

Abstract
Tyrosine (Tyr) sulfation is a common post-translational modification that is implicated in a variety of important biological processes, including the fusion and entry of human immunodeficiency virus type-1 (HIV-1). A number of sulfated Tyr (sTyr) residues on the N-terminus of the CCR5 chemokine receptor are involved in a crucial binding interaction with the gp120 HIV-1 envelope glycoprotein. Despite the established importance of these sTyr residues, the exact structural and functional role of this post-translational modification in HIV-1 infection is not fully understood. Detailed biological studies are hindered in part by the difficulty in accessing homogeneous sulfopeptides and sulfoproteins through biological expression and established synthetic techniques. Herein we describe an efficient approach to the synthesis of sulfopeptides bearing discrete sulfation patterns through the divergent, site-selective incorporation of sTyr residues on solid support. By employing three orthogonally protected Tyr building blocks and a solid-phase sulfation protocol, we demonstrate the synthesis of a library of target N-terminal CCR5(2-22) sulfoforms bearing discrete and differential sulfation at Tyr10, Tyr14, and Tyr15, from a single resin-bound intermediate. We demonstrate the importance of distinct sites of Tyr sulfation in binding gp120 through a competitive binding assay between the synthetic CCR5 sulfopeptides and an anti-gp120 monoclonal antibody. These studies revealed a critical role of sulfation at Tyr14 for binding and a possible additional role for sulfation at Tyr10. N-terminal CCR5 variants bearing a sTyr residue at position 14 were also found to complement viral entry into cells expressing an N-terminally truncated CCR5 receptor.
Tyrosine (Tyr) sulfation is one of the most common post-translational modifications affecting secreted and transmembrane proteins, with estimates that more than 1% of all human proteins contain sulfated Tyr (sTyr) residues.1−5 The sulfation process is mediated by tyrosylprotein sulfotransferase-1 and -2 (TPST-1 and TPST-2, respectively), enzymes located in the trans-Golgi network that catalyze the transfer of sulfate from 3′-phosphoadenosine 5′-phosphosulfate (PAPS) to the phenol moiety of a peptidyltyrosine residue.6−8 sTyr residues are implicated in a number of important physiological processes, including blood coagulation,9−12 cell–cell interactions,13 chemokine signaling through chemokine receptors14−16 and viral entry into host cells.2,17−20 Indeed, Tyr sulfation is known to have a particularly significant role in mediating the fusion and entry of human immunodeficiency virus type-1 (HIV-1) into target cells. Multiple sulfation sites on the N-terminal fragment of the chemokine coreceptor CCR5 are thought to be involved in a high affinity binding interaction with the HIV-1 envelope glycoprotein gp120-cellular CD4 receptor complex that is essential for viral entry.17 The crucial role of sTyr in CCR5-mediated HIV-1 infection is of particular interest for the broader development of CCR5 mimics and coreceptor antagonists as novel antivirals.21−23
Despite the biological importance of sulfation, the precise structural and functional role of sTyr residues in CCR5 and in the majority of other proteins known to be sulfated is not fully understood.3 Detailed studies of the role of Tyr sulfation on biological activity are complicated by difficulties in accessing large quantities of high-purity, homogeneous sulfoproteins. This is owing to the fact that the Tyr sulfation process is not templated but rather is controlled by the relative activities of TPSTs in the cell. Indeed, under the influence of the sulfotransferase enzymes, a heterogeneous mixture of different protein sulfoforms results, varying widely in the number and location of sTyr residues. The highly acid-labile phenolic sulfate ester linkage further complicates the extraction and manipulation of sulfopeptides as well as the ability to access target compounds by standard chemical methods. As such, there is a strong demand for the development of new synthetic routes for the preparation of peptides and proteins bearing discrete Tyr sulfation patterns.
Early chemical methods for accessing sulfopeptides involved global sulfation protocols whereby unprotected Tyr residues were reacted with sulfur trioxide-N,N-dimethylformamide (DMF)24−28 or sulfur trioxide-pyridine complexes.29,30 The incorporation of preformed sTyr amino acid building blocks as the sodium26,31,32 or tetraalkylammonium salts33 in solid-phase peptide synthesis (SPPS) subsequently enabled the site-selective installation of sTyr residues. However, cleavage of the target peptide from the solid support and concomitant global side-chain deprotection under the standard acidic conditions employed in SPPS leads to substantial loss of the acid-labile sulfate monoester.34,35 The orthogonal protection of sTyr residues as robust diesters for use in Fmoc-SPPS has therefore been explored as an alternative approach for cassette strategy incorporation of Tyr sulfation.36 A number of acid-stable aryl sulfate protecting groups, including neopentyl,36,37 2,2,2-trifluoroethyl (TFE),38 2,2,2-trichloroethyl (TCE),39−41 and 2,2-dichlorovinyl (DCV) sulfate esters,42,43 have been employed in the solid-phase synthesis of target sulfopeptides. Liskamp and co-workers have further extended the use of sulfate diesters to encompass the global sulfation of free Tyr residues on resin-bound peptides via treatment with TCE chlorosulfate.44 We have recently explored the use of sulfuryl imidazolium salts,38,45 such as Taylor’s TCE sulfating reagent 1 (Scheme 1A)46 and the corresponding TFE derivative,38 for the total synthesis of the sulfoprotein hirudin P611,12 as well as for the solid-phase sulfation of two Tyr residues within the N-terminal domain of the chemokine receptor CCR2.39 This study enabled subsequent biochemical studies of the role of Tyr sulfation in chemokine recognition.47−49
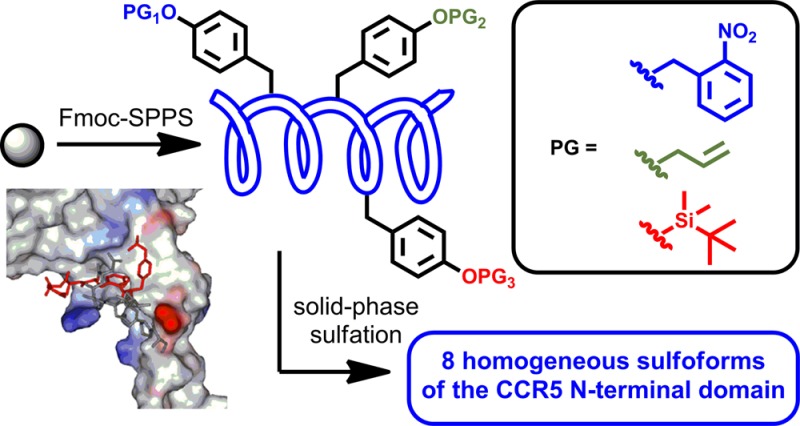
Scheme 1. (A) Divergent Solid-Phase Sulfation Strategy Employing Three Orthogonal Protecting Groups; (B) Target N-Terminal CCR5(2-22) Sulfoforms 2–9.

