

Abstract
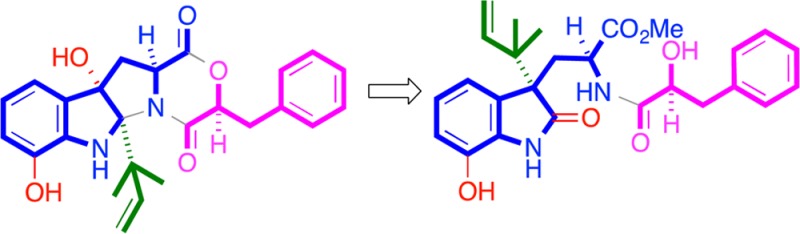
Chemical analysis of an Australian marine sediment-derived Aspergillus sp. (CMB-M081F) yielded the new diketomorpholine (DKM) shornephine A (1) together with two known and one new diketopiperazine (DKP), 15b-β-hydroxy-5-N-acetyladreemin (2), 5-N-acetyladreemin (3), and 15b-β-methoxy-5-N-acetyladreemin (4), respectively. Structure elucidation of 1–4 was achieved by detailed spectroscopic analysis, supported by chemical degradation and derivatization, and biosynthetic considerations. The DKM (1) underwent a facile (auto) acid-mediated methanolysis to yield seco-shornephine A methyl ester (1a). Our mechanistic explanation of this transformation prompted us to demonstrate that the acid-labile and solvolytically unstable DKM scaffold can be stabilized by N-alkylation. Furthermore, we demonstrate that at 20 μM shornephine A (1) is a noncytotoxic inhibitor of P-glycoprotein-mediated drug efflux in multidrug-resistant human colon cancer cells.
Introduction
Diketopiperazines (DKPs) are well represented among natural products, particularly fungal metabolites, with structural diversity extending across fused heterocycles, prenylation, polythio-bridging, dimerization, nitration, halogenation, oxidation, and more. Typically viewed as products of nonribosomal peptide synthases,1 the recent discovery of a DKP cyclase2 suggests a more complex biosynthetic landscape. Indeed, the diverse structural and biological properties exhibited by natural DKPs have attracted much attention, encouraging efforts to probe biosynthetic pathways, develop innovative syntheses, and apply DKPs in human and animal health.3 The 2010 release by Pfizer Animal Health of a new class of anthelmintic inspired by the Penicillium DKP paraherquamide (6) (the first in over two decades) is illustrative of this potential.4 By contrast, reports of the closely related diketomorpholine (DKM) motif are rare, with accounts of the DKM scaffold being rare across natural products, synthetic, and medicinal chemistry.
This report describes our investigation of an Australian marine sediment-derived Aspergillus sp. (CMB-M081F), leading to the isolation of a new DKM, shornephine A (1), plus its methanolysis product seco-shornephine A methyl ester (1a), and three biosynthetically related DKPs, 15b-β-hydroxy-5-N-acetyladreemin (2), 5-N-acetyladreemin (3), and 15b-β-methoxy-5-N-acetyladreemin (4) (Figure 1). Structures were assigned to 1–4 on the basis of detailed spectroscopic analysis, chemical degradation and derivatization, and consideration of biosynthetic relationships. To explore the mechanism behind the solvolysis of 1, we reviewed all known DKM natural products (7–16) (Figure 5), and assessed the chemical stability of a range of synthetic DKMs (17–26) (Figure 6). We also investigated the biological properties of 1 against prokaryotic, eukaryotic, and mammalian cells.
Figure 1.

Aspergillus sp. (CMB-M081F) metabolites 1–4 and methanolysis product 1a.
Figure 5.

Known DKM natural products 7–16.
Figure 6.

Synthetic DKMs 17–26: (a) HBTU, DIPEA, DMF, rt, h; (b) p-TsOH, toluene, microwave 140 °C, 300 W, 3 min.
Results and Discussion
Aspergillus sp. (CMB-M081F), isolated from a marine sediment collected in 2007 at an intertidal depth of 1 m near Shorncliffe, Queensland, Australia, was noteworthy in that cultivations were especially slow at producing secondary metabolites. For example, HPLC-DAD-ESIMS analysis of EtOAc extracts derived from 2-week agar plate cultivations failed to detect any secondary metabolites, with peaks corresponding to 1–4 only appearing post 5-weeks (Supporting Information, Figure S1). To investigate 1–4 further, we subjected a scaled up (15 plate) cultivation to solvent extraction, partitioning, trituration, and C8 reversed-phase HPLC (H2O/MeOH) fractionation, to yield 1a and 2–4, with the unexpected isolation of 1a (m/z 489) rather than 1 (m/z 457) being attributed to methanolysis during fractionation. Detailed spectroscopic analysis readily identified 2 (C28H28N4O4, Δmmu +1.3) and 3 (C28H28N4O3, Δmmu −0.4) as the known DKP fungal metabolites 15b-β-hydroxy-5-N-acetylardeemin5 and 5-N-acetylardeemin,6 respectively, with 4 (C29H30N4O4, Δmmu +0.4) being identified as the new homologue 15b-β-methoxy-5-N-acetylardeemin (Supporting Information, Figures S10–S15). Supportive of this latter assignment, the NMR (CDCl3) data for 4 exhibited resonances for a 15b-β-OMe moiety (δH 2.70; δC 52.1), positioned by HMBC correlations from the OMe to C-15β, and ROESY correlations from the OMe to H-16α and H-17. A detailed account of the structure elucidation, chemistry, and biology of 1 and its methanolysis product 1a is presented below.
