

Abstract
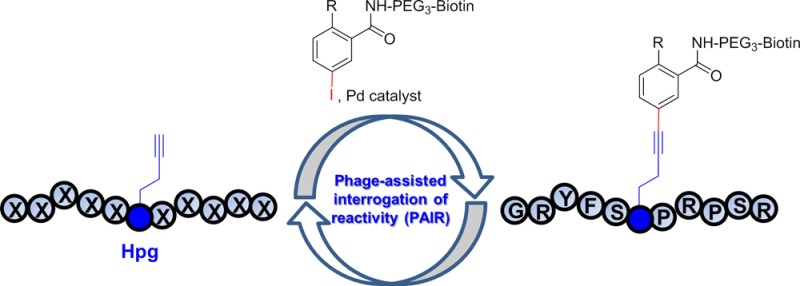
Fast and specific bioorthogonal reactions are highly desirable because they provide efficient tracking of biomolecules that are present in low abundance and/or involved in fast dynamic process in living systems. Toward this end, classic strategy involves the optimization of substrate structures and reaction conditions in test tubes, testing their compatibility with biological systems, devising synthetic biology schemes to introduce the modified substrates into living cells or organisms, and finally validating the superior kinetics for enhanced capacity in tracking biomolecules in vivo—a lengthy process often mired by unexpected results. Here, we report a streamlined approach in which the “microenvironment” of a bioorthogonal chemical reporter is exploited directly in biological systems via phage-assisted interrogation of reactivity (PAIR) to optimize not only reaction kinetics but also specificity. Using the PAIR strategy, we identified a short alkyne-containing peptide sequence showing fast kinetics (k2 = 13 000 ± 2000 M–1 s–1) in a palladium-mediated cross-coupling reaction. Site-directed mutagenesis studies suggested that the residues surrounding the alkyne moiety facilitate the assembly of a key palladium–alkyne intermediate along the reaction pathway. When this peptide sequence was inserted into the extracellular domain of epidermal growth factor receptor (EGFR), this reactive sequence directed the specific labeling of EGFR in live mammalian cells.
Complementary to the development of genetic tags, such as green fluorescent protein and its many variants,1 and fluorescent RNA sensors,2 the advent of bioorthogonal chemistry has offered a chemical reactivity-based tool to study all classes of biomolecules and their posttranslational modifications in their native environs.3 The growing list of bioorthogonal reactions4 and their applications in biological systems are a testament of the power of this approach in understanding the complexity of living systems. These bioorthogonal reactions include Staudinger ligation,5 copper-catalyzed azide–alkyne cycloaddition reaction (“click” chemistry),6,7 and the related copper-free version,8 “photoclick” chemistry,9−11 tetrazine ligation,12,13 thiolvinylether/o-quinone methide ligation,14 and Pictet–Spengler ligation.15 Because of the richness of palladium chemistry, the palladium-mediated bioorthogonal cross-coupling reactions have attracted a lot of interests since the early reports of Mizoroki–Heck and Sonogashira reactions for protein labeling in vitro.16,17 Several recent studies have highlighted the potential of palladium-mediated reactions in biological systems. For example, the Suzuki–Miyaura cross-coupling reaction was performed on proteins in vitro,18 on bacterial cell surface,19 and inside mammalian cells with the cell-permeable nanoparticle-based palladium catalysts;20 we recently reported a Cu-free Sonogashira cross-coupling reaction for functionalizing the alkyne-containing protein in vitro and in bacterial cells.21
It is widely recognized that fast bioorthogonal reactions are most valuable as they allow monitoring of fast biological processes while keeping the reagent concentrations low to minimize potential cytotoxicity. Thus, a major challenge in bioorthogonal reaction development is to identify ways to enhance reaction kinetics without compromising the reaction specificity. Indeed, compared with the enzyme-mediated residue-specific modifications,22,23 which can exhibit a kcat/KM value of ∼106 M–1 s–1, the second-order rate constant (k2) for bioorthogonal reactions typically ranges from 10–3 to 104 M–1 s–1. In optimizing reaction kinetics, the classical strategy involves substrate activation, including the design of strained substrates,11,24 the exploitation of LUMO-lowering or HOMO-lifting effect,25 and the introduction of fluorine substituent for electronic perturbation.26 An embodiment of this approach is shown in Figure 1a, where a fast bioorthogonal inverse-electron-demand Diels–Alder reaction for fluorogenic protein labeling in vivo was accomplished through the design of a genetically encodable, strained bicyclo[6,1,0]non-4-yn-9-ylmethyllysine derivative, and the LUMO-lowered tetrazine derivatives;27 the highest rate ever reported in the tetrazine system was 2.8 × 106 M–1 s–1 measured in PBS at 37 °C.28 A limitation of substrate activation approach is that the activated substrates may become chemically unstable29 or undergo some undesired side reactions.30
Figure 1.

Strategies for optimizing bioorthogonal reactions. (a) Substrate activation strategy in which the ground state energies of reactants are elevated through electronic or strain effect. An inverse electron-demand Diels–Alder reaction between a strained alkyne and a LUMO-lowered tetrazine is shown. (b) Exploiting the microenvironment of a bioorthogonal chemical reporter. Improved sequence-dependent reactivity is achieved by employing a phage-assisted interrogation of reactivity (PAIR) strategy. The optimization of a bioorthogonal, palladium-mediated Cu-free Sonogashira cross-coupling reaction between a homopropargylglycine-encoded peptide and fluorescein iodide is shown.
Taking cues from the enzyme-catalyzed reactions in which substrates are sequestered into the active site of an enzyme via noncovalent interactions, we set out to investigate whether the vicinity of a bioorthogonal chemical reporter, hereafter referred to as “microenvironment”, could be exploited to improve reaction kinetics and specificity of a bioorthogonal reaction. We envisioned that the microenvironment can increase the reaction kinetics by sequestering the reaction partner from the bulk solvent via noncovalent interactions such as chelation,31,32 H-bonding, and π–π and hydrophobic interactions, and facilitate the assembly of key intermediates along the reaction coordinate. To exploit the microenvironment effect, here we report the development of a phage-assisted interrogation of reactivity (PAIR) strategy in which the reactivities of a large peptide library (≥106 diversity) displayed on phage surface are directly assessed en masse (Figure 1b). Since reactivity enhancement is sequence-dependent, we expect a concurrent increase in specificity as well. We employed this PAIR strategy to optimize the Cu-free Sonogashira cross-coupling reaction we reported recently21 and identified a unique alkyne-encoded peptide sequence from a naïve peptide phage library that shows greater than 5-fold increase in reaction rate along with a strong preference for phenyl iodide substrates. On the basis of the truncation and substitution results, we propose a mechanism for this sequence-specific Cu-free cross-coupling reaction in which the adjacent two key residues help to preorganize a key aryl-palladium-alkyne intermediate along the reaction coordinate. In addition, we demonstrate the utility of this sequence-specific bioorthogonal reaction in selectively labeling of a membrane protein encoding the alkyne-peptide tag on live mammalian cell surface.