Despite these advances, there are as yet no divergent synthetic approaches to sulfopeptide libraries of targets bearing more than two sulfation sites. Driven by our interest in the role of the multiply sulfated N-terminal region of CCR5 (containing four possible Tyr sulfation sites, three of which are proposed to be important to CCR5 coreceptor function)17 in binding gp120 and mediating HIV-1 entry, we were interested in developing a divergent, solid-phase sulfation strategy that would allow access to sulfopeptide libraries containing variable sulfation at three distinct sites. To this end, we envisaged the use of three orthogonal side-chain Tyr protecting groups, o-nitrobenzyl (o-Nb), allyl (All), and tert-butyl-dimethylsilyl (TBS) groups, which after installation at the required position in the target peptide sequence can be selectively and differentially deprotected in the presence of one another and all other side-chain protecting groups commonly employed in Fmoc-SPPS (Scheme 1A). We proposed that orthogonal deprotection(s), followed by site-selective solid-phase sulfation of the selectively unmasked Tyr residues with TCE sulfating reagent 1, would enable rapid access to all eight variants of a target sulfopeptide bearing three sulfation sites (Scheme 1A). Importantly, this methodology would enable access to a library of target N-terminal CCR5(2-22) sulfoforms (compounds 2–9, Scheme 1B), bearing discrete and differential sulfation at Tyr10, Tyr14, and Tyr15, from a single resin-bound peptide and without any intermediary purification steps. We describe herein the development of this novel methodology and demonstrate the high-yielding syntheses of eight differentially sulfated CCR5 targets to facilitate a systematic study of the role of sulfation in binding to the HIV-1 envelope glycoprotein gp120. We have also demonstrated that the sulfation pattern on the N-terminus of CCR5 heavily influences whether fragments of the N-terminal CCR5 domain can complement viral entry of an N-terminally truncated CCR5 receptor on a CD4-expressing human cell line.
Results and Discussion
Synthesis of Target CCR5 Sulfopeptides
In order to implement the proposed divergent solid-phase sulfation strategy, we first synthesized three preformed, orthogonally protected Tyr building blocks (10, 11, and 12), bearing side-chain o-Nb, All, and TBS protection of the phenolic Tyr side chain, respectively (Figure 1, see Supporting Information for synthetic details). These building blocks could be accessed in gram quantities and in high enantiopurities from commercially available, protected Tyr precursors. With the requisite Tyr derivatives in hand, we next embarked upon the solid-phase synthesis of the target CCR5 sulfopeptides, composed of residues 2–22 of the N-terminal domain, with a single substitution of C20S to prevent interpeptide disulfide formation, together with the incorporation of three possible sulfation sites at Tyr10, Tyr14, and Tyr15. Synthesis was performed using Rink amide resin and began with the loading of Fmoc-Lys(Boc)-OH. Elongation to residue Thr16 (just prior to the first potential Tyr sulfation site) was accomplished using standard Fmoc-SPPS (see Scheme 2 and Supporting Information). Building block 12 was next incorporated using a slight excess of amino acid (1.5 equiv), in the presence of 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) (1.5 equiv) and N,N-diisopropylethylamine (DIPEA) (3.0 equiv) in DMF. Compound 11 bearing side-chain allylic protection was subsequently incorporated at position 14, using identical coupling conditions. Extension to Asp11 using standard Fmoc-SPPS was followed by the coupling of o-nitrobenzyl Tyr derivative 10 using optimized conditions of (benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP) (2.0 equiv), 1-hydroxybenzotriazole (HOBt) (2.0 equiv), and N-methylmorpholine (NMM) (4.0 equiv) in DMF (see Supporting Information for details). Following incorporation of the final Tyr building block, the peptide was further elongated to CCR5(2-22) 13 and the resin split into eight equal portions to facilitate divergent solid-phase manipulations at Tyr10, Tyr14, and Tyr15.
Figure 1.
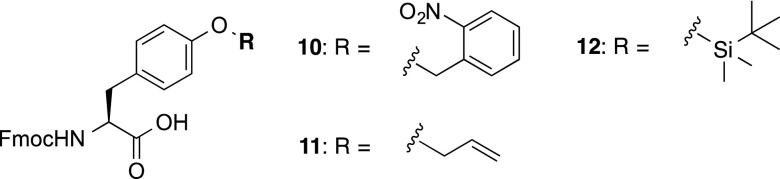
Fmoc-protected Tyr building blocks 10, 11, and 12.
Scheme 2. Divergent Solid-Phase Synthesis of CCR5(2-22) (Sulfo)peptides 2–9.