HRESI(+)MS analysis of 1a returned an adduct ion ([M + Na]+) consistent with a molecular formula (C26H30N2O6, Δmmu +0.6) requiring 13 double-bond equivalents (DBE). The 13C NMR (DMSO-d6) data for 1a (Table 1) revealed resonances for three ester/amide carbonyls (δC 172.5, 173.5 and 179.7) and 14 sp2 carbons (δC 113.8 to 143.5), accounting for 10 DBE and requiring that 1a be tricyclic. Further analysis of the NMR data revealed COSY correlations diagnostic for five isolated spin systems (Figure 2) and 1H NMR resonances indicative of a methyl ester (δH 3.38) and two isolated deshielded exchangeable protons (δH 9.50 and 10.20). A series of HMBC correlations permitted assembly of the complete planar structure for 1a as shown (Figure 2). On the basis of this assignment, we hypothesized that 1a was a methanolysis artifact of solvolytically unstable DKM natural product (i.e., shornephine A (1)).
Table 1. 1H NMR (600 MHz) and 13C (150 MHz) Data of Shornephine A (1) and seco-Shornephine A Methyl Ester (1a).
| pos | δH, mult (J in Hz) for 1 in CDCl3 | δCa | pos | δH, mult (J in Hz) for 1a in DMSO | δCa |
|---|---|---|---|---|---|
| 1 | 167.7 | 1 | 172.5 | ||
| 2 | 4.32, d (11.1) | 57.3 | 1-OMe | 3.38, s | 52.1 |
| 3 | a 3.26, d (13.7) | 36.9 | 2 | 3.79, ddd (9.7, 8.0, 5.4) | 49.8 |
| b 2.81, dd (13.7, 11.1) | 2-NH | 7.35, d (8.0) | |||
| 4 | 88.5 | 3 | a 2.32, dd (14.2, 5.4) | 32.8 | |
| 4-OH | 2.07, br s | b 2.24, dd (14.2, 9.7) | |||
| 5 | 131.5 | 4 | 55.8 | ||
| 6 | 6.91, d (7.7) | 117.1 | 5 | 130.0 | |
| 7 | 6.69b, m | 117.3e | 6 | 6.56, d (7.4) | 117.0 |
| 8 | 6.67b, m | 121.2e | 7 | 6.78, dd (8.0, 7.4) | 121.4 |
| 6 | 6.91, d (7.7) | 117.1 | 8 | 6.70, d (8.0) | 115.6 |
| 9 | 141.4 | 9 | 141.5 | ||
| 9-OH | d | 9-OH | 9.50, br s | ||
| 10 | 135.9 | 10 | 131.0 | ||
| 11-NH | 6.34, s | 11-NH | 10.20, br s | ||
| 12 | 94.9 | 12 | 179.7 | ||
| 13 | 44.9 | 13 | 42.3 | ||
| 14 | 6.39, dd (17.3, 10.6) | 144.1 | 14 | 6.04, dd (17.4, 10.8) | 143.5 |
| 15 | a 5.18, d (17.3) | 113.1 | 15 | a 5.07, dd (10.8, 0.6) | 113.8 |
| b 5.11, d (10.6) | b 4.99, dd (17.4, 0.6) | ||||
| 16 | 1.38, s | 22.9 | 16 | 1.01, s | 22.1 |
| 17 | 1.38, s | 25.8 | 17 | 0.93, s | 21.8 |
| 1′ | 165.9 | 1′ | 173.5 | ||
| 2′ | 4.76, dd (8.8, 1.6) | 78.5 | 2′ | 3.90, dd (9.6, 3.3) | 72.5 |
| 3′ | a 3.32, d (15.1) | 34.4 | 2′-OH | 5.53, br s | |
| b 2.92, dd (15.1, 8.8) | 3′ | a 2.79, dd (13.8, 3.3) | 40.4 | ||
| b 2.61, dd (13.8, 9.6) | |||||
| 4′ | 136.2 | 4′ | 138.8 | ||
| 5′/9′ | 7.20b | 126.7f | 5′/9′ | 7.22, d (7.2) | 129.7 |
| 6′/8′ | 7.20b | 129.3f | 6′/8′ | 7.26, ddd (7.2, 7.2, 0.6) | 128.2 |
| 7′ | 7.20b | 128.5 | 7′ | 7.18, td (7.2, 0.6) | 126.3 |
13C NMR assignments supported by gHSQC and gHMBC data.
Overlapping signals.
Not observed.
Assignments are interchangeable.
Figure 2.

Diagnostic 2D NMR correlations for 1 and 1a.
