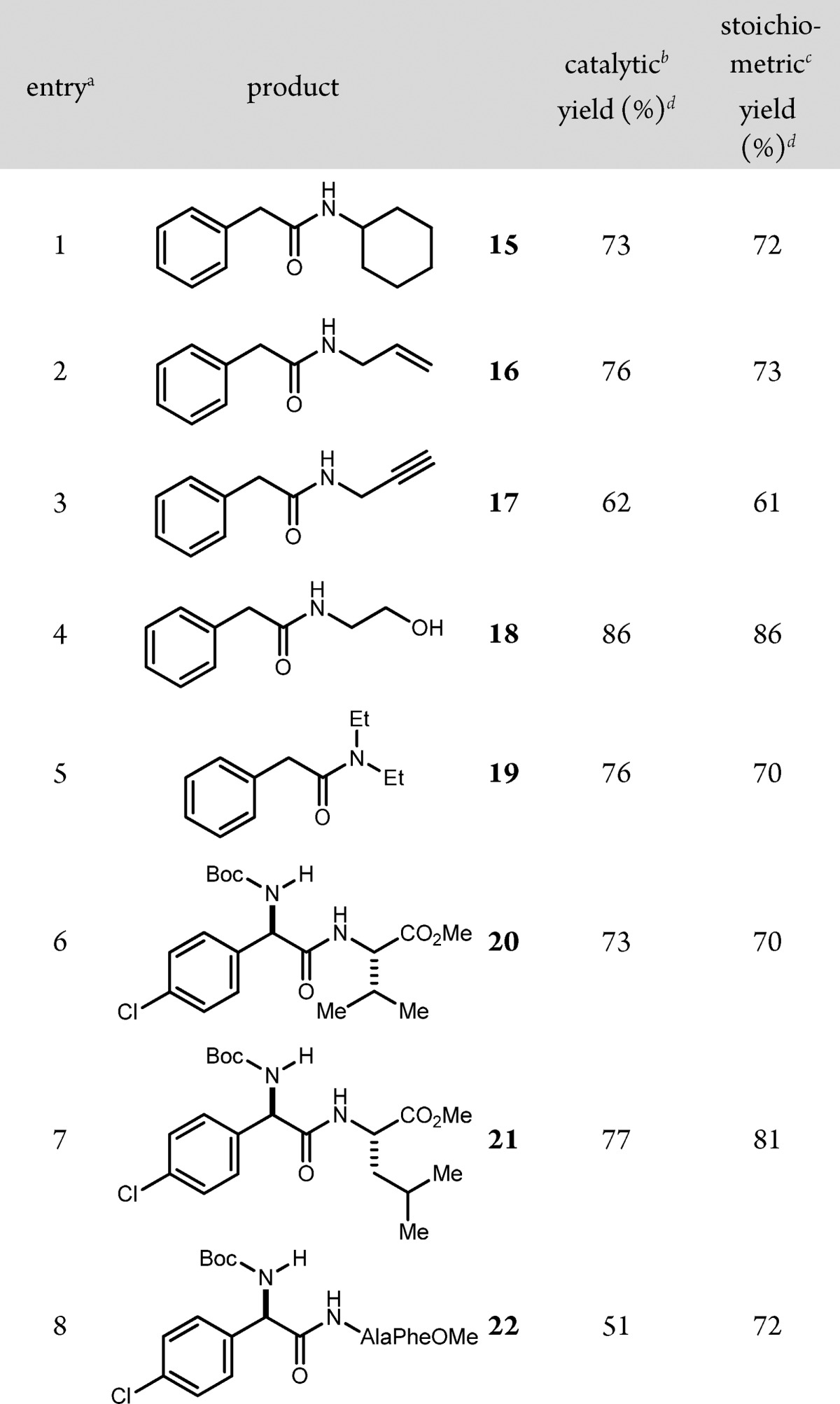
All reactions were conducted using bromonitroalkane (1 equiv, 0.2 M in DME), amine (1.2 equiv), K2CO3 (2 equiv), and H2O (5 equiv) at 0 °C. Entries 6–8 were >20:1 dr.
