

Abstract
This Letter reports a family of novel antimicrobial compounds obtained by combining peptide library screening with structure-based design. Library screening led to the identification of a human LL-37 peptide resistant to chymotrypsin. This d-amino-acid-containing peptide template was active against Escherichia coli but not methicillin-resistant Staphylococcus aureus (MRSA). It possesses a unique nonclassic amphipathic structure with hydrophobic defects. By repairing the hydrophobic defects, the peptide (17BIPHE2) gained activity against the ESKAPE pathogens, including Enterococcus faecium, S. aureus, Klebsiella pneumoniae, Acinetobacter baumanii, Pseudomonas aeruginosa, and Enterobacter species. In vitro, 17BIPHE2 could disrupt bacterial membranes and bind to DNA. In vivo, the peptide prevented staphylococcal biofilm formation in a mouse model of catheter-associated infection. Meanwhile, it boosted the innate immune response to further combat the infection. Because these peptides are potent, cell-selective, and stable to several proteases, they may be utilized to combat one or more ESKAPE pathogens.
A hallmark of emerging difficult-to-treat clinical superbugs is their ability to “escape” the action of multiple traditional antibiotics, in part due to biofilm formation in the host. The ESKAPE pathogens include Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumanii, Pseudomonas aeruginosa, and Enterobacter species.1 According to the Centers for Disease Control and Prevention, the six ESKAPE bacterial species cause two-thirds of health care-associated infections, leading to 99,000 deaths annually in the United States. S. aureus infections alone cause a comparable number of deaths as human immunodeficiency virus type 1 (HIV-1).2 Antimicrobial peptides (AMPs) are host defense molecules that have maintained antimicrobial activity for millions of years by keeping pace with the evolution of bacterial resistance mechanisms. This feature makes them appealing candidates for developing new antibacterial compounds.3−5
The protective effect of human LL-37 against infection is underscored by both clinical observations and recent findings from animal models.6−8 Therefore, there is growing interest in its medical use. Interestingly, sunlight triggers the biosynthesis of dihydroxyvitamin D in skin that binds the vitamin D receptor to initiate peptide expression.6 At the moment, clinical trials of vitamin D as an anti-infective agent have not demonstrated efficacy.9 Another avenue is to administer LL-37 at infected sites. Cationic LL-37 possesses a long amphipathic helical structure covering residues 2–31 that recognizes and disrupts anionic bacterial membranes (Figure 1a).10−12 To minimize the cost for peptide synthesis, several laboratories reported the identification of active fragments, including GF-17 corresponding to the major antimicrobial region (residues 17–32).12−15 Since these natural peptides lack sufficient stability to proteases, this study intends to re-engineer human LL-37 into peptide analogues with potency, stability, and cell selectivity against superbugs.
Figure 1.

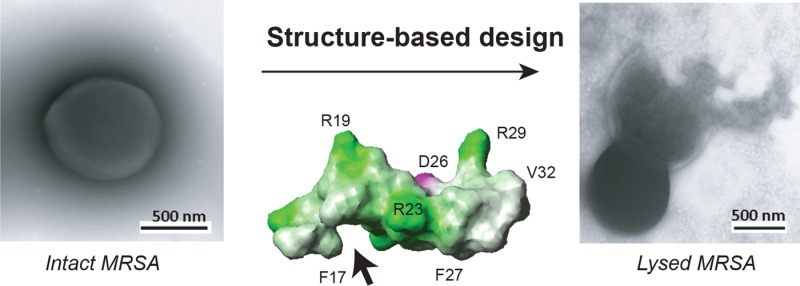
Structure-based design of potent peptides to combat the ESKAPE pathogens. Shown are NMR structures of human LL-37 (A); its peptides GF-17 (B) and GF-17d3 (C and D) bound to membrane-mimetic micelles;10,15,18 peptide degradation kinetics (E);19 flow cytometry of S. aureus USA300 after treatments with 40 μM GF-17, 17BIPHE, or 17BIPHE2 peptides (F);18 transmission electron microscopy images of S. aureus USA300 before (G) and after (H) peptide treatment;14 and DNA retardation (I) by the peptides in Table 1. Each peptide was evaluated by a 1% agarose gel (casted with 1 μg/mL ethidium bromide) at two concentrations, first at 6 and then 12 μM (lanes 1: 75 ng pUC19 DNA plasmid itself; 2 and 3: 17F2; 4 and 5: 17mF-F; 6 and 7: 17F-Naph; 8 and 9: 17mF-Naph; 10 and 11: 17Naph-mF; 12 and 13: 17BIPHE; 14 and 15: 17BIPHE2). Structural images were generated using MOLMOL.29