To test this hypothesis, we repeated the cultivation and employed an alternative isolation strategy, involving sequential trituration with hexane and CH2Cl2, followed by HPLC with an H2O/MeCN gradient, to yield 1−4. HRESI(+)MS analysis of 1 returned an adduct ion ([M + Na]+, C25H26N2O5, Δmmu −0.3) consistent with a DKM, while 1D NMR (CDCl3) data (Table 1) confirmed replacement of the methyl ester and 2′-H hydroxyl methine resonances in 1a, with a deshielded 2′-H lactone methine (δH 4.76) in 1. As predicted, exposure of 1 to MeOH resulted in methanolysis to 1a. Further examination of the 2D NMR data for 1 revealed an ABC heterocyclic ring system similar to that exhibited by cometabolite ardeemins 2–4, but bearing 4-OH, 9-OH, and a C-12 reverse isoprene moiety. Diagnostic ROESY correlations positioned H-2, H-2′, H-3α, 4-OH, H3-16, and H3-17 on the same (α) face of the heterocyclic scaffold and defined the relative configuration for 1 (Figure 2). Analytical acid hydrolysis of 1 (50 μg) followed by esterification of the hydrolysate with (R)-α-methoxy-α-(trifluoromethyl)phenylacetic acid (R-Mosher acid) returned an ester identical with that obtained from authentic (S)-phenyllactic acid (Supporting Information, Figure S2). The observations outlined above confirmed the structures for 1 and 1a as indicated (Figure 1). As the mechanism behind the methanolysis of 1 to 1a was not immediately obvious, we elected to explore this matter further. We hypothesize (Figure 3) that 1 undergoes an auto (C-9 phenol) acid-catalyzed dehydration to yield a C-4 carbocation, which engages in a 1,2-sigmatropic rearrangement and H2O addition to yield seco-shornephine A (1b). Our failure to detect 1b suggests that this intermediate is solvolytically very unstable and rapidly transforms to the methyl ester 1a. Curiously, 1 appeared to be stable to direct solvolysis as no trace of the methanolysis product 1c could be detected (Figure 3).
Figure 3.

Proposed mechanism for methanolysis of 1.
This mechanistic proposal is interesting on two levels. First, it highlights a simple nonenzymatic transformation that could have implications in our understanding of the biosynthesis (and biomimetic synthesis) of DKPs such as notoamide C (5),7 itself biosynthetically related to paraherquamide (6)4 (Figure 4), a topic that has attracted much recent attention.2,8−10 Second, it suggests that with suitable functionalization (e.g., 1), the DKM moiety can be rendered stable to direct solvolysis, which has implications for the possible use of DKMs in drug discovery.
Figure 4.

Biosynthetic relationship between notoamide C (5) and paraherquamide (6).
To explore this latter issue further, we reviewed the literature on known DKM natural products, noting it was limited to the fungal metabolites lateritin (7) from Gibberella lateritium,(11) bassiatin (8) from Beauveria bassiana (K-717),12 three homologous DKMs 9–11 from Fusarium sporotrichioides,(13) the DKM 12 from the Thai Sea hare Bursatella leachii,(14) the fungal mollenines A and B (13 and 14) from Eupenicillium molle,15 and javanicunines A and B (15 and 16) from Eupenicillium javanicum(16) (Figure 5). On close examination, we noted that 7–16 were all N-alkylated, and none were solvolytically unstable. Consistent with these observations, while shornephine A (1) is N-alkylated and stable to direct solvolysis (i.e., does not form 1c), the seco form 1b is not N-alkylated and is rapidly transformed into 1a. To test the hypothesis that solvolytically unstable DKMs can be stabilized by N-alkylation, we prepared the synthetic DKMs 17–24 and the N-methylated analogues 25 and 26. As predicted, on exposure to MeOH, 17–24 underwent rapid (t1/2 < 10 min) methanolysis, while 25 and 26 proved stable even after 48 h.
The Aspergillus sp. (CMB-M081F) metabolites 1–4 and the methanolysis artifact 1a were not cytotoxic (IC50 > 30 μM) against Gram-negative bacteria Escherichia coli (ATCC 11775) and Pseudomonas aeruginosa (ATCC 10145), Gram-positive bacteria Staphylococcus aureus (ATCC 9144 and ATCC 25923) and Bacillus subtilis (ATCC 6633 and ATCC 6051), the fungus Candida albicans (ATCC 90028), or human colon (SW620 and SW620 Ad300) or cervical (KB-3–1 and KB-V1) cancer cell lines (Supporting Information, Figures S36 and S38).
Systemic administration of chemotherapeutic agents (anticancer drugs) is often used for the treatment of human cancers. While clinically successful, this mode of treatment is compromised by multidrug resistant (MDR) cancers that exhibit either high intrinsic or acquired resistance to multiple chemotherapeutic agents. Factors that contribute to MDR include overexpression of membrane spanning adenosine triphosphate binding cassette (ABC) transporter proteins such as P-glycoprotein (P-gp) and associated accelerated drug efflux. Although P-gp inhibitors offer the prospect of reversing the MDR phenotype, no P-gp inhibitors have yet advanced to the clinic.17 To evaluate the P-gp inhibitory properties of 1 and related synthetic DKMs we employed a Calcein AM assay. In this assay, a nonfluorescent reagent calcein AM diffuses into the cellular cytoplasm of P-gp overexpressing human colon cancer (SW620 Ad300) cells, where it undergoes hydrolysis to yield the fluorescent dye calcein. Significantly, calcein AM is a P-gp substrate and the hydrolyzed product calcein is not. In response to functioning P-gp, calcein AM is effluxed prior to hydrolysis, leading to reduced intracellular fluorescence. In the presence of a P-gp inhibitor, calcein AM efflux is blocked and calcein AM undergoes hydrolysis to calcein, leading to increased intracellular fluorescence. Intracellular calcein fluorescence is quantified by cell flow cytometry to arrive at a fluorescence arbitrary ratio (FAR), which measures intracellular calcein fluorescence in cells exposed to a putative P-gp inhibitor, with that from cells not exposed to an inhibitor. The larger the FAR value then (in principle) the more effective the P-gp inhibitor.17 Importantly, all test DKMs were determined to be stable to solvolysis for the 45 min duration of the calcein AM assay. Using this approach we established the P-gp inhibitory properties of 1 (FAR 35.5), 19 (FAR 52.9), 23 (FAR 41.5) and 24 (FAR 40.5) (positive control verapamil FAR = 72.5) (Supporting Information, Figure S37). This observation raises the prospect that the hitherto largely overlooked DKM scaffold may be engineered to deliver a clinically useful inhibitor of P-gp mediated drug efflux. If achieved, such an outcome would greatly improve the prognosis for MDR cancer chemotherapy.