From the fully protected resin-bound peptide 13, we first embarked upon the synthesis of target CCR5(2-22) peptide 2, a triply sulfated variant bearing sTyr residues at positions 10, 14, and 15. As construction of the desired sulfation pattern of peptide 2 would first require the removal of all three Tyr side-chain protecting groups, we hypothesized that this synthesis would enable us to affirm the orthogonality of the three Tyr protecting groups selected (Scheme 2). To probe our protecting group strategy, resin-bound CCR5 peptide 13 was first irradiated with UV light at λ = 365 nm in DMF for 24 h (deprotection condition A). Gratifyingly, these conditions facilitated quantitative and selective removal of the side-chain o-nitrobenzyl group at Tyr10, leaving the remaining phenolic protecting groups intact (as judged by HPLC–MS analysis of the cleavage of a small number of resin beads). Subsequent treatment with Pd(PPh3)4, triethylsilane (TES), and acetic acid in CH2Cl2 (deprotection condition B) effected clean removal of the allyl ether at Tyr14. A final treatment with tetrabutylammonium fluoride (TBAF) buffered with acetic acid39 (deprotection condition C) resulted in the loss of the TBS protecting group at Tyr15, affording a resin-bound peptide bearing three unprotected Tyr residues. It should be noted that the order of incorporation of the protected Tyr building blocks 10–12 into the peptide chain could be altered without affecting the orthogonality of the deprotection steps (see Supporting Information for details). Having demonstrated the iterative and selective deprotection of each Tyr protecting group, we next subjected the immobilized peptide to solid-phase sulfation using TCE imidazolium sulfating reagent 1 (8 equiv per free phenol) in the presence of triethylamine, to afford peptide 14 bearing TCE-protected sulfate diesters at positions 10, 14, and 15. Acidic cleavage of the peptide from the resin was followed by removal of the TCE protecting group via catalytic hydrogenolysis using Pd(OH)2 on carbon.43 HPLC purification in the presence of 0.1 M NH4OAc (to avoid loss of the acid-labile sulfate monoester) afforded the triply sulfated CCR5 variant 2 in 28% yield based on the original resin loading (over 47 linear steps).
With conditions in hand for both the selective, on-resin deprotection of each orthogonal Tyr side-chain protecting group as well as the subsequent installation of the TCE sulfate diester on the solid phase, we proceeded to synthesize the remaining CCR5(2-22) sulfoforms of our proposed library using a combinatorial deprotection–solid-phase sulfation strategy. In each case the initial deprotection(s) (conditions A, B, and/or C, on-resin deprotection step(s) 1, Scheme 2) were followed by solid-phase sulfation with imidazolium sulfating reagent 1 to install the appropriately positioned TCE-protected sulfate diesters onto the unmasked phenolic Tyr side chains. A second round of deprotection reactions (on-resin deprotection step(s) 2, Scheme 2) then facilitated the removal of the remaining Tyr side-chain protecting groups at positions 10 and/or 14. It should be noted that in cases where Tyr15 remained unsulfated (e.g., resin-bound peptides 15, 16, 17, and 19), the side-chain TBS ether was left intact, as loss of this protecting group could be facilitated concomitantly with acidic cleavage from the resin and global side-chain deprotection.39 Following solid-phase manipulations, the resin-bound peptides 15–21 were treated with an acidic cocktail of TFA/iPr3SiH/H2O (90:5:5 v/v/v) to effect cleavage from the resin and removal of all remaining side-chain protecting groups. At this point, unsulfated variant 3 was obtained directly following HPLC purification in 60% yield based on the original resin loading. The crude, differentially TCE-sulfated peptides derived from resin-bound variants 16–21 were then treated with Pd(OH)2 in the presence of H2 to remove the TCE sulfate diester protecting group(s). Purification by HPLC afforded the six remaining sulfoforms 4–9 in good overall yields (20–42%, an average of 97–98% per step). Importantly, this synthetic approach represents the first divergent synthesis of all 8 distinct sulfoforms of a given sulfopeptide bearing three sites of sulfation, in this case the CCR5 N-terminus (bearing sulfation at Tyr10, Tyr14, and Tyr15), from a single-resin bound peptide and serves as a proof of concept for the rapid synthesis of compound libraries of multiply sulfated peptide and protein targets.
Binding Interaction of N-Terminal CCR5 Sulfoforms with gp120
With the eight target sulfoforms of the N-terminal region of CCR5 in hand (compounds 2–9), we were next interested in investigating the effect that discrete CCR5 sulfation patterns had on mediating binding with the HIV-1 Env glycoprotein gp120 and facilitating viral entry. Although previous studies have examined the role of specific sTyr residues in CCR5 binding,17,19,20 we were interested in carrying out the first systematic screen of multiple sulfation patterns, including a detailed analysis of the role of sulfation at Tyr15. To evaluate the ability of our synthetic CCR5 peptides to bind gp120, we first carried out a competitive binding assay between peptides 2–9 and the anti-gp120 monoclonal antibody 17b50 (Figure 2A). In this ELISA-based assay, recombinant gp120 was captured on plates coated with recombinant human CD4 lacking its transmembrane domain (rsCD4), which exposes the binding site on gp120 for CCR5. The protocol for producing rsCD4 is provided in detail in the Methods section. Conveniently, the epitope on gp120 for 17b overlaps with the CCR5 binding site, such that the displacement of antibody 17b from gp120 upon treatment with the sulfopeptide is indicative of the ability of the sulfopeptide to bind to gp120. This method for measuring the binding interaction between the N-terminal CCR5 (sulfo)peptides and gp120 is preferable to a direct binding assay because the binding of 17b to gp120, unlike the interaction between CCR5 and gp120, has known stoichiometry and is a well characterized process, both structurally and biochemically.51,52 The results in Figure 2A show that unsulfated peptide 3 could not displace the binding of 17b to gp120, confirming that sulfation is essential for gp120 binding to CCR5. Only peptides sulfated at Tyr10, Tyr14, and Tyr15 (peptide 2), Tyr10 and Tyr14 (peptide 7), Tyr14 and Tyr15 (peptide 9), and Tyr14 (peptide 5) could inhibit 17b binding to gp120, albeit to variable levels, whereas peptides sulfated at Tyr10 and Tyr15 (peptide 8) and Tyr15 alone (peptide 6) could not. However, the most potent inhibition of antibody binding was exhibited by triply sulfated peptide 2. Together, these results suggest that Tyr14 is the most critical sulfation site for gp120 binding to CCR5, but that sulfation of Tyr10 and Tyr15 may further enhance gp120 binding affinity.