Results and Discussion
Generation of an Alkyne-Encoded Peptide Phage Library via Methionine Biosynthesis Inhibition
To identify reactive alkyne-encoded peptides from a large alkyne-encoded peptide library, we decided to use phage display for selection because (1) several unnatural amino acids have been displayed on filamentous phage surface using amber codon suppression approach;33,34 (2) a few bioorthogonal reactions have been performed on phage surface including click chemistry33 and Staudinger ligation;35 and (3) phage display has been used to evolve reactive peptide sequences capable of forming covalent bonds.34,36,37 To ascertain whether the reaction conditions for Cu-free Sonogashira cross-coupling is compatible with phage display, M13KE phage was incubated with the palladium catalyst at 37 °C for 30 min and the phage titer was then determined. No significant decrease in phage titer was detected (Table S1 in the Supporting Information), suggesting that phage was tolerant to the palladium treatment.
To study the microenvironment effect in an unbiased manner, we designed an 11-mer peptide phage library by placing the myc-tagged randomized peptides at the N-terminus of the truncated pIII protein (residues 198–406) in the pComb3HSS vector (Figure 2a). A single methionine was placed in the middle of the sequence (X5MX5) to serve as the incorporation site for the methionine surrogate homopropargylglycine (Hpg). To preclude additional methionine, NNC degenerate codons (N = A, C, T, G) were used in the synthetic oligonucleotides; as a result, only 15 amino acids (excluding M, E, Q, W, K) were encoded in the randomized positions with a measured library size of 1.0 × 106. It should be noted that while methionines are also present in the wild-type pIII proteins as well as the minor coat proteins pVII and pIX, they will serve as a constant background during the selection. Importantly, a single Met is encoded at position-28 of the major coat protein pVIII, which is packed inside the coat and not accessible to the reaction.38
Figure 2.

Validation of phage-assisted interrogation of reactivity strategy. (a) Schematic diagram for the use of PAIR strategy in identifying reactive Hpg-encoded peptide sequences for Cu-free Sonogashira cross-coupling. M stands for methionine, and X stands for one of the following 15 amino acids: A, C, D, F, G, H, I, L, N, P, R, S, T, V, and Y (amino acid one letter code). (b) Verifying the incorporation of Hpg into phage coat protein pIII through selective reactivity in Cu-free Sonogashira cross-coupling reaction. Western blot analyses of Met-or Hpg-encoded phage lysates expressing myc-PMP-pIII proteins. 1011 cfu phage was loaded into each lane based on UV–vis measurement. In anti-pIII Western blot, * denotes the full-length pIII protein while # denotes the truncated pIII. In probing biotin conjugation (panel 1), the membrane was blotted using the preformed avidin/biotinylated enzyme complex. (c) Selective enrichment of Hpg-phage after Pd-mediated cross-coupling with the biotinylated fluorescein iodide. The phage was probed with anti-pVIII-HRP antibody and quantified by adding ABTS substrate and measuring absorbance at 416 nm. ABTS = 2,2-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid).
We chose Hpg as the bioorthogonal chemical reporter in our peptide phage library because (1) this unsaturated alkyne amino acid is an excellent methionine surrogate by virtue of substrate promiscuity of methionyl-tRNA synthetase;39 (2) Hpg has been shown to be a good substrate for Cu-free Sonogashira cross-coupling reaction when incorporated into proteins in bacterial cells;21 and (3) other alkyne chemistries such as click chemistry can be performed with Hpg as well. To introduce Hpg into the peptide phage library, we employed the methionine biosynthesis inhibition (MBI) technique40 in which high concentrations of six amino acids (Leu, Ile, Val, Thr, Phe, Lys) known to inhibit aspartokinase—the first enzyme involved in methionine biosynthesis pathway—were added to the growth medium along with Hpg (see Methods for details). The use of MBI technique obviates the need of a phage-infectible methionine auxotroph and permits the use of robust XL1-Blue strain in phage propagation. Based on liquid chromatography–mass spectrometry (LC-MS) analysis, the replacement of methionine by Hpg in a model protein using MBI technique gave occupancy of 64% (Figure S1 in the SI), slightly lower than 88% reported with the use of methionine auxotroph.39 To maintain high library diversity, the peptide phage library was first propagated in a rich medium to yield the wild-type peptide library followed by second-round propagation in a methionine-deficient Hpg-supplemented medium containing the methionine biosynthesis inhibitors to generate the Hpg-encoded peptide phage library.
Characterization of Hpg Reactivity on Phage Surface in Cu-Free Sonogashira Cross-Coupling
To verify the expression of Hpg on phage surface, we constructed two phagemids, myc-PMP and myc-M4, by fusing the myc-tagged peptide sequence THDYPMPGANP and MMPGMM, respectively, to the N-terminus of the truncated pIII. A single colony harboring the desired phagemid was cultured in a rich medium first before switching to the methionine-deficient Hpg-supplemented medium containing the methionine biosynthesis inhibitors. Helper phage was then added to allow phage assembly and packaging for the generation of myc-PMP-Hpg and myc-M4-Hpg phages. As a control, we also generated the wild-type phage, designated as myc-PMP-Met and myc-M4-Met, by propagating the phage in the methionine-containing rich medium. The yields of the Hpg-encoded phage were comparable to those of the Met-encoded wild-type phage (Figure S2 in SI).
To confirm the presence of Hpg on phage surface, myc-PMP-Met or myc-PMP-Hpg phage was incubated with the preactivated biotinylated fluorescein iodide–palladium complex at 37 °C for 30 min in PBS, pH 7.4, and the biotinylation of phage was probed with a preformed avidin/biotinylated enzyme complex. To our satisfaction, only myc-PMP-Hpg phage showed selective biotinylation while myc-PMP-Met did not (panel 1 in Figure 2b). The Western blots with anti-pIII (panel 2) and antimyc (panel 3) antibodies confirmed that the biotinylated product is myc-PMP-fused pIII protein. Importantly, the reaction did not proceed with the wild-type phage or in the absence of the Pd catalyst (panel 1), indicating that the reaction occurred exclusively with Hpg and was mediated by palladium. Furthermore, myc-M4-Hpg phage showed significantly higher absorbance at 416 nm than myc-M4-Met phage after treatment with the preactivated biotinylated fluorescein iodide-palladium complex in the ELISA experiment (Figure 2c). The increased background seen with wild-type phage after palladium treatment is likely due to nonspecific binding to palladium by polar residues on phage surface.41,42
Identification and Evaluation of the Reactive Alkyne-Encoded Peptide Sequences
To identify Hpg-encoded peptide sequences that are highly reactive toward fluorescein iodide in Cu-free Sonogashira cross-coupling, we performed three rounds of reaction-based panning with the freshly prepared Hpg-encoded peptide phage library and representative clones that survived each round of selection were sequenced (Tables S2 and S3 in SI). While sequence alignment of 24 selected clones revealed no consensus, several trends were noted: (1) 8 out of 24 clones encode Cys, likely due to its strong binding to the palladium; (2) Pro was enriched in the selected sequences, frequently appearing in tandem, which can be attributed to its flat hydrophobic surface that may interact with the fluorescein moiety; and (3) polar residues such as Ser, Thr, Asn, and His were found in the Hpg vicinity (i–2, i–1, i+1, i+2) in 16 clones. This is not a result of codon bias because among the 16 degenerate NNC codons, only Ser is represented twice; indeed, sequencing of the naïve library revealed no codon bias for any residues except Ser (Table S3 in SI).