Experimental Section
Collection and Isolation of Aspergillus sp. (CMB-M081F)
Strain CMB-M081F was isolated from marine sediment collected collected in 2007 at an intertidal depth of 1 m near Shorncliffe, Queensland, Australia. The freshly collected sediment was sealed in a Falcon tube (50 mL) and transferred at rt to the laboratory, where it was stored in the dark at −30 °C for 1 week. A sample (1 g) was thawed, suspended in sterile 0.9% saline (8 mL), and subjected to heat-shock (60 °C for 30 min), and an aliquot (100 μL) was used to prepare three 10-fold serial dilutions. Aliquots (50 μL) from the saline solution and serial dilutions were applied to M1 agar plates (comprising 2% agar in artificial ocean sea salt (3.3%; 25 mL), starch (1%), yeast extract (0.4%), peptone (0.2%), and rifampicin (0.0005%) and incubated at 27 °C for 4–5 weeks. Pure strains of individual bacterial and fungal colonies, including CMB-M081F, were obtained by standard microbiological techniques and were grown to dense colonies on single agar plates. Taxonomic analysis identified CMB-M081F as an Aspergillus sp. (Supporting Information, section 1.1).
Analytical Cultivation and Chemical Analysis of Aspergillus sp. (CMB-M081F)
A single colony of Aspergillus sp. (CMB-M081F) applied to an M1 agar plate was incubated at 27 °C for 5 weeks, after which the agar was diced and extracted with EtOAc (100 mL), and the organic phase was concentrated in vacuo. The extract (5.6 mg) was analyzed by HPLC-DAD-ESI(±)MS (Zorbax SB-C8 5 μm 150 × 4.6 mm column, 1 mL/min gradient elution from 90% H2O:MeCN to 100% MeCN over 15 min with isocratic 0.05% HCO2H modifier) to reveal noteworthy peaks at 11.1 min (m/z 435 [M + H]+, 1), 12.2 min (m/z 485 [M + H]+, 2), 12.4 min (m/z 469 [M + H]+, 3), and 12.8 min (m/z 499 [M + H]+, 4) (Supporting Information, Figure S1).
Preparative Cultivation and Fractionation of Aspergillus sp. (CMB-M081F)
Method 1: Fifteen M1 agar plates were cultivated and processed as described above to yield an extract (83.7 mg) that was sequentially triturated (10 mL aliquots) to yield hexane (33.8 mg), CH2Cl2 (25.9 mg) and MeOH (7.8 mg) soluble fractions. The CH2Cl2 solubles were fractionated by semipreparative reversed-phase HPLC (Zorbax SB-C8 5 μm 250 × 9.4 mm column, 3 mL/min gradient elution from 90% H2O/MeOH to 100% MeOH over 30 min) to yield seco-shornephine A methyl ester (1a) (tR = 23.9 min, 1.5 mg, 1.8%), 15b-β-hydroxy-5-N-acetylardeemin (2) (tR = 25.5 min, 1.9 mg, 2.2%), 5-N-acetylardeemin (3) (tR = 27.0 min, 2.1 mg, 2.5%), and 15b-β-methoxy-5-N-acetylardeemin (4) (tR = 28.0 min, 2.5 mg, 3.0%). Method 2: Ten M1 agar plates were cultivated and processed as described above to yield an extract (60.2 mg) that was sequentially partitioned into hexane (2 mg) and CH2Cl2 (60 mg) soluble fractions. The CH2Cl2 solubles were fractionated by semipreparative reversed-phase HPLC (Zorbax SB-C8 5 μm column 250 × 9.4 mm column, 3 mL/min gradient elution from 90% H2O/MeCN to 100% MeCN over 30 min) to yield shornephine A (1) (tR = 23.9 min, 2.0 mg, 1.8%), 15b-β-hydroxy-5-N-acetylardeemin (2) (tR = 25.5 min, 1.5 mg, 2.2%), 5-N-acetylardeemin (3) (tR = 27.0 min, 1.9 mg, 2.5%), and 15b-β-methoxy-5-N-acetylardeemin (4) (tR = 28.0 min, 1.5 mg, 3.0%). (Note: All % yields were determined on a mass-to-mass measure against the crude EtOAc extract.)
Characterization of Aspergillus sp. (CMB-M081F) Metabolites and Solvolysis Products
Shornephine A (1):
pale yellow oil; [α]D22 +22 (c 0.05, CHCl3); UV (MeCN) λmax (log ε): 210 (4.54), 242 (3.79), 301 (3.37) nm; NMR (CDCl3) see Table 1 and Supporting Information Table S1 and Figures S6 and S7; HRMS(ESI-TOF) m/z [M + Na]+ calcd for C25H26N2O5Na+ 457.1734, found 457.1731.
seco-Shornephine A methyl ester (1a):
pale yellow oil; [α]D23 −73 (c 0.02, CHCl3); UV (MeCN) λmax (log ε): 218 (3.94), 256 (3.75), 310 (3.59) nm; NMR (DMSO-d6) see Table 1 and Supporting Information Table S2 and Figures S8–S9; HRMS(ESI-TOF) m/z [M + Na]+ calcd for C26H30N2O6Na+ 489.2009, found 489.2015.