Figure 2.

(A) CCR5 sulfoforms inhibit binding of 17b to gp120. gp120 (recombinant) from the HIV-1 Bal strain was incubated on plates coated with recombinant soluble CD4 prior to addition of CCR5 sulfoforms. 17b was then added to the wells, followed by sheep anti-human horseradish peroxidase (HRP) and A450 values were read. Binding curves were constructed where % binding = 100 – [(Abs17b + peptide – Abs17b + no peptide) × 100]. Data points represent the mean and standard error of the mean from 5 independent experiments. B) CCR5 sulfoforms can reconstitute HIV-1 entry in cells expressing N-terminally truncated CCR5. U87-CD4 cells expressing an N-terminally truncated CCR5 (Δ2-17) were incubated with CCR5 peptide sulfoforms (100 μM concentration) for 30 min prior to infection with luciferase reporter viruses pseudotyped with HIV-1 YU-2 Env. Entry is expressed as a percentage of entry obtained in U87-CD4 cells expressing an equivalent amount of wild type CCR5. The data is the mean and standard deviation of triplicate infections from two independent experiments.
To determine whether the pattern of sulfated Tyr residues in CCR5 that are implicated in gp120 binding are also functionally important for HIV-1 entry into cells, we next undertook a series of CCR5 N-terminus complementation assays using luciferase reporter HIV-1 pseudotyped with gp120 from the YU-2 HIV-1 strain (Figure 2B). These assays are based on those previously described by Farzan and co-workers.20 Briefly, cells expressing CD4 together with N-terminally truncated (and thus nonfunctional) CCR5 (Δ2-17) were inoculated with virus in the presence or absence of each sulfopeptide. Any reconstitution of HIV-1 entry into the cells by addition of a synthetic sulfopeptide to the media would signify a “functional” sulfopeptide that is capable of mediating HIV-1 entry into cells after gp120 binding. When compared to cells expressing wild type (WT) CCR5, only HIV-1 that was treated with CCR5 variants sulfated at Tyr10, Tyr14, and Tyr15 (peptide 2), Tyr10 and Tyr14 (peptide 7), and Tyr14 (peptide 5) could restore HIV-1 entry, with the triply sulfated peptide 2 and peptide 7 sulfated at Tyr10 and Tyr14 showing the greatest levels of restoration (Figure 2B). Not unexpectedly, the overall levels of restored virus entry were markedly lower than those mediated by WT CCR5. Together, the results of these functional studies largely mirror those from the gp120 binding studies (Figure 2A) and suggest a critical role for sTyr14 in CCR5 in mediating HIV-1 entry and a possible additional role for sTyr10 for gp120 binding and entry. The observed importance of Tyr sulfation at positions 10 and 14 is corroborated by the original complementation assays performed by Farzan and co-workers,20 which demonstrated that a CCR5 N-terminal peptide bearing sTyr residues at positions 10 and 14 could functionally reconstitute a CCR5 variant lacking the critical N-terminal region. Dragic and co-workers have additionally demonstrated that CCR5 N-terminal peptides bearing sTyr residues at positions 10 and 14 (but not the singly sulfated variants) were capable of binding gp120-CD4 complexes.19 The observed binding motifs may be a direct result of the structural conformation of CCR5 when in complex with gp120 and CD4 (the complex necessary for host entry), which has recently been interrogated using NMR techniques.53,54 In order to probe the potential binding interactions of the eight differentially sulfated CCR5 peptides synthesized in this study, we used molecular modeling to characterize the intermolecular interactions to a YU-2 gp120 protein structure that had been previously docked with an NMR structure of an N-terminal fragment of CCR5 by Huang et al. (see Supporting Information).53 Analysis of the potential ionic and hydrogen bonding interactions of the different CCR5 peptide sulfoforms with gp120 showed that sTyr at position 14 promotes interactions with both the ascending (Asn300, Asn302, Thr303) and the descending strand of the V3 loop and may explain why the presence of sTyr14 alone or in combination with sulfation at other residues has the greatest effect on binding. sTyr at position 15 is oriented away from the V3 loop and appears to be important for interactions with the C4 region of gp120. This is consistent with saturation transfer difference (STD) NMR studies that have shown that sTyr14 is positioned closer to gp120 than sTyr10 and is thought to make a number of important hydrogen-bonding interactions through its appended sulfate group.53,54 In contrast, Tyr15 was shown to be closely packed with neighboring hydrophobic residues and pointed toward the host cell membrane.53 Therefore, it is possible that the enhanced binding and inhibition observed with triply sulfated peptide 2 relative to doubly sulfated peptide 7 bearing sTyr residues at positions 10 and 14 may occur via an indirect structural effect.
Conclusion
In summary, we have developed the first solid-phase sulfation strategy to allow for the divergent and site-selective incorporation of sTyr at three possible sulfation sites in a target peptide. By incorporating three orthogonally protected Tyr residues into a single resin-bound peptide, we were able to access a library of eight sulfoforms of an N-terminal fragment of the HIV-1 coreceptor CCR5 (bearing differential sulfation at Tyr10, Tyr14, and Tyr15). This was achieved through iterative solid-phase deprotection(s), followed by solid-phase sulfation. This technique enabled the rapid and high-yielding construction of CCR5(2-22) variants 2–9. These distinct sulfoforms were subsequently utilized in the first comprehensive interrogation of the ability of CCR5(2-22) variants bearing discrete Tyr sulfate modifications at positions 10, 14, and 15 to bind gp120 and mediate HIV-1 entry into host cells. A number of sulfated CCR5 variants, particularly those bearing a sTyr residue at position 14, were shown to bind gp120 and enhance viral entry into cells expressing a truncated CCR5 coreceptor that lacks the crucial N-terminus. The results of these studies, which were corroborated by molecular modeling studies, provide important insight into the functional role of site-specific Tyr sulfation and should prove useful in the development of CCR5 mimetics and antagonists as novel antiviral therapies. In addition, the generality of the solid-phase synthetic methodology should facilitate the construction of other sulfopeptide and sulfoprotein libraries to interrogate the importance of Tyr sulfate modifications in a number of diverse systems, including chemokine-chemokine receptor interactions (e.g. CCR5/RANTES),56 decoy receptors, and complement proteins,55 in the future.