Because of the lack of sequence convergence, we decided to examine the reactivity of the individual sequences identified from the selection. Nine representative sequences were fused to the C-terminus of a small protein, ubiquitin, to ensure Hpg accessibility by the palladium complex. The ubiquitin–peptide fusions were expressed in a methionine auxotroph in the presence of Hpg and their identities were confirmed by mass spectrometry (Table S5 in SI). A previously reported Hpg-encoded ubiquitin–peptide fusion, Ub-G4, in which Hpg is flanked by 2 Gly residues on each side was used as a benchmark21 on the basis that it represents a sterically least hindered microenvironment for the Pd-mediated reaction. To our surprise, the highest yielding peptide sequence, GRYFSΘPRPSR, hereafter referred to as R1–4, was identified from the first round of selection. Based on the ion counts in the LC-MS,21 Ub-R1–4 gave 99% yield with no detectable starting materials remaining after the reaction, whereas the reference Ub-G4 afforded 89% yield under identical conditions (entries 1 and 2 in Table 1). Importantly, the control reaction with the methionine-encoded fusion protein, Ub-R1–4-Met, did not give any product under identical conditions, confirming that the reaction is specific toward Hpg (Figure S3 in SI). On the other hand, most proline-rich sequences (contains ≥3 Pro with at least two adjacent to each other) gave modest to excellent yields (52–90%, entries 5–8). It is known that proline-rich regions (PRRs) adopt the polyproline helix as a result of proline’s restricted conformation.43 While PRRs appear in proteins mostly as structural elements, there are several examples where they were implicated in binding to flat aromatic residues with high affinity, albeit little specificity.43−45 On the other hand, despite having three noncontinuous Pro residues, Ub-R1–8 showed poor reactivity (45% yield, entry 3), possibly due to palladium binding by Cys.41,42 The same is true with Ub-R3–20 (29% yield, entry 9) in which the two His residues adjacent to Hpg may provide strong coordination with the palladium and thus inhibit the cross-coupling reaction. Intriguingly, despite having identical immediate adjacent residues, Ub-R1–9 afforded a much lower yield compared to Ub-R1–4 (compare entry 4 to 2), indicating that the distal residues also play a role in the cross-coupling reaction.
Table 1. Reactivity Study of the Ubiquitin-Fused Peptides Selected from Phage Display in Cu-Free Sonogashira Cross-Coupling Reactiona.
| entry | name | C-terminal peptide sequence | yield (%) |
|---|---|---|---|
| 1 | G4 | GGΘGG | 89 |
| 2 | R1–4 | GRYFSΘPRPSR | 99 |
| 3 | R1–8 | PSLPCΘYSFPD | 45 |
| 4 | R1–9 | ALFLPΘSRVHD | 73 |
| 5 | R2–6 | DPPPTΘDVPPH | 52 |
| 6 | R3–3 | SPPHSΘVPTPA | 84 |
| 7 | R3–8 | NVPPLΘGTPSL | 62 |
| 8 | R3–9 | FPRPPΘTPPPH | 90 |
| 9 | R3–20 | FPTNHΘHRHPT | 29 |
| 10 | R3–24 | DGRDAΘSPHYY | 79 |
A preactivated fluorescein iodide–Pd complex (8 equiv) was incubated with 2.5 μM of ubiquitin-peptide fusion proteins in PBS buffer at 37 °C for 30 min. The yields of the products were determined by LC-MS based on ion counts and calculated using the following equation: yield % = Iproduct/(IUb-peptide + Iproduct + Iside product), where IUb-peptide, Iproduct, and Iside product represent the ion counts of the remaining Ub-peptide, and the formed product and side products (if present), respectively. Θ = Hpg.
To gain an insight into the reaction kinetics, we compared the initial reaction rate of Ub-R1–4 to that of Ub-G4. While both substrates displayed clean and time-dependent conversions, there was a rapid product formation with Ub-R1–4 in as short as 2 s in contrast to Ub-G4, which showed significant product accumulation only after 1 min (Figure 3). Fitting the data gave rise to the second-order rate constant k2 of 13 000 ± 2000 M–1 s–1 for Ub-R1–4 and 2500 ± 400 M–1 s–1 for Ub-G4 (Table 2, Figure S4 in SI), indicating that R1–4 is greater than 5 times more reactive than the benchmark sequence. Considering the small increase in reaction yield, the effect of microenvironment on Hpg reactivity appears to be mostly kinetic in nature.
Figure 3.

Kinetic characterization of the sequence-specific Cu-free Sonogashira cross-coupling. Time courses of the cross-coupling reaction between the preactivated fluorescein iodide–Pd complex and the fusion protein Ub-G4 (a) or Ub-R1–4 (b) as monitored by LC-MS. 2.5 μM of the fusion protein was incubated with 20 μM of the preactivated fluorescein iodide–palladium complex in PBS at 37 °C. SM = starting material; Prod = cross-coupling product.
Table 2. Reactivity Study of R1–4 Truncation and Substitution Mutantsa.
| entry | name | C-terminal peptide sequence | yield (%) | k2 (M–1 s–1)b |
|---|---|---|---|---|
| 1 | G4 | GGΘGG | 89 | 2500 ± 400 |
| 2 | R1–4 | GRYFSΘPRPSR | 99 | 13 000 ± 2000 |
| 3 | R1–4–5 | FSΘPR | 91 | 8500 ± 1500 |
| 4 | R1–4–3 | SΘP | 12 | ND |
| 5 | R1–4-F4A | GRYASΘPRPSR | 8 | ND |
| 6 | R1–4-S5A | GRYFAΘPRPSR | 71 | 4600 ± 550 |
| 7 | R1–4-P7A | GRYFSΘARPSR | 51 | 4000 ± 700 |
| 8 | R1–4-R8A | GRYFSΘPAPSR | 31 | ND |
| 9 | R1–4-F4Y | GRYYSΘPRPSR | 98 | 6000 ± 1000 |
| 10 | R1–4-F4W | GRYWSΘPRPSR | 85 | 8000 ± 1500 |
| 11 | R1–4-R8K | GRYFSΘPKPSR | 97 | 7500 ± 500 |
| 12 | R1–4-R8E | GRYFSΘPEPSR | 31 | ND |
| 13 | R1–4-R8H | GRYFSΘPHPSR | 51 | ND |
20 μM of the preactivated fluorescein iodide–Pd complex was incubated with 2.5 μM of the ubiquitin-peptide fusion protein in PBS buffer at 37 °C for 30 min. The product yield was determined by LC-MS (average of at least 2 trials) as described previously.
Average kinetic constants derived from at least 2 trials. Θ = Hpg; ND = not determined.
Truncation and Site-Directed Mutagenesis Studies of R1–4 Sequence
To better understand the microenvironment effect of R1–4 sequence, we constructed two truncation mutants by appending a pentameric or trimeric peptide comprising of only the immediate residues flanking Hpg to the ubiquitin C-terminus. We found that the pentamer fusion protein, R1–4–5, gave 91% yield in the cross-coupling reaction (entry 3, Table 2) while the trimer fusion protein, R1–4–3, gave drastically lower yield of 12% (entry 4, Table 2). This result indicates that the surrounding residues Phe-4 and Arg-8 are critical for the observed high reactivity. To probe the contribution of each residue, we performed an alanine scan of the central four residues in the R1–4 sequence and found that all four alanine mutants exhibited reduced reactivity (entries 5–8), with F4A mutant showing the largest reduction followed by R8A mutant. Kinetic analyses of the S5A and P7A mutants revealed that these two mutants gave faster reactions than the benchmark G4 sequence despite their lower yields after 30 min incubation (Table 2; Figure S4 in SI). One explanation for this apparent discrepancy is that after the initial “burst” of product generation, there is increased product inhibition because of competing sequestration of the aryl-palladium complex by the product, leading to overall lower yields at longer incubation time.