15b-β-Hydroxy-5-N-acetyladreemin (2):5,6
pale yellow oil; [α]D23 −19 (c 0.05, MeOH); NMR (CDCl3) see Supporting Information Table S3 and Figures S10–S11; HRMS(ESI-TOF) m/z [M + Na]+ calcd for C28H28N4O4Na+ 507.2003, found 507.2016.
5-N-Acetyladreemin (3):5,6
pale yellow oil; [α]D23 −21 (c 0.05, MeOH); NMR (CDCl3) see Supporting Information Table S4 and Figures S12–S13; HRMS(ESI-TOF) m/z: [M + Na]+ calcd for C28H28N4O3Na+ 491.2054, found 491.2050.
15b-β-Methoxy-5-N-acetyladreemin (4):
pale yellow oil; [α]D23 −16 (c 0.05, MeOH); UV (MeCN) λmax (log ε) 219 (3.97), 259 (3.78), 307 (3.62) nm; NMR (CDCl3) see Supporting Information Table S5 and Figures S14–S15; HRMS(ESI-TOF) m/z [M + Na]+ calcd for C29H30N4O4Na+ 521.2159, found 521.2163.
Synthesis of DKMs 17–26
The DKMs 17–26 were all prepared using a common two-step method. Step 1: An amino acid methyl ester (1 equiv) was treated with HBTU (1.2 equiv) and DIPEA (2.8 equiv) in the presence of either (S)-phenyllactic acid (1 equiv) or (S)-mandelic acid (1 equiv), in anhydrous DMF (5 mL). The resulting reaction mixture was stirred at rt for 3 h under argon, concentrated in vacuo, and partitioned between EtOAc (2 × 50 mL) and 1 M HCl (50 mL), and the combined organic layers were dried with anhydrous MgSO4 and concentrated in vacuo. Step 2: The amide product from step 1 (1 equiv) was treated with p-TsOH (1.5 equiv) in anhydrous toluene (5 mL), heated in a microwave reactor at 140 °C/300 W for 3 min, and concentrated in vacuo, and the residue was purified by C8 reversed-phase HPLC (H2O/MeCN). Overall yields: 17 (75%), 18 (72%), 19 (75%), 20 (75%), 21 (65%), 22 (65%), 23 (62%), 24 (69%), 25 (50%), and 26 (41%).
Characterization of Synthetic DKMs 17–26
cyclo-(l-Phenylalanine-l-mandelic acid) (17):
colorless oil (3.0 mg, 75%); [α]D23 −74 (c 0.04, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.39 (m, H-4′/8′), 7.37–7.42 (m, H-5′/6′/7′), 7.31 (m, H-6/8), 7.27 (m, H-7), 7.07 (d, J = 7.1 Hz, H-5/9), 5.83 (s, H-2′), 4.41 (br d, J = 11.4 Hz, H-2), 3.29 (dd, J = 13.9, 3.1 Hz, H-3a), 2.62 (dd, J = 13.9, 11.4 Hz, H-3b); 13C NMR (150 MHz, CDCl3) δ 166.2 (C-1′), 134.7 (C-4), 134.1 (C-3′), 129.8 (C-5/6/8/9), 129.6 (C-5′/6′/7′), 128.4 (C-7), 127.1 (C-4′/8′), 79.7 (C-2′), 55.4 (C-2), 39.3 (C-3); HRMS(ESI-TOF) m/z [M + H]+ calcd for C17H16NO3+ 282.1125, found 282.1117.
cyclo-(l-Phenylalanine-l-phenyllactic acid) (18):
white solid (2.5 mg, 72%); [α]D23 −446 (c 0.05, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.31 (d, J = 7.3 Hz, H-5′/9′), 7.18–7.27 (m, H-6/7/8/6′/7′/8′), 6.93 (d, J = 7.1 Hz, H-5/9), 5.67 (br s, 2-NH), 5.02 (dd, J = 5.3, 4.1 Hz, H-2′), 4.11 (br d, J = 11.0 Hz, H-2), 3.20 (dd, J = 14.3, 4.1 Hz, H-3′a), 3.08 (dd, J = 14.3, 5.3 Hz, H-3′b), 3.02 (dd, J = 13.9, 3.4 Hz, H-3a), 1.58 (dd, J = 13.9, 11.0 Hz, H-3b); 13C NMR (150 MHz, CDCl3) δ 167.1 (C-1), 166.3 (C-1′), 135.4 (C-4′), 135.1 (C-4), 128.8 (C-5/9), 128.3 (C-5′/9′), 127.8–130.8 (C-6/7/8/6′/7′/8′), 78.1 (C-2′), 54.7 (C-2), 39.0 (C-3), 38.1 (C-3′); HRMS(ESI-TOF) m/z [M + H]+ calcd for C18H18NO3+ 296.1281, found 296.1277.