5 mol % NIS was used in the presence of an O2 balloon.
1 equiv of NIS was used.
Isolated yields.
In conclusion, consideration of a mechanistic hypothesis that Umpolung Amide Synthesis under aerobic conditions provides a pathway to electrophilic halonium regeneration resulted in the development of a protocol using substoichiometric N-iodosuccinimide. The protocol described here does not require reagent excess and avoids the production of coproducts that present purification issues. The fact that the overall route from bromonitromethane to chiral nonracemic α-amino amide is substoichiometric in all components, except for the base (K2CO3) and oxygen, provides a two-step solution for a catalytic, enantioselective synthesis of α-amino amides and peptides.
Acknowledgments
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health (GM 063557). Predoctoral fellowship support by the National Science Foundation (J.P.S.) is gratefully acknowledged.
Supporting Information Available
Experimental procedures and spectroscopic data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Constable D. J. C.; Dunn P. J.; Hayler J. D.; Humphrey G. R.; Leazer J. J. L.; Linderman R. J.; Lorenz K.; Manley J.; Pearlman B. A.; Wells A.; Zaks A.; Zhang T. Y. Green Chem. 2007, 9, 411–420. [Google Scholar]
- Pattabiraman V. R.; Bode J. W. Nature 2011, 480, 471–479. [DOI] [PubMed] [Google Scholar]; Nilsson B. L.; Soellner M. B.; Raines R. T. Annu. Rev. Biophys. Biomol. Struct. 2005, 34, 91–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrifield R. B. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar]; Valeur E.; Bradley M. Chem. Soc. Rev. 2009, 38, 606–631. [DOI] [PubMed] [Google Scholar]
- Dawson P. E.; Muir T. W.; Clarklewis I.; Kent S. B. H. Science 1994, 266, 776–779. [DOI] [PubMed] [Google Scholar]; Haase C.; Seitz O. Angew. Chem., Int. Ed. 2008, 47, 1553–1556. [DOI] [PubMed] [Google Scholar]
- Wu J.; Ruiz-Rodriguez J.; Comstock J. M.; Dong J. Z.; Bode J. W. Chem. Sci. 2011, 2, 1976–1979. [Google Scholar]; Bode J. W.; Fox R. M.; Baucom K. D. Angew. Chem., Int. Ed. 2006, 45, 1248–1252. [DOI] [PubMed] [Google Scholar]
- Zheng J.-S.; Yu M.; Qi Y.-K.; Tang S.; Shen F.; Wang Z.-P.; Xiao L.; Zhang L.; Tian C.-L.; Liu L. J. Am. Chem. Soc. 2014, 136, 3695–3704. [DOI] [PubMed] [Google Scholar]
- Zheng J.-S.; Tang S.; Huang Y.-C.; Liu L. Acc. Chem. Res. 2013, 46, 2475–2484. [DOI] [PubMed] [Google Scholar]
- Wang T.; Danishefsky S. J. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 11708–11713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg H.; Tinnis F.; Selander N.; Adolfsson H. Chem. Soc. Rev. 2014, 43, 2714–2742. [DOI] [PubMed] [Google Scholar]; Singh C.; Kumar V.; Sharma U.; Kumar N.; Singh B. Curr. Org. Synth. 2013, 10, 241–264. [Google Scholar]
- Ishihara K.; Kubota M.; Kurihara H.; Yamamoto H. J. Org. Chem. 1996, 61, 4560–4567. [DOI] [PubMed] [Google Scholar]; Marcelli T. Angew. Chem., Int. Ed. 2010, 49, 6840–6843. [DOI] [PubMed] [Google Scholar]; Al-Zoubi R. M.; Marion O.; Hall D. G. Angew. Chem., Int. Ed. 2008, 47, 2876–2879. [DOI] [PubMed] [Google Scholar]; Charville H.; Jackson D.; Hodges G.; Whiting A. Chem. Commun. (Cambridge, U. K.) 2010, 46, 1813–1823. [DOI] [PubMed] [Google Scholar]