Since NNC degenerate codons exclude Met, Glu, Gln, Trp, and Lys from the coded peptide sequences, we investigated whether substitution of Phe-4 and Arg-8 by amino acids of similar characteristics would lead to further enhancement in reaction rate. As expected, substitution of Phe-4 to either Tyr or Trp, and Arg-8 to Lys, afforded good to excellent yields (98%, 85%, and 97%, respectively, Table 2), indicating that the aromatic character at i–2 position and the basic side chain at i+2 are important for retaining reactivity. However, the kinetic constants of these three mutants were lower than that of R1–4 (compare entries 9–11 to 2, Table 2), highlighting the structural subtlety in determining reaction kinetics.
Substrate Specificity of Aryl Iodide–Palladium Complex
One of the hallmarks of enzyme-catalyzed reactions is that enzymes typically exhibit high substrate specificity. To probe whether R1–4 sequence prefers certain aryl iodide substrates, an array of aryl/vinyl iodides were examined in the palladium-mediated cross-coupling reaction with Ub-R1–4. To our surprise, all substituted phenyl iodides gave excellent yields regardless of their substitution pattern and electronic property (Figure 4, Table S7 in SI), suggesting that the xanthene moiety of fluorescein iodide is not necessary. Importantly, Ub-R1–4 furnished higher yields for all substituted phenyl iodides than Ub-G4 even though lower amounts of the preactivated aryl-palladium reagents were used (8 equiv for Ub-R1–4 vs 50 equiv for Ub-G4), indicating sequence-dependent reactivity enhancement. On the other hand, heterocyclic thiophene iodide and coumarin iodide gave modest yields (36% and 54%, respectively), which were also lower than those for the corresponding reactions with Ub-G4 (Figure 4). While the decrease in yield can be partially attributed to their lower intrinsic reactivity, the absence of sequence-dependent reactivity enhancement suggests that the microenvironment surrounding Hpg disfavors heteroaromatic and vinyl iodides to some extent. Presumably, the preference for substituted phenyl iodides is a result of strong π–π interactions between the phenyl group in the aryl–palladium complex and the phenyl ring of Phe-4 in R1–4 sequence (vide infra). This observation also implies that detection modalities other than the fluorescent xanthene moiety can be attached to phenyl iodide with minimum effect on reactivity.
Figure 4.

Small molecule substrate specificity in the Cu-free Sonogashira cross-coupling reaction with Ub-R1–4. Top, reaction scheme; bottom, small-molecule substrate structures and their respective yields in the Cu-free Sonogashira cross-coupling reactions toward Ub-R1–4 and Ub-G4 (listed in parentheses; data taken from ref (21)). The product yield was determined using LC-MS as described previously. For Ub-G4, 50 equiv of the aryl/vinyl iodide-palladium complexes was used.
Mechanism of Sequence-Specific Palladium-Mediated Cross-Coupling Reaction
For palladium-catalyzed, Cu-free Sonogashira cross-coupling in organic solvents, base-promoted deprotonation of the terminal alkyne was proposed to be the rate-limiting step in the catalytic cycle.46,47 It is plausible that the same mechanism may also operate in our sequence-specific cross-coupling reaction in aqueous medium. As shown in Figure 5, the proline residue at i+1 position in R1–4 helps the short peptide to adopt a turn structure such that the side chains of the three key residues, Phe-4, Hpg-6, and Arg-8, are clustered into the same side of the peptide chain to form a binding pocket for the incoming aryl–Pd complex. The flat aromatic surface of Phe-4 assists in the recruitment by interacting with the phenyl ring via π–π stacking48 or hydrophobic interaction, which in turn allows facile complexation of Pd with the alkyne side chain of Hpg. Upon Pd complexation, the pKa of alkyne terminal hydrogen is lowered considerably47 such that the deprotonation can proceed in aqueous medium at neutral pH, possibly with assistance from Arg-8.49 The overall effect is that the deprotonation of alkyne is accelerated, leading to faster product formation. This model is consistent with our mutagenesis results (Table 2) in which Phe-4 can be replaced by Tyr and Trp but not Ala, and Arg-8 can be replaced by Lys, but not Glu and His, as well as the result from the substrate scope study in which coumarin iodide gave a lower yield (Figure 4).
Figure 5.
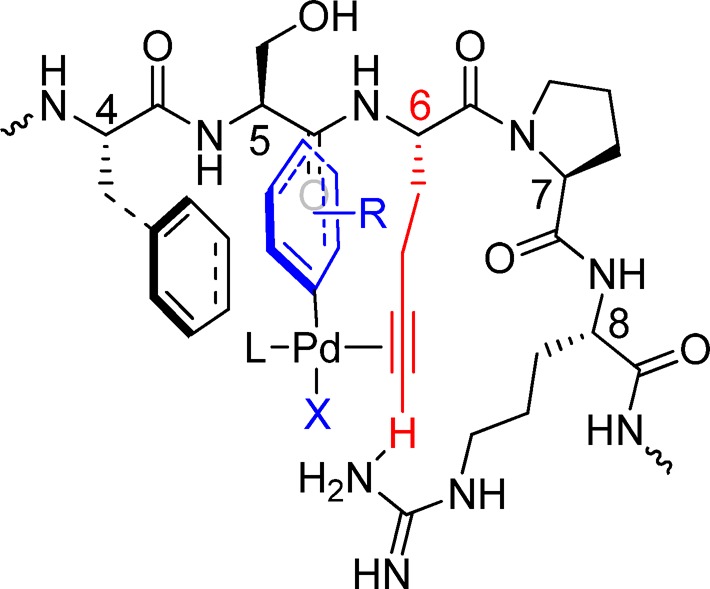
Proposed mechanism for the palladium-mediated sequence-specific cross-coupling reaction. A possible stabilized R1–4/aryl–Pd complex is shown in which Phe-4 and Arg-8 of R1–4 are preorganized to facilitate alkyne deprotonation, the rate-limiting step in Cu-free Sonogashira cross-coupling.