cyclo-(L-Tryptophan-l-mandelic acid) (19):
colorless oil (4.0 mg, 75%); [α]D23 −5 (c 0.14, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.12 (br s, H-9), 7.47 (d, J = 8.4 Hz, H-10), 7.04–7.47 (m, H-5/6/7/8/5′/6′/7′), 5.77 (s, H-2′), 4.41 (br d, J = 10.5 Hz, H-2), 3.47 (dd, J = 14.8, 3.4 Hz, H-3a), 3.24 (br s, 2-NH), 2.87 (dd, J = 14.8, 10.5 Hz, H-3b); 13C NMR (150 MHz, CDCl3) δ 167.1 (C-1′), 166.2 (C-1), 136.6 (C-8a), 134.5 (C-3′), 129.5 (C-5′/7′), 129.0 (C-6′), 127.1 (C-4a), 127.1 (C-4′/8′), 126.8 (C-5), 126.7 (C-6), 124.0 (C-7), 118.7 (C-10), 111.7 (C-8), 108.8 (C-4), 79.5 (C-2′), 54.7 (C-2), 29.4 (C-3); HRMS(ESI-TOF) m/z [M + Na]+ calcd for C19H16N2O3Na+ 343.1053, found 343.1055.
cyclo-(L-Tryptophan-l-phenyllactic acid) (20):
colorless oil (4.0 mg, 75%); [α]D23 −76 (c 0.05, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.12 (br s, H-9), 7.48 (d, J = 7.6 Hz, H-5), 7.35 (d, J = 7.3 Hz, H-8), 7.35 (t, J = 7.3 Hz, H-6′/8′), 7.28 (m, H-5′/9′), 7.26 (m, H-7′), 7.23 (dd, J = 7.6, 7.3 Hz, H-7), 7.14 (dd, J = 7.6, 7.6 Hz, H-6), 6.90 (br s, H-10), 5.67 (br s, 2-NH), 5.05 (dd, J = 5.3, 4.0 Hz, H-2′), 4.26 (br d, J = 10.1 Hz, H-2), 3.31 (m, H-3a), 3.28 (m, H-3b), 3.27 (dd, J = 14.4, 4.0 Hz, H-3′a), 3.15 (dd, J = 14.4, 5.3 Hz, H-3′b); 13C NMR (150 MHz, CDCl3) δ 166.8 (C-1), 166.3 (C-1′), 137.1 (C-8a), 135.1 (C-4′), 131.3 (C-5′/9′), 128.6 (C-7′), 126.8 (C-4a), 126.8 (C-6′/8′), 124.0 (C-10), 123.6 (C-7), 120.6 (C-6), 119.0 (C-5), 112.5 (C-8), 109.4 (C-4), 79.6 (C-2′), 53.6 (C-2), 38.6 (C-3′), 30.1 (C-3); HRMS(ESI-TOF) m/z [M + H]+ calcd for C20H19N2O3+ 335.1390, found 335.1376.
cyclo-(l-Alanine-l-mandelic acid) (21):
colorless oil (2.5 mg, 62%); [α]D23 +29 (c 0.05, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.38 (m, H-4′/8′), 7.28–7.35 (m, H-5′/6′/7′), 5.11 (s, H-2′), 4.50 (m, H-2), 3.42 (br s, 2-NH), 1.48 (d, J = 6.8 Hz, H-3); 13C NMR (150 MHz, CDCl3) δ 175.8 (C-1), 173.8 (C-1′), 138.9 (C-3′), 126.8 (C-4′/8′), 126.1–130.2 (C-5′/6′/7′), 75.7 (C-2′), 49.7 (C-2), 19.1 (C-3); HRMS(ESI-TOF) m/z [M + Na]+ calcd for C11H11NO3Na+ 228.0631, found 228.0633.
cyclo-(l-Alanine-l-phenyllactic acid) (22):
colorless oil (3.1 mg, 69%); [α]D23 +85 (c 0.05, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.19–7.31 (m, H-5′/6′/7′/8′/9′), 4.45 (br s, H-2), 4.29 (br s, H-2′), 4.22 (br s, 2-NH), 3.14 (d, J = 13.3 Hz, H-3′a), 2.85 (dd, J = 13.3, 7.6 Hz, H-3′b), 1.33 (br s, H-3); 13C NMR (150 MHz, CDCl3) δ 174.7 (C-1), 174.5 (C-1′), 136.9 (C-4′), 127.2–129.3 (C-5′/6′/7′/8′/9′), 72.8 (C-2), 50.2 (C-2′), 40.5 (C-3′), 17.8 (C-3); HRMS(ESI-TOF) m/z [M + Na]+ calcd for C12H13NO3Na+ 242.0788, found 242.0790.
cyclo-(l-Tyrosine-l-mandelic acid) (23):
colorless oil (2.5 mg, 65%); [α]D23 +2 (c 0.2, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.38–7.41 (m, H-4′/5′/6′/7′/8′), 6.95 (d, J = 8.1 Hz, H-6/8), 6.76 (d, J = 8.1 Hz, H-5/9), 5.82 (s, H-2′), 5.78 (br s, 7-OH), 4.76 (br s, 2-NH), 4.36 (br d, J = 10.4 Hz, H-2), 3.21 (dd, J = 14.0, 3.4 Hz, H-3a), 2.57 (dd, J = 14.0, 10.4 Hz, H-3b); 13C NMR (150 MHz, CDCl3) δ 131.1 (C-6/8), 129.9 (C-5′/7′), 129.7 (C-6′), 127.0 (C-4′/8′), 116.7 (C-5/9), 80.6 (C-2′), 55.4 (C-2), 38.8 (C-3); HRMS(ESI-TOF) m/z [M + Na]+ calcd for C17H15N1O4Na+ 320.0893, found 320.0895.