- Bode J. W.; Sohn S. S. J. Am. Chem. Soc. 2007, 129, 13798–13799. [DOI] [PubMed] [Google Scholar]; Vora H. U.; Rovis T. J. Am. Chem. Soc. 2007, 129, 13796–13797. [DOI] [PMC free article] [PubMed] [Google Scholar]; De Sarkar S.; Studer A. Org. Lett. 2010, 12, 1992–1995. [DOI] [PubMed] [Google Scholar]; Chiang P.-C.; Kim Y.; Bode J. W. Chem. Commun. (Cambridge, U. K.) 2009, 4566–4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunanathan C.; Ben-David Y.; Milstein D. Science 2007, 317, 790–792. [DOI] [PubMed] [Google Scholar]; Nordstrøm L. U.; Vogt H.; Madsen R. J. Am. Chem. Soc. 2008, 130, 17672–17673. [DOI] [PubMed] [Google Scholar]; Muthaiah S.; Ghosh S. C.; Jee J.-E.; Chen C.; Zhang J.; Hong S. H. J. Org. Chem. 2010, 75, 3002–3006. [DOI] [PubMed] [Google Scholar]; Saha B.; Sengupta G.; Sarbajna A.; Dutta I.; Bera J. K.. J. Organomet. Chem., DOI: 10.1016/j.jorganchem.2013.12.051. [DOI] [Google Scholar]; Srimani D.; Balaraman E.; Hu P.; Ben-David Y.; Milstein D. Adv. Synth. Catal. 2013, 355, 2525–2530. [Google Scholar]; Cho D.; Ko K. C.; Lee J. Y. Organometallics 2013, 32, 4571–4576. [Google Scholar]
- Kang B.; Fu Z.; Hong S. H. J. Am. Chem. Soc. 2013, 135, 11704–11707. [DOI] [PubMed] [Google Scholar]
- Gunanathan C.; Milstein D. Science 2013, 341, 1229712. [DOI] [PubMed] [Google Scholar]; Chen C.; Hong S. H. Org. Biomol. Chem. 2011, 9, 20–26. [DOI] [PubMed] [Google Scholar]
- Shangguan N.; Katukojvala S.; Greenberg R.; Williams L. J. J. Am. Chem. Soc. 2003, 125, 7754–7755. [DOI] [PubMed] [Google Scholar]
- Shen B.; Makley D. M.; Johnston J. N. Nature 2010, 465, 1027–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For an umpolung N-phenyl hydroxamic acid synthesis, see:Wong F. T.; Patra P. K.; Seayad J.; Zhang Y.; Ying J. Y. Org. Lett. 2008, 10, 2333–2336. [DOI] [PubMed] [Google Scholar]
- Williams R. M.; Hendrix J. A. Chem. Rev. 1992, 92, 889–917. [Google Scholar]
- Leighty M. W.; Shen B.; Johnston J. N. J. Am. Chem. Soc. 2012, 134, 15233–15236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackleford J. P.; Shen B.; Johnston J. N. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 44–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior to the work reported here, aside from the careful mechanistic studies (ref (20)), a typical reaction setup involved a vial or flask capped with a septum, creating a static atmosphere. In these cases, exposure to atmospheric oxygen is best described as variable.
- THF was originally used as the solvent, but its oxidation by NIS to the corresponding lactol and lactone was observed when amide formation was slow. Dimethoxyethane (DME) was identified as the optimal solvent for these studies since it did not suffer from detectable oxidation.
- Hodges G. R.; Ingold K. U. J. Am. Chem. Soc. 1999, 121, 10695–10701. [Google Scholar]; Goldstein S.; Czapski G. J. Am. Chem. Soc. 1998, 120, 3458–3463. [Google Scholar]
- Finkbeiner P.; Nachtsheim B. J. Synthesis 2013, 45, 979–999. [Google Scholar]
- Papayannis D. K.; Kosmas A. M. Chem. Phys. 2005, 315, 251–258. [Google Scholar]
- NBS is a competent halonium source for the reaction. However, the conversion to amide is slower and hindered by the formation of the dibrominated nitroalkane.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.