To identify protease-resistant peptide templates, we screened 30 synthetic peptides. A standard microdilution assay protocol15 was modified to include proteases into the duplicated wells with high concentrations of peptide so that both antimicrobial activity and protease stability could be evaluated simultaneously. In this setup, most of the LL-37 peptides showed bactericidal effects in the absence of the protease but became inactive in the presence of chymotrypsin after overnight incubation at 37 °C (peptide:protease molar ratio of 40:1). However, GF-17d3, a GF-17-derived peptide with incorporation of three d-amino acids at positions 20, 24, and 28 (numbered as in LL-37),12 retained antimicrobial activity against Escherichia coli K12 (Supplementary Table 1). In contrast, another two peptide analogues containing one to two d-amino acids at positions 20 or both 20 and 24 were not stable. Shai and colleagues also found peptide stability improvement due to the incorporation of d-amino acids into other peptides.16 Compared to GF-17, GF-17d3 lost antibacterial activity against S. aureus USA300 LAC.17,18 This protease-resistant template was selected for subsequent engineering of peptides for activity against S. aureus as well as other ESKAPE pathogens.
We conducted a rational design on the basis of the 3D structure of GF-17d3. NMR structural analysis revealed that the three d-amino acids distorted the regular helical backbone structure of GF-17 (Figure 1b)14 into a novel nonclassic amphipathic structure of GF-17d3 (Figure 1c).15 The noncoherent packing of the GF-17d3 side chains caused a hydrophobic defect in the structure (Figure 1c,d, arrow), leading to reduced hydrophobicity. We hypothesized that the antimicrobial activity of this peptide 17F2 (sequence GX1KRLVQRLKDX2LRNLV-amide, see Supplementary Table 1) could be enhanced by filling in the hydrophobic cavity (Figure 1d, arrow) with a larger hydrophobic side chain. The antimicrobial activities of these peptide analogues were evaluated using the standard microdilution assays,19 and the minimal inhibitory concentrations (MIC) against a panel of the ESKAPE pathogens are provided in Table 1. 17F2 inhibited E. coli, A. baumannii, and E. cloacae (MIC 6.2–25 μM) but not S. aureus, E. faecium, K. pneumoniae, or P. aeruginosa until 100 μM or higher. When X1 was replaced with 4-trifluoromethylphenylalanine (mF), the resultant peptide 17mF-F displayed MICs in the range of 25–50 μM against most of the ESKAPE strains and 3.1–6.2 μM against A. baumannii. We also aimed to identify a second site for activity enhancement. When X2 of 17F2 was mutated to 2-naphthylalanine (Naph), the new peptide 17F-Naph became active against E. faecium and A. baumannii (MIC 3.1 μM). When both the above substitutions were made at X1 and X2, the activity of the resultant peptide 17mF-Naph was further increased, especially against MRSA USA300 (MIC 6.2 μM). To compare the two sites, we swapped these two substitutions at positions X1 and X2 to obtain 17Naph-mF. Interestingly, 17mF-Naph and 17Naph-mF have an essentially identical antibacterial activity spectrum based on MICs (Table 1). In addition, we also incorporated biphenylalanines at position X1 alone or at both X1 and X2 of the 17F2 template. While 17BIPHE with one replacement was more active against S. aureus than 17mF-F, 17BIPHE2 with two replacements showed the highest antibacterial activity against all of the ESKAPE pathogens (MIC 3.1–6.2 μM). Killing kinetics assays revealed that 17BIPHE2 was able to eliminate P. aeruginosa PAO1 in 30 min and S. aureus USA300 in 90 min (Supplementary Figure 1). The killing abilities of these peptides are essentially proportional to their MIC values as well as retention times (a measure of hydrophobicity) on a reverse-phase HPLC column.19 Hence, our structure-based design produced a family of antimicrobials against the ESKAPE pathogens (Table 1).