Methods
Synthesis of N-Terminal CCR5 (Sulfo)peptides
For the synthesis of protected Tyr derivatives 10, 11, and 12 and detailed solid-phase peptide synthesis (SPPS) protocols for the construction of (sulfo)peptides 2–9, see Supporting Information.
Production of Recombinant Soluble CD4 (rsCD4)
The CD4deltaTM plasmid expresses recombinant soluble human CD4 lacking the transmembrane domain but which retains the cytoplasmic domain that facilitates solid-phase binding assays (rsCD4, aa 1–395, 421–458). CD4deltaTM was produced by splice overlap extension PCR from the full-length clone of CD4, T4pMV7 (NIH), removing the region encoding the TM domain of CD4. Two intermediate clones were produced. The first clone encoding the CD4 extracellular domain was generated using the RL1: CGG GAA TTC ACA ATG GAC CGG GGA GTC CC and RL3: G CCT TCG GTG CCG GCA CCT CTG CAC CGG GGT GGA CC primer pair. The second clone encoding the CD4 cytoplasmic tail with a 6×His tag at the C-terminus was generated using the RL4: GG TCC ACC CCG GTG CAG AGG TGC CGG CAC CGA AGG C and RL5: GGG CTC GAG TCA ATG ATG ATG ATG ATG ATG AGA ACC ACC AAT GGG GCT ACA TGT CTT CTG AA primer pair. Each clone was created with an overlapping region. The two clones were combined using PCR with the outermost 5′ and 3′ primers, RL1 and RL5. The construct was then subcloned into the eukaryotic expression vector pcDNA 3.1 Zeocin (Invitrogen). CD4deltaTM clones were confirmed by DNA sequencing. To produce rsCD4, HEK 293T cells were transfected with 70 μg of CD4deltaTM plasmid using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Cell culture supernatants containing sCD4 were collected after 48–72 h and clarified by centrifugation to remove cellular debris.
Competitive Binding Assay between Peptides 2–9 and Anti-gp120 Monoclonal Antibody 17b
All incubation steps were carried out using 50 μL solutions for 2 h at RT, except where stated otherwise. Every step was followed by four washes with 0.05% (v/v) PBS/Tween (pH 7.4). Microtiter plates (Maxisorp; Nunc) were coated overnight at 4 °C with soluble CD4 diluted in coating buffer (18.2 mM Na2CO3, 45 mM NaHCO3, pH 9.6). The wells were blocked for 2 h with 230 μL 3% (w/v) skim milk in PBS (blocking solution). HIV-1BaL gp120 recombinant was then diluted in PBS/Tween 0.05% (v/v) at 1 μg mL–1. Dilutions of synthetic CCR5 sulfopeptides were added to the plates and incubated for 1 h. 17b antibody (SDIX) was then added in the presence of sulfopeptides at a final concentration of 15 nM for 30 min. After washing, plates were incubated with sheep anti-human/HRP 1:2000 in PBS/Tween 0.05% (v/v) solution. Control samples of CD4 bound alone were detected with mouse anti-CD4 RPA-T4 (Sigma) at 1:1000 and anti-mouse/HRP at 1:2000. Bound enzyme was quantified using the 3,3′5,5′-tetramethylbenzidine/H2O2 colorimetric assay (Sigma), which was quenched with 0.5 M H2SO4, and A450 values were measured. The A450 values were corrected for the background absorbance of substrate alone. Data points were analyzed using GraphPad Prism (Version 5.00 for Windows, GraphPad Software, San Diego, USA, www.graphpad.com) using a single-site binding equation: Y = BmaxX/(KD + X). This equation describes the binding of ligand to a receptor that follows the law of mass action. Bmax represents the maximal binding, and KD is the concentration of ligand required to reach one-half maximal binding.
Complementation Assays
The production and titration of luciferase reporter viruses and infection of cells expressing CCR5 mutants have been described previously.21 Briefly, U87-CD4 cells were transfected with 4 μg of pcDNA3-CCR5 (Wild type-WT) or CCR5 (Δ2-17) using lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. The WT CCR5 plasmid was serially diluted (2-fold) to create populations of cells expressing a range of CCR5. These cells were used to create a standard curve of CCR5 expression, to which the expression of CCR5 (Δ2-17) could be matched. Expression of CCR5 was determined by flow cytometry 48 h post transfection with the CCR5 specific antibody 2D7 (BD Pharmingen). At 48 h post transfection, the cells were either left untreated (WT and Δ2-17) or incubated for 30 min with 100 μM CCR5 sulfopeptides (Δ2-17 only) prior to infection with 200 TCID50 of YU-2 Env pseudotyped luciferase reporter virus. At 72 h post infection, the cells were lysed, and luciferase activity was read according to the manufacturer’s protocol (Promega). The luciferase activity in CCR5 Δ2-17 cells incubated with CCR5 sulfopeptides was expressed as a percentage of that obtained in cells expressing an equivalent amount of WT CCR5.