cyclo-(l-Tyrosine-l-phenyllactic acid) (24):
colorless oil (3.1 mg, 65%); [α]D23 −89 (c 0.1, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.31 (t, J = 7.5 Hz, H-5′/9′), 7.23 (m, H-6′/8′), 7.23 (m, H-7′), 6.75 (d, J = 8.2 Hz, H-6/8), 6.67 (d, J = 8.2 Hz, H-5/9), 6.05 (s, 2-NH), 5.03 (dd, J = 5.0, 4.6 Hz, H-2), 4.01 (br d, J = 10.5 Hz, H-2′), 3.20 (dd, J = 14.5, 4.6 Hz, H-3a), 3.09 (dd, J = 14.5, 5.0 Hz, H-3b), 2.90 (dd, J = 14.1, 3.4 Hz, H-3′a), 1.43 (dd, J = 14.1, 10.5 Hz, H-3′b); 13C NMR (150 MHz, CDCl3) δ 170.6 (C-1′), 170.0 (C-1), 155.5 (C-7), 134.6 (C-4), 134.2 (C-4′), 131.1 (C-6/8), 130.6 (C-7′), 129.2 (C-5′/9′), 128.2 (C-6′/8′), 116.5 (C-5/9), 78.7 (C-2), 55.4 (C-2′), 38.6 (C-3′), 38.2 (C-3); HRMS(ESI-TOF) m/z [M + Na]+ calcd for C18H17N1O4Na+ 334.1050, found 334.1044.
cyclo-(N-Methyl-l-tyrosine-l-mandelic acid) (25):
colorless oil (0.9 mg, 41%); [α]D23 +38 (c 0.05, CHCl3); 1H NMR (600 MHz, DMSO-d6) δ 7.23 (m, H-5′/6′/7′), 7.05 (d, J = 7.4 Hz, H-4′/8′), 6.84 (d, J = 8.1 Hz, H-6/8), 6.55 (d, J = 8.1 Hz, H-5/9), 5.26 (s, H-2′), 4.43 (dd, J = 10.1, 5.3 Hz, H-2), 2.95 (dd, J = 14.7, 5.3 Hz, H-3a), 2.64 (dd, J = 14.7, 10.1 Hz, H-3b), 2.60 (s, 2-Me); 13C NMR (150 MHz, DMSO-d6) δ 172.2 (C-1′), 172.0 (C-1), 155.3 (C-7), 141.4 (C-3′), 129.8 (C-4), 129.7 (C-6/8), 127.4 (C-4′/8′), 126.7–128.2 (C-5′/6′/7′), 115.1 (C-5/9), 70.5 (C-2′), 63.5 (C-2), 34.8 (C-3), 28.6 (2-Me); HRMS(ESI-TOF) m/z [M + Na]+ calcd for C18H17NO4Na+ 334.1050, found 334.1059.
cyclo-(N-methyl-l-tyrosine-l-phenyllactic acid) (26):
colorless oil (1.1 mg, 50%); [α]D23 +37 (c 0.05, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.29 (m, H-7′), 7.19 (m, H-6′/8′), 7.04 (d, J = 7.6 Hz, H-5′/9′), 6.97 (d, J = 8.3 Hz, H-6/8), 6.81 (d, J = 8.3 Hz, H-5/9), 5.23 (br s, 7-OH), 4.83 (dd, J = 9.3, 3.0 Hz, H-2′), 4.28 (dd, J = 4.7, 4.7 Hz, H-2), 2.96 (s, 2-NMe), 2.92 (dd, J = 14.7, 4.7 Hz, H-3a), 2.87 (dd, J = 14.5, 3.0 Hz, H-3′a), 2.79 (dd, J = 14.7, 4.7 Hz, H-3b), 1.70 (dd, J = 14.5, 9.3 Hz, H-3′b); 13C NMR (150 MHz, CDCl3) δ 164.8 (C-1′), 164.0 (C-1), 155.3 (C-7), 135.3 (C-4′), 131.0 (C-6/8), 129.2 (C-5′/9′), 128.3 (C-7′), 126.9 (C-6′/8′), 126.3 (C-4), 115.8 (C-5/9), 79.5 (C-2′), 61.7 (C-2), 39.4 (C-3′), 36.7 (C-3), 32.7 (2-Me); HRMS(ESI-TOF) m/z [M + Na]+ calcd for C19H19NO4Na+ 348.1206, found 348.1208.
Methanolysis of Shornephine A (1)
Solutions of 1 (100 μg in 100 μL MeOH) were stirred at rt for 30 min, 8 and 24 h, respectively, after which they were analyzed by HPLC-DAD-ESIMS (Zorbax SB-C8 5 μm 150 × 4.6 mm column, 1.0 mL/min, gradient elution 90% H2O:MeCN to 100% MeCN over 15 min with isocratic 0.05% HCO2H modifier) (Supporting Information, Figure S3).
Mosher Ester Analysis of Shornephine A (1)
A solution of 1 (50 μg) in 6 M HCl (200 μL) was stirred overnight in a sealed vial at 110 °C, after which it was evaporated to dryness under N2 at 40 °C. Diastereomeric Mosher ester derivatives were prepared for both the hydrolysate of 1, and an authentic sample of (S)-phenyllactic acid, according to protocol of Hoye et al.18 Briefly, samples of analyte in dry CH2Cl2 (150 μL) were treated with 3 eq. of either (R)-α-methoxy-α-trifluoromethylphenylacetic acid [R-MTPA-OH] or (S)-α-methoxy-α-(trifluoromethyl)phenylacetic acid [S-MTPA-OH] in dry CH2Cl2 (150 μL), followed by 3 equiv of dicyclohexylcarbodiimide (DCC) and 4-(dimethylamino)pyridine (DMAP) in dry CH2Cl2 (200 μL). The resulting mixtures were stirred at rt overnight, after which they were filtered, dried under N2 at 40 °C, redissolved in MeOH (100 μL), and analyzed by HPLC-ESIMS (Zorbax SB-C8 5 μm 150 × 4.6 mm column, 1.0 mL/min, gradient elution from 90% H2O/MeCN to 100% MeCN over 15 min followed by a 5 min hold at 100% MeCN, with isocratic 0.05% HCO2H modifier) (Supporting Information, Figure S2).