Table 1. Minimal Inhibitory Concentration (MIC), Cytotoxicity, and Hydrophobicity of a Series of Peptides Designed Based on Human Cathelicidin LL-37.
| MIC (μM) |
||||||||
|---|---|---|---|---|---|---|---|---|
| peptide | EFa | SA | KP | AB | PA | EC | HL50 (μM)b | tR (min)c |
| 17F2 | >100 | >100 | >100 | 6.2–12.5 | 100 | 25 | >900 | 9.95 |
| 17mF-F | 25–50 | 25 | 50 | 3.1–6.2 | 25 | 25 | >900 | 10.32 |
| 17F-Naph | 3.1 | 25 | 25 | 3.1 | 12.5 | 12.5 | >900 | 10.56 |
| 17mF-Naph | 3.1 | 6.2 | 12.5 | 3.1 | 6.2–12.5 | 6.2 | 500 | 10.99 |
| 17Naph-mF | 3.1 | 6.2 | 12.5 | 3.1 | 6.2–12.5 | 6.2–12.5 | 950 | 10.94 |
| 17BIPHE | 12.5 | 12.5 | 25 | 3.1 | 12.5 | 12.5 | >900 | 10.55 |
| 17BIPHE2 | 3.1 | 3.1 | 3.1 | 3.1 | 6.2 | 3.1 | 225 | 11.26 |
Abbreviations used: EF, E. faecium ATCC51559; SA, S. aureus USA300 LAC; KP, K. pneumonia ATCC13883; AB, A. baumannii B2367-12; PA, P. aeruginosa PAO1; EC, E. cloacae B2366-12.
The peptide concentration (μM) that causes 50% lysis of human erythrocytes.
HPLC retention time of the peptide.
In developing peptide-based antimicrobials, it is essential to minimize potential cytotoxicity to human cells. We found that 17F2 and various single substitution variants displayed poor hemolytic ability, with a 50% hemolysis concentration (HL50) greater than 900 μM (Table 1). However, the substitution swapped pair, 17mF-Naph and 17Naph-mF, gave different HL50 concentrations of 500 and 950 μM, respectively. 17BIPHE2, the most potent peptide, showed an HL50 of 225 μM. Relative to the MIC value (3.1 μM) that inhibited the majority of these ESKAPE pathogens, we obtained a cell selectivity index (i.e., the ratio between HL50 and MIC) of 72, meaning that 17BIPHE2 is not cytotoxic at the MIC. If the same MIC of 3.1 μM was used, the cell selectivity indices for 17mF-Naph and 17Naph-mF were 161 and 306, respectively. This is extremely interesting considering that 17Naph-mF, with a better selectivity index, is also more efficient in killing MRSA than 17mF-Naph (Supplementary Figure 1). Thus, there is an excellent therapeutic window for us to utilize these novel compounds to treat infections caused by the ESKAPE pathogens.
To validate the stability of the engineered peptides, we followed the degradation kinetics of 17BIPHE2 in the presence of chymotrypsin by SDS-PAGE. While a GF-17 analogue18 was rapidly digested in 4 h (t1/2 < 0.5 h), the level of intact 17BIPHE2 remained constant during this time period (Figure 1e). Furthermore, 17BIPHE2, but not GF-17, was also stable for at least 24 h in the presence of S. aureus V8 protease or fungal protease K (Supplementary Figure 2). We conclude that 17BIPHE2 is stable to the action of several proteases.
To shed light on the killing mechanism of 17BIPHE2, we conducted flow cytometry studies. When bacterial membranes are compromised, flow cytometry can follow the cellular entry of the nonpermeable dye TO-PRO3, which upon binding bacterial DNA leads to a rapid increase in fluorescence.18 At 80 μM, this was indeed the case with 17BIPHE and 17BIPHE2 (Figure 1f), which were comparable to GF-17, a peptide known to target bacterial membranes. At a lower peptide concentration (20 μM), the killing ability of peptides was found to be inversely proportional to the MIC (Supplementary Figure 3). To validate membrane damage, we used transmission electron microscopy (TEM) to directly visualize possible damage to bacteria.19 While untreated bacteria possessed a uniform membrane architecture (Figure 1g), the addition of 17BIPHE2 caused bacterial lysis (Figure 1h).