Acknowledgments
We thank J. Sodroski for providing the pSVIII-YU-2, pcDNA3-CCR5, pCMVΔP1ΔenvpA, and pHIV-1Luc plasmids and CCR5 (Δ2-17) mutant. The following reagents were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: HIV-1BaL gp120 from DAIDS, NIAID; U87-CD4 cells from HongKui Deng and D. R. Littman. We would also like to thank the ARC Discovery Project Scheme (DP130101984) for funding and the Australian Postgraduate Award Scheme (X.L.) and the International Postgraduate Research Scholarship Scheme (L.R.M.) for Ph.D. funding. P.R.G. and R.J.P. are supported by Australian Research Council Future Fellowships. M.R., J.S., R.D., M.L.G., N.C.B., D.A.A., and P.R.G. gratefully acknowledge the contribution to this work of the Victorian Operational Infrastructure Support Program received by the Burnet Institute.
Supporting Information Available
Detailed experimental procedures and analytical data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Huttner W. B. (1982) Sulfation of tyrosine residues-a widespread modification of proteins. Nature 299, 273–276. [DOI] [PubMed] [Google Scholar]
- Stone M. J.; Chuang S.; Hou X.; Shoham M.; Zhu J. Z. (2009) Tyrosine sulfation: an increasingly recognised post-translational modification of secreted proteins. New Biotechnol. 25, 299–317. [DOI] [PubMed] [Google Scholar]
- Kehoe J. W.; Bertozzi C. R. (2000) Tyrosine sulfation: a modulator of extracellular protein–protein interactions. Chem. Biol. 7, R57–R61. [DOI] [PubMed] [Google Scholar]
- Moore K. L. (2003) The biology and enzymology of protein tyrosine O-sulfation. J. Biol. Chem. 278, 24243–24246. [DOI] [PubMed] [Google Scholar]
- Moore K. L. (2009) Protein tyrosine sulfation: a critical posttranslation modification in plants and animals. Proc. Natl. Acad. Sci. U.S.A. 106, 14741–14742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee R. W.; Huttner W. B. (1985) Glu62, Ala30, Tyr8)n serves as high-affinity substrate for tyrosylprotein sulfotransferase: a Golgi enzyme. Proc. Natl. Acad. Sci. U.S.A. 82, 6143–6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee R. W.; Huttner W. B. (1983) Tyrosine-O-sulfated proteins of PC12 pheochromocytoma cells and their sulfation by a tyrosylprotein sulfotransferase. J. Biol. Chem. 258, 11326–11334. [PubMed] [Google Scholar]
- Baeuerle P. A.; Huttner W. B. (1987) Tyrosine sulfation is a trans-Golgi-specific protein modification. J. Cell. Biol. 105, 2655–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyte A.; van Schijndel H. B.; Niehrs C.; Huttner W. B.; Verbeet M. P.; Mertens K.; van Mourik J. A. (1991) Sulfation of Tyr1680 of human blood coagulation factor VIII is essential for the interaction of factor VIII with von Willebrand factor. J. Biol. Chem. 266, 740–746. [PubMed] [Google Scholar]
- Liu C. C.; Brustad E.; Liu W.; Schultz P. G. (2007) Crystal structure of a biosynthetic sulfo-hirudin complexed to thrombin. J. Am. Chem. Soc. 129, 10648–10649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh Y. S. Y.; Taleski D.; Wilkinson B. L.; Wijeyewickrema L. C.; Adams T. E.; Pike R. N.; Payne R. J. (2012) Effect of O-glycosylation and tyrosine sulfation of leech-derived peptides on binding and inhibitory activity against thrombin. Chem. Commun. 48, 1547–1549. [DOI] [PubMed] [Google Scholar]
- Hsieh Y. S.; Wijeyewickrema L. C.; Wilkinson B. L.; Pike R. N.; Payne R. J. (2014) Total synthesis of homogeneous variants of hirudin P6: a post-translationally modified anti-thrombotic leech-derived protein. Angew. Chem., Int. Ed. 53, 3947–3951. [DOI] [PubMed] [Google Scholar]
- Fong A. M.; Alam S. M.; Imai T.; Haribabu B.; Patel D. D. (2002) CX3CR1 tyrosine sulfation enhances fractalkine-induced cell adhesion. J. Biol. Chem. 277, 19418–19423. [DOI] [PubMed] [Google Scholar]
- Preobrazhensky A. A.; Dragan S.; Kawano T.; Gavrilin M. A.; Gulina I. V.; Chakravarty L.; Kolattukudy P. E. (2000) Monocyte chemotactic protein-1 receptor CCR2B is a glycoprotein that has tyrosine sulfation in a conserved extracellular N-terminal region. J. Immunol. 165, 5295–5303. [DOI] [PubMed] [Google Scholar]
- Choe H.; Moore M. J.; Owens C. M.; Wright P. L.; Vasilieva N.; Li W.; Singh A. P.; Shakri R.; Chitnis C. E.; Farzan M. (2005) Sulphated tyrosines mediate association of chemokines and Plasmodium vivax Duffy binding protein with the Duffy antigen/receptor for chemokines (DARC). Mol. Microbiol. 55, 1413–1422. [DOI] [PubMed] [Google Scholar]
- Ludeman J. P.; Stone M. J. (2014) The structural role of receptor tyrosine sulfation in chemokine recognition. Br. J. Pharmacol. 171, 1167–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farzan M.; Mirzabekov T.; Kolchinsky P.; Wyatt R.; Cayabyab M.; Gerard N. P.; Gerard C.; Sodroski J.; Choe H. (1999) Tyrosine sulfation of the amino terminus of CCR5 facilitates HIV-1 entry. Cell 96, 667–676. [DOI] [PubMed] [Google Scholar]