Site-Specific Labeling of Hpg-Encoded Proteins on Live Mammalian Cell Surface
One major limitation of residue-specific incorporation of unnatural amino acids into proteins via methionine biosynthesis pathway is that this metabolic approach lacks target specificity as the methionine surrogates are incorporated into other methionine-containing proteins. To overcome this limitation, we envision that a methionine surrogate present in a specific microenvironment can direct a target-specific, sequence-specific bioorthogonal reaction. To this end, we tested whether R1–4 identified from phage display can direct selective site-specific labeling of a transmembrane protein on live mammalian cells via palladium-mediated cross-coupling. Thus, R1–4 sequence was inserted in-between residues Ala-21 and Ser-22 of epidermal growth factor receptor (EGFR) with EGFP fused at its C-terminus to generate R1–4-EGFR-EGFP. Since this particular site is known to be solvent exposed,50 we expect that the Hpg-encoded R1–4 should exhibit enhanced reactivity in the palladium-mediated cross-coupling reaction compared to Hpg’s that are displayed at other methionine sites of R1–4-EGFR-EGFP as well as other endogenous proteins. The expression of the Hpg-encoded R1–4-EGFR-EGFP in HEK293T cells was accomplished by growing the cells in Met-deficient DMEM medium for 30 min to deplete the intracellular Met stock before adding 1 mM Hpg into the medium.51 After 6 h of expression, the cells were treated with 50 μM of the preactivated biotinylated phenyl iodide (3)–palladium complex (Figure 6a) in PBS at 37 °C for 30 min. The biotinylation of EGFR was monitored by confocal microscopy after treating the cells with Alexa 568-conjugated streptavidin. To our satisfaction, cells expressing Hpg-encoded R1–4-EGFR-EGFP showed specific cell surface labeling in Alexa 568 channel, coinciding with the image in EGFP channel (Figure 6b). No biotinylation was detected in Alexa 568 channel for cells expressing either Met-encoded R1–4-EGFR-EGFP (Figure 6c) or Hpg-encoded EGFR-EGFP that lacks R1–4 moiety (Figure 6d), indicating that the cross-coupling reaction was highly specific toward the Hpg-encoded R1–4.
Figure 6.

Site-specific labeling of HPG-encoded, R1–4-tagged EGFR on live mammalian cell surface. (a) Structure of biotinylated phenyl iodide. Human embryonic kidney (HEK) 293T cells expressing R1–4-EGFR-EGFP (b, c) or EGFR-EGFP (d) were cultured in Met-deficient DMEM medium supplemented with 1 mM Hpg (b, d) or 1 mM Met (c), and the cells were treated with 50 μM of the preactivated biotinylated phenyl iodide–palladium complex for 30 min before washing with PBS and treating with streptavidin-Alexa 568 for 20 min. Scale bar = 50 μm.
Collectively, our data demonstrated that the microenvironment surrounding a bioorthogonal alkyne reporter can be exploited to optimize reaction kinetics and specificity. While no consensus sequence emerged from the selection, the divergent sequences identified from the peptide phage library nonetheless exhibited good-to-excellent reactivity, suggesting multiple solutions to the kinetics problem. The truncation and mutagenesis study of the most reactive sequence identified, R1–4, revealed that the microenvironment effect is likely due to the selective recruitment of the aryl–palladium complex via noncovalent interactions such as π–π and hydrogen bonding interactions, which may facilitate the assembly of a key intermediate along the reaction coordinate. Compared to other peptide tags that serve as substrates for the various enzymes including biotin ligase,50 sortase,52 formylglycine-generating enzyme,53 phosphopantetheinyl transferase,54 and lipoic acid ligase,55 our alkyne-encoded peptide tag exhibits intrinsically high chemical reactivity without the need of an enzyme. Compared to the substrate activation approach where the modified substrates are synthesized and evaluated one at a time, the PAIR approach is performed directly in biological systems, shortening the development timeline. In principle, the PAIR strategy can be extended to other bioorthogonal ligation reactions as long as one reactant can be metabolically encoded and the reaction conditions are compatible with phage.
In summary, we have deployed a phage-assisted interrogation of reactivity strategy to optimize a bioorthogonal, palladium-mediated Cu-free Sonogashira cross-coupling reaction. Using a methionine biosynthesis inhibitor cocktail, we generated a peptide phage library containing a methionine surrogate, homopropargylglycine, in the middle of the randomized 11-mer peptide sequences. From the reactivity-based selection, a number of Hpg-containing peptide sequences were identified that exhibit high reactivity and specificity toward fluorescein iodide in the palladium-mediated cross-coupling reaction. The majority of the selected peptides contain single or multiple proline residues, possibly due to their structural effect in organizing the microenvironment for improved binding to the reaction partner. Compared to the naïve G4 sequence, the most reactive peptide sequence R1–4, GRYFSΘPRPSR (Θ = Hpg), gave greater than 5-fold enhancement in kinetic constant when measured in fusion protein context. The mutagenesis studies of R1–4 indicated that Phe-4 and Arg-8 contribute critically to the enhanced reactivity by providing π–π and hydrogen-bonding interactions, respectively, with the aryl–palladium complex. Additional aryl iodide substrate studies suggested that the substituted phenyl iodides are preferred substrates for R1–4 in the cross-coupling reaction. When R1–4 sequence was inserted into the solvent-exposed loop region of the membrane protein EGFR and expressed in HEK293T cells in the Hpg-supplemented culture medium, selective cross-coupling reaction proceeded on live mammalian cell surface. Since no exogenous proteins are needed for the labeling reaction and only 11-mer peptide sequence is required, the R1–4 sequence identified herein should offer a convenient peptide tag for tracking proteins in living cells.
Methods
Generation of Hpg-Encoded Phage Library via Methionine Biosynthesis Inhibition
An oligonucleotide library encoding 11-mer randomized peptide, X5MX5 (M stands for Met while X stands for any of the 15 amino acids coded by NNC degenerate codons) was prepared by running extension PCR with a pair of primers: FWD: 5′-CAGGCGGCCGAGCTCGAACAGAAGTTGATTTCCGAAGA AGACCTCGGTACC-3′; and REV: 5′-CGGCCTGGCCACTAGTGTCGAC(GNN)5CAT(GNN)5GGT ACCGAGGTCTTCTTCGGAAATCAAC-3′ (the underlined oligonucleotide sequences denote SacI and SpeI restriction digestion sites, respectively). The oligonucleotide library was digested with SacI and SpeI, and purified by agarose gel. A suitable amount of the digested oligonucleotides was ligated into the SacI/SpeI–digested pComb3HSS phagemid vector using T4 DNA ligase (Invitrogen). The ligation mixture was purified using PCR purification kit (Qiagen), and the purified phagemid DNA was divided into several 0.2 cm cuvettes (Invitrogen) each containing 200 μL of electrocompetent XL1-Blue cells for electroporation. The transformed cells were immediately recovered with SOC medium, pooled together, diluted to 25 mL SOC medium, and incubated at 37 °C for 15 min. An aliquot of the SOC outgrowth was serially diluted and then plated on LB plates containing 100 μg/mL ampicillin to determine the library complexity, while the rest of the SOC outgrowth was used to inoculate 500 mL 2YT medium containing 100 μg/mL ampicillin and helper phage (final concentration: 1 × 1010 phage/mL). After incubation at 37 °C for 90 min, kanamycin was added (final concentration = 10 μg/mL) and the culture was continued for 16 h. Phage isolation and purification was carried out by following New England Biolab’s (NEB) recommended protocol to yield the methionine-encoded peptide phage library, whose titer was then determined.
A fresh culture of XL1-Blue cells in 2 mL rich medium containing 20 μg/mL tetracycline was spun down and the pellet was resuspended in 1 mL SelenoMet plus nutrient mix medium (Molecular Dimensions) supplemented with 1 g/L dextrose and 100 μg/mL ampicillin. A 100-μL aliquot was withdrawn and added into 100 mL of the same medium, and the culture was continued at 37 °C for 16 h. After adjusting OD600 to 0.5 with SelenoMet plus nutrient mix medium, cells were infected with the methionine-encoded peptide phage library (1:1.2 cells/virion) at 37 °C and 200 rpm for 30 min. Afterward, a methionine biosynthesis inhibitor cocktail comprised of 100 mg/L Lys/Phe/Thr, and 50 mg/L Iso/Leu/Val was added. Twenty minutes later, 1 mM Hpg and 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) were added. Phage packaging and propagation was initiated by adding helper phage (final concentration = 1 × 1010 phage/mL) followed by the addition of 10 μg/mL kanamycin 1.5 h later with continuous shaking at 30 °C and 200 rpm for 16 h. Phage isolation and purification was carried out by following NEB’s protocol to generate the Hpg-encoded peptide phage library.