Bacterial membrane damage raised the possibility that 17BIPHE2 might also enter cells and bind DNA. To provide evidence for this, we used gel retardation experiments.20 While 17F2 did not bind to DNA in this assay, 17BIPHE2 at 12 μM was trapped in the wells (Figure 1i). At the same concentration, both 17BIPHE and 17F-Naph, with a single substitution, retarded DNA to a similar degree. 17mF-Naph and 17Naph-mF, with the two substitutions swapped, behaved similarly. Thus, this family of peptide analogues possesses a wide range of DNA retarding abilities. Interestingly, the extent of DNA retardation (presumably the amount of peptide associated with DNA) is proportional to bacterial killing ability, with 17BIPHE2 being the strongest and 17F2 the poorest. A similar trend was observed using the DNA purified from P. aeruginosa PAO1 (Supplementary Figure 4). These correlations suggest that DNA binding could be part of the mechanism of bacterial killing.
The in vivo efficacy of 17BIPHE2 against S. aureus USA300 LAC was examined using a mouse model of catheter-associated biofilm infection as previously described.21−24 Animals used in this study were cared for by following institutional guidelines. Peptide treatment was initiated at the time of infection (time 0), by directly injecting 200 μg peptide, dissolved in PBS, into the catheter followed by additional administration of 200 μg peptide injected subcutaneously at four different sites surrounding the catheter at 24 and 48 h postinfection. Bacterial titers associated with the catheter and surrounding host tissue were evaluated at day 3 (Figure 2a–c) and day 14 postinfection (Figure 2c–e) to determine the impact of peptide treatment on bacterial burdens. Mice treated with 17BIPHE2 exhibited a significant decrease in bacterial titers on the catheter surfaces (Figure 2a,d) as well as surrounding tissues compared to vehicle-treated animals (Figure 2b,e). Importantly, early peptide treatment was key to preventing S. aureus biofilm establishment, since minimal bacterial growth was detected at day 14 following infection even though the last dosing occurred at 48 h (Figure 2b,e). As a negative control, LL-23V9, a peptide designed based on the N-terminal 23 residues of LL-37, which is inactive against S. aureus USA300 in vitro,25 was unable to decrease bacterial burdens on biofilm-infected catheters in vivo (Figure 2c).
Figure 2.

Prophylactic 17BIPHE2 treatment reduced bacterial burdens in S. aureus catheter-associated biofilms in vivo (A–E), down-regulated IL-10 (F), up-regulated CCL2 (G) and CXCL10 (H), and recruited monocytes (I). Mice received 200 μg injections of 17BIPHE2 (A, B, D, and E) and LL-23V9 (C) directly into the catheter at the time of infection (time 0) followed by 200 μg peptide injected subcutaneously at four different sites around the catheter, 24 and 48 h following infection. Animals were sacrificed at days 3 (A, B, and C) and 14 (D and E) following S. aureus exposure, whereupon catheters (A, C, and D) and surrounding host tissues (B and E) were recovered to quantitate bacterial burdens. Results are expressed as the number of CFU/mL for catheters or CFU/mg tissue, to correct for differences in tissue sampling size. Significant differences in bacterial burdens between PBS and peptide-treated mice are denoted by asterisks (*, p < 0.05; **, p < 0.01) with results presented from individual animals and bars representing the mean of each group (A and B: n = 16/group; C, D, and E: n = 8/group).