- Farzan M.; Vasilieva N.; Schnitzler C. E.; Chung S.; Robinson J.; Gerard N. P.; Gerard C.; Choe H.; Sodroski J. (2000) A tyrosine-sulfated peptide based on the N terminus of CCR5 interacts with a CD4-enhanced epitope of the HIV-1 gp120 envelope glycoprotein and inhibits HIV-1 entry. J. Biol. Chem. 275, 33516–33521. [DOI] [PubMed] [Google Scholar]
- Cormier E. G.; Persuh M.; Thompson D. A.; Lin S. W.; Sakmar T. P.; Olson W. C.; Dragic T. (2000) Specific interaction of CCR5 amino-terminal domain peptides containing sulfotyrosines with HIV-1 envelope glycoprotein gp120. Proc. Natl. Acad. Sci. U.S.A. 97, 5762–5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farzan M.; Chung S.; Li W.; Vasilieva N.; Wright P. L.; Schnitzler C. E.; Marchione R. J.; Gerard C.; Gerard N. P.; Sodroski J.; Choe H. (2002) Tyrosine-sulfated peptides functionally reconstitute a CCR5 variant lacking a critical amino-terminal region. J. Biol. Chem. 277, 40397–40402. [DOI] [PubMed] [Google Scholar]
- Roche M.; Salimi H.; Duncan R.; Wilkinson B. L.; Chikere K.; Moore M. S.; Webb N. E.; Zappi H.; Sterjovski J.; Flynn J. K.; Ellett A.; Gray L. R.; Lee B.; Jubb B.; Westby M.; Ramsland P. A.; Lewin S. R.; Payne R. J.; Churchill M. J.; Gorry P. R. (2013) A common mechanism of clinical HIV-1 resistance to the CCR5 antagonist maraviroc despite divergent resistance levels and lack of common gp120 resistance mutations. Retrovirology 10, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong J. A.; Dorfman T.; Quinlan B. D.; Chiang J. J.; Ahmed A. A.; Choe H.; Farzan M. (2011) A tyrosine-sulfated CCR5-mimetic peptide promotes conformational transitions in the HIV-1 envelope glycoprotein. J. Virol. 85, 7563–7571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang J. J.; Gardner M. R.; Quinlan B. D.; Dorfman T.; Choe H.; Farzan M. (2012) Enhanced recognition and neutralization of HIV-1 by antibody-derived CCR5-mimetic peptide variants. J. Virol. 86, 12417–12421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futaki S.; Taike T.; Akita T.; Kitagawa K. (1992) Syntheses of two tyrosine-sulphate containing peptides. Leucosulfakinin (LSK)-II and cholecystokinin (CCK)-12, using the O-p-(methylsulphinyl)benzyl serine for the selective sulphation of tyrosine. Tetrahedron 48, 8899–8914. [Google Scholar]
- Futaki S.; Taike T.; Yagami T.; Ogawa T.; Akita T.; Kitagawa K. (1990) Use of dimethylformamide-sulphur trioxide complex as a sulphating agent of tyrosine. J. Chem. Soc., Perkin Trans. 1, 1739–1744. [Google Scholar]
- Kitagawa K.; Futaki S.; Yagami T.; Sumi S.; Inoue K. (1994) Solid-phase synthesis of cionin, a protochordate-derived octapeptide related to the gastrin/cholecystokinin family of peptides, and its mono-tyrosine-sulfate-containing derivatives. Int. J. Pept. Protein Res. 43, 190–200. [DOI] [PubMed] [Google Scholar]
- Young T.; Kiessling L. L. (2002) A strategy for the synthesis of sulfated peptides. Angew. Chem., Int. Ed. 41, 3449–3451. [DOI] [PubMed] [Google Scholar]
- Campos S. V.; Miranda L. P.; Meldal M. (2002) Preparation of novel O-sulfated amino acid building blocks with improved acid stability for Fmoc-based solid-phase peptide synthesis. J. Chem. Soc., Perkin Trans. 1, 682–686. [Google Scholar]
- Vázquez-Campos S.; St Hilaire P. M.; Damgaard D.; Meldal M. (2005) GAG mimetic libraries: sulphated peptide as heparin-like glycosaminoglycan mimics in their interaction with FGF-1. QSAR Comb. Sci. 24, 923–942. [Google Scholar]
- De Luca S.; Morelli G. (2004) Synthesis and characterization of a sulfated and a non-sulfated cyclic CCK8 analogue functionalized with a chelating group for metal labelling. J. Pept. Sci. 10, 265–273. [DOI] [PubMed] [Google Scholar]
- Kitagawa K.; Aida C.; Fujiwara H.; Yagami T.; Futaki S. (1997) Efficient solid-phase synthesis of sulfated tyrosine-containing peptides using 2-chlorotrityl resin: facile synthesis of gastrin/cholecystokinin peptides. Tetrahedron Lett. 38, 599–602. [Google Scholar]
- Kitagawa K.; Aida C.; Fujiwara H.; Yagami T.; Futaki S.; Kogire M.; Ida J.; Inoue K. (2001) Facile solid-phase synthesis of sulfated tyrosine-containing peptides: total synthesis of human big gastrin-II and cholecystokinin (CCK)-39. J. Org. Chem. 66, 1–10. [DOI] [PubMed] [Google Scholar]
- Ueki M.; Watanabe S.; Ishii Y.; Okunaka O.; Uchino K.; Saitoh T.; Higashi K.; Nakashima H.; Yamamoto N.; Ogawara H. (2001) Synthesis and anti-HIV activity of nonatyrosine N- and O1–9-decasulfate. Bioorg. Med. Chem. 9, 477–486. [DOI] [PubMed] [Google Scholar]
- Balsved D.; Bundgaard J. R.; Sen J. W. (2007) Stability of tyrosine sulfate in acidic solutions. Anal. Biochem. 363, 70–76. [DOI] [PubMed] [Google Scholar]
- Huttner W. B. (1984) Determination and occurrence of tyrosine O-sulfate in proteins, in Methods in Enzymology (Finn Wold K. M., Ed.), pp 200–223, Academic Press, New York. [DOI] [PubMed] [Google Scholar]
- Simpson L. S.; Widlanski T. S. (2006) A comprehensive approach to the synthesis of sulfate esters. J. Am. Chem. Soc. 128, 1605–1610. [DOI] [PubMed] [Google Scholar]