Solution-Phase Reactivity-Based Panning
To a 0.6 mL microcentrifuge tube was added 142 μL water, 2 μL of 10 mM biotinylated fluorescein iodide, 2 μL of 80 mM sodium ascorbate, and 2 μL of 10 mM Pd-cat-2 (Table S1 in SI). The mixture was heated at 37 °C for 1 h with vigorous stirring before cooling down to RT. To a 1.7 mL microcentrifuge tube containing 1 mL Hpg-encoded phage library in PBS was added 3.75 μL of the preactivated biotinylated fluorescein iodide–palladium complex (final concentration = 1 μM) to initiate the reaction at 37 °C. After incubation for 30 min, 20% PEG/NaCl (20% of the reaction volume) was added, and the precipitated phage was collected by centrifugation at 13,500 rpm and 4 °C for 10 min. The phage pellet was then resuspended in 200 μL PBST (PBS containing 0.05% Tween-20) and the suspension was added to a streptavidin-coated microtiter plate (Thermo Scientific) that was preblocked with BSA and washed with PBST (6×). The affinity capture of the biotinylated phage proceeded at RT with rocking for 1 h, followed by 10× PBST washing (0.05% Tween-20 for round 1, 0.1% Tween-20 for rounds 2 and 3). The bound phage was eluted out by addition of 1 mM biotin (200 μL/well in PBS), and the rocking continued at RT for 1 h. The eluted phage was pooled and subsequently used to infect 10× volume of the freshly grown XL1-Blue cells (OD600 = 0.5) at 37 °C and 225 rpm for 30 min. A small aliquot was diluted and used in determining the phage titer. Separately, the infected culture was diluted into 25 mL 2YT medium containing ampicillin and M13KO7 helper phage (final concentration = 1 × 1010 phage/mL) before overnight incubation at 37 °C and 200 rpm. The wild-type phage produced after this round of amplification was again titered to determine the amount of infective phage needed for the production of the Hpg-encoded phage library via methionine biosynthesis inhibition strategy described above. Subsequently, the newly generated Hpg-encoded phage was titered to determine the amount of phage needed for the next round of selection.
Expression and Purification of the Hpg-Encoded Ubiquitin-Peptide Fusion Proteins
A single colony of bacteria expressing the desired ubiquitin-peptide fusion protein was grown in LB medium at 37 °C overnight. This starter culture was used to inoculate a larger culture with 100-fold dilution in SelenoMet Plus Nutrient Mix medium supplemented with 1 mM methionine and 100 μg/mL ampicillin. After OD600 reached 0.8, cells were harvested by centrifugation and washed 3× with 0.9% NaCl solution. The cell pellet was resuspended in SelenoMet Plus Nutrient Mix medium containing ampicillin, and the incubation was continued at 37 °C for 1 h to deplete the intracellular methionine stock. Then, 1 mM Hpg and 1 mM IPTG were added to the culture flask to initiate protein expression at 25 °C for 12 h. Next, cells were pelleted by centrifugation and lysed by sonication on ice. The fusion proteins were purified by Ni-NTA affinity chromatography before the treatment with PreScission protease (GE Healthcare) to cleave His6-tag along with the N-terminal Met/Hpg, affording a singly Hpg-encoded ubiquitin-peptide fusion protein.
Assessing the Reactivity of the Selected Peptide in Cu-Free Sonogashira Cross-Coupling
To a 1.50 mL glass vial containing 6.36 mg sodium carbonate (0.06 mmol) was added 100 μL of 200 mM N,N-dimethyl-2-amino-4,6-dihydroxypyrimidine (Pd-cat-3) (0.02 mmol), 50 μL of 200 mM Pd(OAc)2 solution (0.01 mmol) and 100 μL DMSO. The mixture was stirred at 65 °C for 30 min before addition of 100 μL fluorescein iodide solution (0.01 mmol), 50 μL of 200 mM ascorbic acid (0.01 mmol), and 100 μL DMSO. The mixture was stirred for another 30 min to obtain the preactivated reagent cocktail. To 48 μL of 2.5 μM fusion proteins in PBS in a 0.6 mL microcentrifuge tube was added 8 equiv of the preactivated fluorescein iodide–palladium complex (500 μM, 2 μL, 1.0 nmol), and the mixture was stirred at 37 °C for 30 min before quenching with 10 μL 3-mercaptopropanoic acid (4% v/v in water). The product mixture was analyzed directly by LC-MS.
Protein Kinetic Studies
A 0.6 mL eppendorf tube containing 48 μL of 2.5 μM fusion proteins in PBS was prewarmed to 37 °C. With vigorous stirring, 2 μL of the 500 μM preactivated aryl-palladium complex (8 equiv) was added to the eppendorf tube, and the mixture was mixed for 2, 5, 10, 15, 20, or 30 s before quenching by quick addition of 10 μL 3-mercaptopropanoic acid (4% v/v in water). The quenched reaction mixtures were analyzed directly by LC-MS.
Aryl/Vinyl Iodide Substrate Specificity Study
A mixture of 48 μL of 2.5 μM Ub-R1–4 in PBS and 2 μL of 500 μM preactivated aryl/vinyl–palladium complex (8 equiv) in a 0.6 mL eppendorf tube was stirred at 37 °C for 30 min. Afterward, the reaction was quenched with 10 μL of 3-mercaptopropanoic acid (4% v/v in water), and the mixture was analyzed directly by LC-MS.
Live Cell Surface Labeling of R1–4-Tagged Membrane Protein
HEK293T cells were seeded at 25% confluency in a 35 mm glass-bottom tissue culture plate and allowed to grow to 60–70% confluence in 2 mL DMEM medium supplemented with 10% FBS in a humidified 37 °C, 5% CO2 incubator. Afterward, cells were transfected with either 1 μg R1–4-EGFR-EFGP plasmid or 1 μg EGFR-EFGP plasmid together with 6 μL Lipofectamine 2000 (Invitrogen) in OPTI-MEM medium. After incubation at 37 °C for 6 h, cells were washed twice with 2 mL prewarmed PBS before switching to a Met-deficient DMEM medium and additional incubation at 37 °C for 30 min to deplete intracellular methionine stock. The cells were incubated in Met-deficient DMEM medium supplemented with 10% FBS and 1 mM Hpg at 37 °C for 6–7 h. For the control plate, Met-deficient DMEM medium supplemented with 10% FBS and 1 mM Met was used. After washing the cells twice with 2 mL prewarmed PBS, cells were treated with 50 μM of the preactivated biotinylated phenyl iodide–palladium complex at 37 °C for 30 min. Cells were washed twice again with 2 mL prewarmed PBS before treatment with Alexa 568-conjugated streptavidin in PBS (1:500 dilution) at 37 °C for 20 min. The excess reagents were removed and the cells were washed with prewarmed PBS twice before placing cells in PBS for confocal laser scanning microscopy.