We were also interested in determining whether the peptide had any impact on the host immune response by measuring chemokine levels with and without peptide treatment. 17BIPHE2 significantly increased CCL2 (MCP-1) and CXCL10 (IP-10) expression and reduced IL-10 at day 3 postinfection (Figure 2f–h). This observation is remarkable given our previous findings that bacterial biofilm formation strongly suppressed CCL2 and increased IL-10 in the same animal model.24 Increased CCL2 expression coincided with significantly heightened monocyte recruitment at day 3, which returned to baseline at day 14 postinfection (Figure 2i). Thus, one mechanism whereby 17BIPHE2 treatment may deter S. aureus biofilm development is through rapid monocyte recruitment. In addition, the expression of CXCL10 might also directly inhibit MRSA based on its known antimicrobial activity.26,27
In conclusion, we succeeded in engineering human cathelicidin LL-37 into potent compounds by combining peptide library screening with structure-based design (Figure 1). Our engineered peptides have multiple desired features: in vitro and in vivo efficacy against community-associated MRSA USA300, stability to the action of multiple proteases, and good cell selectivity indices (72–306). The most potent compound 17BIPHE2 appears to kill bacteria by damaging bacterial membranes, which may also involve the access and binding to the intracellular bacterial DNA. Apart from bacterial killing in vivo, 17BIPHE2 can augment host innate immunity to further combat bacterial infection. Combined, these properties make 17BIPHE2 an attractive candidate for the treatment of complicated clinical infections involving biofilms of one or more ESKAPE pathogens, especially in implanted medical devices that cost millions of dollars per year in the USA alone.28
Methods
Peptides and Hydrophobicity Measurements
All peptides were chemically synthesized at purity of >95% (Genemed Synthesis Inc.). Peptide hydrophobicity was estimated by measuring its retention time on a Waters HPLC system equipped with an analytical reverse-phase Vydac C18 column (250 mm × 4.6 mm) as described.19
Antibacterial Activity Assays
The antibacterial activities of the peptides were determined using the standard broth microdilution method as described.19
Hemolytic Assays
Peptide hemolysis was assayed using an established protocol.21
Transmission Electron Microscopy
S. aureus USA300 were grown in LB medium at 37 °C to the mid-logarithmic phase. The samples were prepared as described previously19 and observed in an FEI Technai G2 TEM operated at 80 kV accelerating voltage at the University of Nebraska Medical Center (UNMC).
Flow Cytometry
Membrane permeation analysis was performed by using the BacLight bacterial membrane potential kit (Invitrogen) as described on a FACSAria flow cytometer.18 In short, logarithmic growth-phase cultures of S. aureus USA300 (OD600, 0.6–1.0), after being washed twice and resuspended in PBS (0.2 μm filter-sterilized) (∼5 × 108 CFU/mL), were treated at 80 μM concentration of the peptides in Figure 1f and 20 μM in Supplementary Figure 3. Bacteria were then treated for 10 min with 3,3′-diethyloxacarbocyanine iodide [DiOC2(3)] (30 μM)/TO-PRO-3 (100 nM) dye mixture. Data were analyzed with FlowJo software.
Peptide Stability to the Action of Proteases
Peptide stability was evaluated based on our published protocol19 with one modification. Aliquots (10 μL) of the reaction solutions (peptide/protease molar ratio, 40:1) in 10 mM PBS buffer (pH 8) at 37 °C were taken at 0, 6, and 24 h and immediately mixed with 10 μL of 2× SDS loading buffer followed by boiling for 5 min to stop the reaction. Samples were analyzed using 5% stacking/18% resolving tricine gel.
S. aureus Biofilm Infection Model
Male C57BL/6 mice, 6–8 weeks old, were anesthetized with tribromoethanol (avertin), and the skin was shaved and scrubbed with povidone-iodine. A small subcutaneous (sc) incision was made in the left flank and a blunt probe was used to create a pocket for insertion of a sterile 14-gauge Teflon catheter 1 cm in length. The incision was sealed and 1,000 CFU USA300 LAC::lux in 20 μL of sterile PBS was slowly injected through the skin into the infected catheter lumen. Animals were initially treated with 200 μg peptide, dissolved in PBS, injected into the catheter at the time of infection (time 0) followed by 200 μg peptide injected subcutaneously at four different sites surrounding the catheter at 24 and 48 h postinfection. At days 3 and 14 postinfection catheters were removed in order to quantitate bacterial burdens. Catheters were sonicated in 1 mL of PBS to dissociate the biofilm from the intra- and extraluminal surfaces. The tissue surrounding the catheter was also removed, weighed, and homogenized in 500 μL homogenization buffer. Bacterial titers associated with catheters and surrounding tissues were quantified by plating on blood agar plates. To compare the expression of inflammatory mediators associated with biofilm-infected tissues treated with our novel antimicrobial peptides, a mouse microbead array or ELISA detection kit was utilized according to the manufacturer’s instructions. Results were normalized to the amount of total protein recovered to correct for differences in tissue sampling size.