Methanolysis of DKMs 7–26
Solutions of 7–26 (100 μg in 100 μL of MeOH) were treated as described above for the methanolysis of 1 (Supporting Information, Figures S4 and S5).
Acknowledgments
We thank S. Bates and R. Robey (NCE, NIH) for providing SW620 and SW620 Ad300 cell lines and M. M. Gottesman (NIH) for providing KB-3-1 and KB-V1 cell lines. Z.G.K. and X.-C.H. acknowledge the provision of UQ International Postgraduate Scholarships, and R.R. acknowledges the provision of an Australian Postgraduate Award. This research was funded in part by the Institute for Molecular Bioscience, The University of Queensland, and the Australian Research Council (LP120100088).
Supporting Information Available
Full tabulated 2D NMR data including HSQC, HMBC, gCOSY, gROESY, and 1H and 13C NMR spectra, HPLC chromatograms, taxonomic analysis, and bioassay data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† Department of Chemistry & Biomolecular Sciences, Macquarie University, Sydney, NSW 2109, Australia.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Mootz H. D.; Marahiel M. A. Curr. Opin. Chem. Biol. 1997, 1, 543–551. [DOI] [PubMed] [Google Scholar]
- Finefield J. M.; Sherman D. H.; Tsukamoto S.; Williams R. M. J. Org. Chem. 2011, 76, 5954–5958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimaraes D. O.; Borges W. S.; Vieira N. J.; de Oliveira L. F.; da Silva C. H. T. P.; Lopes N. P.; Dias L. G.; Duran-Patron R.; Collado I. G.; Pupo M. T. Phytochemistry 2010, 71, 1423–1429. [DOI] [PubMed] [Google Scholar]
- Little P. R.; Hodge A.; Watson T. G.; Seed J. A.; Maeder S. J. N. Z. Vet. J. 2010, 58, 121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karwowski J. P.; Jackson M.; Rasmussen R. R.; Humphrey P. E.; Poddig J. B.; Kohl W. L.; Scherr M. H.; Kadam S.; McAlpine J. B. J. Antibiot. 1993, 46, 374–379. [DOI] [PubMed] [Google Scholar]
- Hochlowski J. E.; Mullally M. M.; Spanton S. G.; Whittern D. N.; Hill P.; McAlpine J. B. J. Antibiot. 1993, 46, 380–386. [DOI] [PubMed] [Google Scholar]
- Li S.; Finefield J. M.; Sunderhaus J. D.; McAfoos T. J.; Williams R. M.; Sherman D. H. J. Am. Chem. Soc. 2012, 134, 788–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H.; Nakamura Y.; Finefield J. M.; Umaoka H.; Nakahara T.; Williams R. M.; Tsukamoto S. Tetrahedron Lett. 2011, 52, 6923–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finefield J. M.; Greshock T. J.; Sherman D. H.; Tsukamoto S.; Williams R. M. Tetrahedron Lett. 2011, 52, 1987–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.; Shao C.-L.; Fu X.-M.; Xu R.-F.; Zheng J.-J.; Zhao D.-L.; She Z.-G.; Wang C.-Y. J. Nat. Prod. 2013, 76, 547–553. [DOI] [PubMed] [Google Scholar]
- Hasumi K.; Shinohara C.; Iwanaga T.; Endo A. J. Antibiot. 1993, 46, 1782–1787. [DOI] [PubMed] [Google Scholar]
- Kagamizono T.; Nishino E.; Matsumoto K.; Kawashima A.; Kishimoto M.; Sakai N.; He B.-M.; Chen Z.-X.; Adachi T.; et al. J. Antibiot. 1995, 48, 1407–1412. [DOI] [PubMed] [Google Scholar]
- Smelcerovic A.; Yancheva D.; Cherneva E.; Petronijevic Z.; Lamshoeft M.; Herebian D. J. Mol. Struct. 2011, 985, 397–402. [Google Scholar]
- Suntornchashwej S.; Chaichit N.; Isobe M.; Suwanborirux K. J. Nat. Prod. 2005, 68, 951–955. [DOI] [PubMed] [Google Scholar]
- Wang H.-j.; Gloer J. B.; Wicklow D. T.; Dowd P. F. J. Nat. Prod. 1998, 61, 804–807. [DOI] [PubMed] [Google Scholar]
- Nakadate S.; Nozawa K.; Horie H.; Fujii Y.; Nagai M.; Komai S.-i.; Hosoe T.; Kawai K.-i.; Yaguchi T.; Fukushima K. Heterocycles 2006, 68, 1969–1972. [Google Scholar]
- Huang X.-c.; Sun Y.-L.; Salim A. A.; Chen Z.-S.; Capon R. J. Biochem. Pharmacol. 2013, 85, 1257–1268. [DOI] [PubMed] [Google Scholar]
- Hoye T. R.; Jeffrey C. S.; Shao F. Nat. Protoc. 2007, 2, 2451–2458. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.