Acknowledgments
pCombHSS vector was obtained from Dr. C. Barbas’s lab at The Scripps Research Institute, and pcDNA3-EGFR-EGFP was a gift from Dr. J. Koland at the University of Iowa. We gratefully acknowledge the National Institutes of Health (GM 085092) and National Science Foundation (CHE-1305826) for financial support.
Supporting Information Available
Supplemental figures and tables, experimental procedures, and characterization of all new compounds. These materials are available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† California Institute for Biomedical Research, 11119 N. Torrey Pines Rd #100, La Jolla, CA 92037, U.S.A.
Author Present Address
‡ School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi’an, 710062, China
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Tsien R. Y. (1998) The green fluorescent protein. Annu. Rev. Biochem. 67, 509–544. [DOI] [PubMed] [Google Scholar]
- Paige J. S.; Wu K. Y.; Jaffrey S. R. (2011) RNA mimics of green fluorescent protein. Science 333, 642–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sletten E. M.; Bertozzi C. R. (2009) Bioorthogonal chemistry: Fishing for selectivity in a sea of functionality. Angew. Chem., Int. Ed. 48, 6974–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramil C. P.; Lin Q. (2013) Bioorthogonal chemistry: Strategies and recent developments. Chem. Commun. 49, 11007–11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxon E.; Bertozzi C. R. (2000) Cell surface engineering by a modified Staudinger reaction. Science 287, 2007–2010. [DOI] [PubMed] [Google Scholar]
- Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. (2002) A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective ″ligation″ of azides and terminal alkynes. Angew. Chem., Int. Ed. 41, 2596–2599. [DOI] [PubMed] [Google Scholar]
- Tornoe C. W.; Christensen C.; Meldal M. (2002) Peptidotriazoles on solid phase: [1,2,3]-Triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 67, 3057–3064. [DOI] [PubMed] [Google Scholar]
- Agard N. J.; Prescher J. A.; Bertozzi C. R. (2004) A strain-promoted [3+2] azide–alkyne cycloaddition for covalent modification of biomolecules in living systems. J. Am. Chem. Soc. 126, 15046–15047. [DOI] [PubMed] [Google Scholar]
- Song W.; Wang Y.; Qu J.; Madden M.; Lin Q. (2008) A photoinducible 1,3-dipolar cycloaddition reaction for rapid, selective modification of tetrazole-containing proteins. Angew. Chem., Int. Ed. 47, 2832–2835. [DOI] [PubMed] [Google Scholar]
- Song W.; Wang Y.; Qu J.; Lin Q. (2008) Selective functionalization of a genetically encoded alkene-containing protein via ″photoclick chemistry″ in bacterial cells. J. Am. Chem. Soc. 130, 9654–9655. [DOI] [PubMed] [Google Scholar]
- Yu Z.; Lin Q. (2014) Design of spiro[2.3]hex-1-ene, a genetically encodable double-strained alkene for superfast photoclick chemistry. J. Am. Chem. Soc. 136, 4153–4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackman M. L.; Royzen M.; Fox J. M. (2008) Tetrazine ligation: Fast bioconjugation based on inverse-electron-demand Diels–Alder reactivity. J. Am. Chem. Soc. 130, 13518–13519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj N. K.; Weissleder R.; Hilderbrand S. A. (2008) Tetrazine-based cycloadditions: Application to pretargeted live cell imaging. Bioconjugate Chem. 19, 2297–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q.; Dong T.; Liu X.; Lei X. (2013) A bioorthogonal ligation enabled by click cycloaddition of o-quinolinone quinone methide and vinyl thioether. J. Am. Chem. Soc. 135, 4996–4999. [DOI] [PubMed] [Google Scholar]
- Agarwal P.; van der Weijden J.; Sletten E. M.; Rabuka D.; Bertozzi C. R. (2013) A Pictet–Spengler ligation for protein chemical modification. Proc. Natl. Acad. Sci. U.S.A. 110, 46–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodama K.; Fukuzawa S.; Nakayama H.; Kigawa T.; Sakamoto K.; Yabuki T.; Matsuda N.; Shirouzu M.; Takio K.; Tachibana K.; Yokoyama S. (2006) Regioselective carbon–carbon bond formation in proteins with palladium catalysis; new protein chemistry by organometallic chemistry. ChemBioChem 7, 134–139. [DOI] [PubMed] [Google Scholar]
- Kodama K.; Fukuzawa S.; Nakayama H.; Sakamoto K.; Kigawa T.; Yabuki T.; Matsuda N.; Shirouzu M.; Takio K.; Yokoyama S.; Tachibana K. (2007) Site-specific functionalization of proteins by organopalladium reactions. ChemBioChem 8, 232–238. [DOI] [PubMed] [Google Scholar]
- Chalker J. M.; Wood C. S.; Davis B. G. (2009) A convenient catalyst for aqueous and protein Suzuki–Miyaura cross-coupling. J. Am. Chem. Soc. 131, 16346–16347. [DOI] [PubMed] [Google Scholar]
- Spicer C. D.; Triemer T.; Davis B. G. (2012) Palladium-mediated cell-surface labeling. J. Am. Chem. Soc. 134, 800–803. [DOI] [PubMed] [Google Scholar]
- Yusop R. M.; Unciti-Broceta A.; Johansson E. M. V.; Sanchez-Martin R. M.; Bradley M. (2011) Palladium-mediated intracellular chemistry. Nat. Chem. 3, 239–243. [DOI] [PubMed] [Google Scholar]
- Li N.; Lim R. K. V.; Edwardraja S.; Lin Q. (2011) Copper-free Sonogashira cross-coupling for functionalization of alkyne-encoded proteins in aqueous medium and in bacterial cells. J. Am. Chem. Soc. 133, 15316–15319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoelson S. E.; Chatterjee S.; Chaudhuri M.; White M. F. (1992) YMXM motifs of IRS-1 define substrate specificity of the insulin receptor kinase. Proc. Natl. Acad. Sci. U.S.A. 89, 2027–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas N. E.; Bramson H. N.; Nairn A. C.; Greengard P.; Fry D. C.; Mildvan A. S.; Kaiser E. T. (1987) Distinguishing among protein kinases by substrate specificities. Biochemistry 26, 4471–4474. [DOI] [PubMed] [Google Scholar]