Acknowledgments
This work was supported by NIAID/NIH grant R56AI81975 and in part by Nebraska state funding to G.W., P01 AI083211 to K.W.B., and P01 AI083211 Project 4 to T.K. We thank P. Fey for providing the ESKAPE pathogens used in this study, T. Bargar for conducting electron microscopy, and D. L. Vidlak for chemokine assays.
Supporting Information Available
This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Boucher H. W.; Talbot G. H.; Bradley J. S.; Edwards J. E.; Gilbert D.; Rice L. B.; Scheld M.; Spellberg B.; Bartlett J. (2009) Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 48, 1–12. [DOI] [PubMed] [Google Scholar]
- Klevens R. M.; Morrison M. A.; Nadle J.; Petit S.; Gershman K.; Ray S.; Harrison L. H.; Lynfield R.; Dumyati G.; Townes J. M.; Craig A. S.; Zell E. R.; Fosheim G. E.; McDougal L. K.; Carey R. B.; Fridkin S. K. (2007) Invasive methicillin-resistant Staphylococcus aureus infections in the United States. J. Am. Med. Assoc. 298, 1763–1771. [DOI] [PubMed] [Google Scholar]
- Zasloff M. (2002) Antimicrobial peptides of multicellular organisms. Nature 415, 389–395. [DOI] [PubMed] [Google Scholar]
- Lai Y.; Gallo R. L. (2009) AMPed up immunity: how antimicrobial peptides have multiple roles in immune defense. Trends Immunol. 30, 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock R. E.; Sahl H. G. (2013) New strategies and compounds for anti-infective treatment. Curr. Opin. Microbiol. 16, 519–521. [DOI] [PubMed] [Google Scholar]
- van der Does A. M.; Bergman P.; Agerberth B.; Lindbom L. (2012) Induction of the human cathelicidin LL-37 as a novel treatment against bacterial infections. J. Leukocyte Biol. 92, 735–742. [DOI] [PubMed] [Google Scholar]
- Putsep K.; Carsson G.; Boman H. G.; Andersson M. (2002) Deficiency of antibacterial peptides in patients with morbus Kostmann: an observation study. Lancet 360, 1144–1149. [DOI] [PubMed] [Google Scholar]
- Chromek M.; Slamová Z.; Bergman P.; Kovács L.; Podracká L.; Ehrén I.; Hökfelt T.; Gudmundsson G. H.; Gallo R. L.; Agerberth B.; Brauner A. (2006) The antimicrobial peptide cathelicidin protects the urinary tract against invasive bacterial infection. Nat. Med. 12, 636–641. [DOI] [PubMed] [Google Scholar]
- Nakatsuj T.; Gallo R. L. (2012) Antimicrobial peptides: old molecules with new ideas. J. Invest. Dermatol. 132, 887–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G. (2008) Structures of human host defense cathelicidin LL-37 and its smallest antimicrobial peptide KR-12 in lipid micelles. J. Biol. Chem. 283, 32637–32643. [DOI] [PubMed] [Google Scholar]
- Oren Z.; Lerman J. C.; Gudmundsson G. H.; Agerberth B.; Shai Y. (1999) Structure and organization of the human antimicrobial peptide LL-37 in phospholipid membranes: relevance to the molecular basis for its non-cell-selective activity. Biochem. J. 341(Pt 3), 501–513. [PMC free article] [PubMed] [Google Scholar]
- Wang G.; Mishra B.; Epand R. F.; Epand R. M. (2014) High-quality 3D structures shine light on antibacterial, anti-biofilm and antiviral activities of human cathelicidin LL-37 and its fragments. Biochim. Biophys. Acta 1838, 2160–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braff M. H.; Hawkins M. A.; Di Nardo A.; Lopez-Garcia B.; Howell M. D.; Wong C.; Lin K.; Streib J. E.; Dorschner R.; Leung D. Y.; Gallo R. L. (2005) Structure-function relationships among human cathelicidin peptides: dissociation of antimicrobial properties from host immunostimulatory activities. J. Immunol. 174, 4271–4278. [DOI] [PubMed] [Google Scholar]