- Simpson L. S.; Zhu J. Z.; Widlanski T. S.; Stone M. J. (2009) Regulation of chemokine recognition by site-specific tyrosine sulfation of receptor peptides. Chem. Biol. 16, 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desoky A. Y.; Hendel J.; Ingram L.; Taylor S. D. (2011) Preparation of trifluoroethyl- and phenyl-protected sulfates using sulfuryl imidazolium salts. Tetrahedron 67, 1281–1287. [Google Scholar]
- Taleski D.; Butler S. J.; Stone M. J.; Payne R. J. (2011) Divergent and site-selective solid-phase synthesis of sulfopeptides. Chem.—Asian J. 6, 1316–1320. [DOI] [PubMed] [Google Scholar]
- Ali A. M.; Hill B.; Taylor S. D. (2009) Trichloroethyl group as a protecting group for sulfonates and its application to the synthesis of a disulfonate analog of the tyrosine sulfated PSGL-143–50 peptide. J. Org. Chem. 74, 3583–3586. [DOI] [PubMed] [Google Scholar]
- Taylor S. D.; Desoky A. (2011) Rapid and efficient chemoselective and multiple sulfations of phenols using sulfuryl imidazolium salts. Tetrahedron Lett. 52, 3353–3357. [Google Scholar]
- Ali A. M.; Taylor S. D. (2009) Efficient solid-phase synthesis of sulfotyrosine peptides using a sulfate protecting-group strategy. Angew. Chem., Int. Ed. 48, 2024–2026. [DOI] [PubMed] [Google Scholar]
- Ali A. M.; Taylor S. D. (2010) Synthesis of disulfated peptides corresponding to the N-terminus of chemokines receptors CXCR6 (CXCR6 1–20) and DARC (DARC 8–42) using a sulfate-protecting group strategy. J. Pept. Sci. 16, 190–199. [DOI] [PubMed] [Google Scholar]
- Bunschoten A.; Kruijtzer J. A. W.; Ippel J. H.; de Haas C. J. C.; van Strijp J. A. G.; Kemmink J.; Liskamp R. M. J. (2009) A general sequence independent solid phase method for the site specific synthesis of multiple sulfated-tyrosine containing peptides. Chem. Commun. 2999–3001. [DOI] [PubMed] [Google Scholar]
- Ingram L. J.; Taylor S. D. (2006) Introduction of 2,2,2-trichloroethyl-protected sulfates into monosaccharides with a sulfuryl imidazolium salt and application to the synthesis of sulfated carbohydrates. Angew. Chem., Int. Ed. 45, 3503–3506. [DOI] [PubMed] [Google Scholar]
- Ingram L. J.; Desoky A.; Ali A. M.; Taylor S. D. (2009) O- and N-sulfations of carbohydrates using sulfuryl imidazolium salts. J. Org. Chem. 74, 6479–6485. [DOI] [PubMed] [Google Scholar]
- Tan J. H.; Canals M.; Ludeman J. P.; Wedderburn J.; Boston C.; Butler S. J.; Carrick A. M.; Parody T. R.; Taleski D.; Christopoulos A.; Payne R. J.; Stone M. J. (2012) Design and receptor interactions of obligate dimeric mutant of chemokine monocyte chemoattractant protein-1 (MCP-1). J. Biol. Chem. 287, 14692–14702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J. H.; Ludeman J. P.; Wedderburn J.; Canals M.; Hall P.; Butler S. J.; Taleski D.; Christopoulos A.; Hickey M. J.; Payne R. J.; Stone M. J. (2013) Tyrosine sulfation of chemokine receptor CCR2 enhances interactions with both monomeric and dimeric forms of the chemokine monocyte chemoattractant protein-1 (MCP-1). J. Biol. Chem. 288, 10024–10034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huma Z. E.; Ludeman J. P.; Wilkinson B. L.; Payne R. J.; Stone M. J. (2014) NMR characterization of cooperativity: fast ligand binding coupled to slow protein dimerization. Chem. Sci. 5, 2783–2788. [Google Scholar]
- Thali M.; Moore J. P.; Furman C.; Charles M.; Ho D. D.; Robinson J.; Sodroski J. (1993) Characterization of conserved human immunodeficiency virus type 1 gp120 neutralization epitopes exposed upon gp120-CD4 binding. J. Virol. 67, 3978–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong P. D.; Wyatt R.; Robinson J.; Sweet R. W.; Sodroski J.; Hendrickson W. A. (1998) Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393, 648–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Bartesaghi A.; Borgnia M. J.; Sapiro G.; Subramaniam S. (2008) Molecular architecture of native HIV-1 gp120 trimers. Nature 455, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C. C.; Lam S. N.; Acharya P.; Tang M.; Xiang S. H.; Hussan S. S.; Stanfield R. L.; Robinson J.; Sodroski J.; Wilson I. A.; Wyatt R.; Bewley C. A.; Kwong P. D. (2007) Structures of the CCR5 N terminus and of a tyrosine-sulfated antibody with HIV-1 gp120 and CD4. Science 317, 1930–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam S. N.; Acharya P.; Wyatt R.; Kwong P. D.; Bewley C. A. (2008) Tyrosine-sulfate isosteres of CCR5 N-terminus as tools for studying HIV-1 entry. Bioorg. Med. Chem. 16, 10113–10120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnur E.; Kessler N.; Zherdev Y.; Noah E.; Scherf T.; Ding F.; Rabinovich S.; Arshava B.; Kurbatska V.; Leoniks A.; Tsimanis A.; Rosen O.; Naider F.; Anglister J. (2013) FEBS J. 280, 2068–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan R. C.; Mohlin F.; Taleski D.; Coetzer T. H.; Huntington J. A.; Payne R. J.; Blom A. M.; Pike R. N.; Wijeyewickrema L. C. (2012) Identification of a catalytic exosite for complement component C4 on the serine protease domain of C1s. J. Immunol. 189, 2365–2373. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.