- de Almeida G.; Sletten E. M.; Nakamura H.; Palaniappan K. K.; Bertozzi C. R. (2012) Thiacycloalkynes for copper-free click chemistry. Angew. Chem., Int. Ed. 51, 2443–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Song W.; Hu W.; Lin Q. (2009) Fast alkene functionalization in vivo by photoclick chemistry: HOMO lifting of nitrile imine dipoles. Angew. Chem., Int. Ed. 48, 5330–5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskin J. M.; Prescher J. A.; Laughlin S. T.; Agard N. J.; Chang P. V.; Miller I. A.; Lo A.; Codelli J. A.; Bertozzi C. R. (2007) Copper-free click chemistry for dynamic in vivo imaging. Proc. Natl. Acad. Sci. U.S.A. 104, 16793–16797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang K.; Davis L.; Wallace S.; Mahesh M.; Cox D. J.; Blackman M. L.; Fox J. M.; Chin J. W. (2012) Genetic encoding of bicyclononynes and trans-cyclooctenes for site-specific protein labeling in vitro and in live mammalian cells via rapid fluorogenic Diels–Alder reactions. J. Am. Chem. Soc. 134, 10317–10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossin R.; van den Bosch S. M.; Ten Hoeve W.; Carvelli M.; Versteegen R. M.; Lub J.; Robillard M. S. (2013) Highly reactive trans-cyclooctene tags with improved stability for Diels–Alder chemistry in living systems. Bioconjugate Chem. 24, 1210–1217. [DOI] [PubMed] [Google Scholar]
- Sletten E. M.; Nakamura H.; Jewett J. C.; Bertozzi C. R. (2010) Difluorobenzocyclooctyne: Synthesis, reactivity, and stabilization by β-cyclodextrin. J. Am. Chem. Soc. 132, 11799–11805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debets M. F.; van Berkel S. S.; Dommerholt J.; Dirks A. T.; Rutjes F. P.; van Delft F. L. (2011) Bioconjugation with strained alkenes and alkynes. Acc. Chem. Res. 44, 805–815. [DOI] [PubMed] [Google Scholar]
- Brotherton W. S.; Michaels H. A.; Simmons J. T.; Clark R. J.; Dalal N. S.; Zhu L. (2009) Apparent copper(II)-accelerated azide–alkyne cycloaddition. Org. Lett. 11, 4954–4957. [DOI] [PubMed] [Google Scholar]
- Kuang G. C.; Michaels H. A.; Simmons J. T.; Clark R. J.; Zhu L. (2010) Chelation-assisted, copper(II)-acetate-accelerated azide–alkyne cycloaddition. J. Org. Chem. 75, 6540–6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian F.; Tsao M. L.; Schultz P. G. (2004) A phage display system with unnatural amino acids. J. Am. Chem. Soc. 126, 15962–15963. [DOI] [PubMed] [Google Scholar]
- Liu C. C.; Mack A. V.; Brustad E. M.; Mills J. H.; Groff D.; Smider V. V.; Schultz P. G. (2009) Evolution of proteins with genetically encoded ″chemical warheads″. J. Am. Chem. Soc. 131, 9616–9617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao M. L.; Tian F.; Schultz P. G. (2005) Selective Staudinger modification of proteins containing p-azidophenylalanine. ChemBioChem 6, 2147–2149. [DOI] [PubMed] [Google Scholar]
- Tanaka F.; Fuller R.; Asawapornmongkol L.; Warsinke A.; Gobuty S.; Barbas C. F. 3rd. (2007) Development of a small peptide tag for covalent labeling of proteins. Bioconjugate Chem. 18, 1318–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldridge G. M.; Weiss G. A. (2011) Hydrazide reactive peptide tags for site-specific protein labeling. Bioconjugate Chem. 22, 2143–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Held H. A.; Sidhu S. S. (2004) Comprehensive mutational analysis of the M13 major coat protein: Improved scaffolds for C-terminal phage display. J. Mol. Biol. 340, 587–597. [DOI] [PubMed] [Google Scholar]
- van Hest J. C. M.; Kiick K. L.; Tirrell D. A. (2000) Efficient incorporation of unsaturated methionine analogues into proteins in vivo. J. Am. Chem. Soc. 122, 1282–1288. [Google Scholar]
- Doublie S. (2007) Production of selenomethionyl proteins in prokaryotic and eukaryotic expression systems. Methods Mol. Biol. 363, 91–108. [DOI] [PubMed] [Google Scholar]
- Milovic N. M.; Dutca L. M.; Kostic N. M. (2003) Combined use of platinum(II) complexes and palladium(II) complexes for selective cleavage of peptides and proteins. Inorg. Chem. 42, 4036–4045. [DOI] [PubMed] [Google Scholar]
- Krooglyak E. V.; Kazankov G. M.; Kurzeez S. A.; Polyakov V. A.; Semenov A. N.; Ryabov A. D. (1996) Targeting of pallada- and platinacycles to the N-((tert-Butyloxy)carbonyl)-l-methionine p-nitrophenyl ester for promotion of the ester cleavage. Inorg. Chem. 35, 4804–4806. [Google Scholar]
- Williamson M. P. (1994) The structure and function of proline-rich regions in proteins. Biochem. J. 297, 249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrinpar A.; Bhattacharyya R. P.; Lim W. A. (2003) The structure and function of proline recognition domains. Sci. STKE 2003, RE8. [DOI] [PubMed] [Google Scholar]
- Crespo L.; Sanclimens G.; Montaner B.; Pérez-Tomás R.; Royo M.; Pons M.; Albericio F.; Giralt E. (2002) Peptide dendrimers based on polyproline helices. J. Am. Chem. Soc. 124, 8876–8883. [DOI] [PubMed] [Google Scholar]
- Ljungdahl T.; Bennur T.; Dallas A.; Emtenäs H.; Mårtensson J. (2008) Two competing mechanisms for the copper-free sonogashira cross-coupling reaction. Organometallics 27, 2490–2498. [Google Scholar]
- Tougerti A.; Negri S.; Jutand A. (2007) Mechanism of the copper-free palladium-catalyzed Sonagashira reactions: Multiple role of amines. Chem.—Eur. J. 13, 666–676. [DOI] [PubMed] [Google Scholar]
- Hunter C. A.; Singh J.; Thornton J. M. (1991) π–π interactions: The geometry and energetics of phenylalanine–phenylalanine interactions in proteins. J. Mol. Biol. 218, 837–846. [DOI] [PubMed] [Google Scholar]
- Guillen Schlippe Y. V.; Hedstrom L. (2005) A twisted base? The role of arginine in enzyme-catalyzed proton abstractions. Arch. Biochem. Biophys. 433, 266–278. [DOI] [PubMed] [Google Scholar]
- Chen I.; Howarth M.; Lin W.; Ting A. Y. (2005) Site-specific labeling of cell surface proteins with biophysical probes using biotin ligase. Nat. Methods 2, 99–104. [DOI] [PubMed] [Google Scholar]
- Song W.; Wang Y.; Yu Z.; Vera C. I.; Qu J.; Lin Q. (2010) A metabolic alkene reporter for spatiotemporally controlled imaging of newly synthesized proteins in mammalian cells. ACS Chem. Biol. 5, 875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp M. W.; Antos J. M.; Grotenbreg G. M.; Spooner E.; Ploegh H. L. (2007) Sortagging: A versatile method for protein labeling. Nat. Chem. Biol. 3, 707–708. [DOI] [PubMed] [Google Scholar]
- Carrico I. S.; Carlson B. L.; Bertozzi C. R. (2007) Introducing genetically encoded aldehydes into proteins. Nat. Chem. Biol. 3, 321–322. [DOI] [PubMed] [Google Scholar]
- Yin J.; Liu F.; Li X.; Walsh C. T. (2004) Labeling proteins with small molecules by site-specific posttranslational modification. J. Am. Chem. Soc. 126, 7754–7755. [DOI] [PubMed] [Google Scholar]
- Uttamapinant C.; White K. A.; Baruah H.; Thompson S.; Fernandez-Suarez M.; Puthenveetil S.; Ting A. Y. (2010) A fluorophore ligase for site-specific protein labeling inside living cells. Proc. Natl. Acad. Sci. U.S.A. 107, 10914–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.