- Nagaoka I.; Kuwahara-Arai K.; Tamura H.; Hiramatsu K.; Hirata M. (2005) Augmentation of the bactericidal activities of human cathelicidin CAP18/LL-37-derived antimicrobial peptides by amino acid substitutions. Infammation. Res. 54, 66–73. [DOI] [PubMed] [Google Scholar]
- Li X.; Li Y.; Han H.; Miller D. W.; Wang G. (2006) Solution structures of human LL-37 fragments and NMR-based identification of a minimal membrane-targeting antimicrobial and anticancer region. J. Am. Chem. Soc. 128, 5776–5785. [DOI] [PubMed] [Google Scholar]
- Papo N.; Shai Y. (2003) New lytic peptides based on the D,L-amphipathic helix motif preferentially kill tumor cells compared to normal cells. Biochemistry 42, 9346–9354. [DOI] [PubMed] [Google Scholar]
- Epand R. F.; Wang G.; Berno B.; Epand R. M. (2009) Lipid segregation explains selective toxicity of a series of fragments derived from the human cathelicidin LL-37. Antimicrob. Agents Chemother. 53, 3705–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G.; Epand R. F.; Mishra B.; Lushnikova T.; Thomas V. C.; Bayles K. W.; Epand R. M. (2012) Decoding the functional roles of cationic side chains of the major antimicrobial region of human cathelicidin LL-37. Antimicrob. Agents Chemother. 56, 845–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra B.; Wang G. (2012) Ab initio design of potent anti-MRSA peptides based on database filtering technology. J. Am. Chem. Soc. 134, 12426–12429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C. B.; Kim H. S.; Kim S. C. (1998) Mechanism of action of the antimicrobial peptide buforin II: buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem. Biophys. Res. Commun. 244, 253–257. [DOI] [PubMed] [Google Scholar]
- Menousek J.; Mishra B.; Hanke M. L.; Heim C. E.; Kielian T.; Wang G. (2012) Database screening and in vivo efficacy of antimicrobial peptides against methicillin-resistant Staphylococcus aureus USA300. Int. J. Antimicrob. Agents 39, 402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassat J. E.; Lee C. Y.; Smeltzer M. S. (2007) formation in clinical isolates of Staphylococcus aureus. Methods Mol. Biol. 391, 127–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim C. E.; Hanke M. L.; Kielian T. (2014) A mouse model of Staphylococcus catheter-associated biofilm infection. Methods Mol. Biol. 1106, 183–191. [DOI] [PubMed] [Google Scholar]
- Thurlow L. R.; Hanke M. L.; Fritz T.; Angle A.; Aldrich A.; Williams S. H.; Engebretsen I. L.; Bayles K. W.; Horswill A. R.; Kielian T. (2011) Staphylococcus aureus biofilms prevent macrophage phagocytosis and attenuate inflammation in vivo. J. Immunol. 186, 6585–6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G.; Elliott M.; Cogen A. L.; Ezell E. L.; Gallo R. L.; Hancock R. E. W. (2012) Structure, dynamics, and antimicrobial and immune modulatory activities of human LL-23 and its single-residue variants mutated on the basis of homologous primate cathelicidins. Biochemistry 51, 653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung S. C.; Parenti D.; Murphy P. M. (2011) Host chemokines bind to Staphylococcus aureus and stimulate protein A release. J. Biol. Chem. 286, 5069–5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D.; Chen Q.; Hoover D. M.; Staley P.; Tucker K. D.; Lubkowski J.; Oppenheim J. J. (2003) Many chemokines including CCL20/MIP-3alpha display antimicrobial activity. J. Leukocyte Biol. 74, 448–455. [DOI] [PubMed] [Google Scholar]
- Darouiche R. O. (2004) Treatment of infections associated with surgical implants. N. Engl. J. Med. 350, 1422–1429. [DOI] [PubMed] [Google Scholar]
- Koradi R.; Billeter M.; Wüthrich K. (1996) MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graphics 14, 51–55. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.