

Abstract

Mcl-1 is overexpressed in many cancers and can confer resistance to cell-death signaling in refractory disease. Molecules that specifically inhibit Mcl-1 hold potential for diagnosing and disrupting Mcl-1-dependent cell survival. We selected three peptides from a yeast-surface display library that showed moderate specificity and affinity for binding to Mcl-1 over Bfl-1, Bcl-xL, Bcl-2, and Bcl-w. Specificity for Mcl-1 was improved by introducing threonine at peptide position 2e. The most specific peptide, MS1, bound Mcl-1 with 40-fold or greater specificity over four other human Bcl-2 paralogs. In BH3 profiling assays, MS1 caused depolarization in several human Mcl-1-dependent cell lines with EC50 values of ∼3 μM, contrasted with EC50 values of >100 μM for Bcl-2-, Bcl-xL-, or Bfl-1-dependent cell lines. MS1 is at least 30-fold more potent in this assay than the previously used Mcl-1 targeting reagent NoxaA BH3. These peptides can be used to detect Mcl-1 dependency in cells and provide leads for developing Mcl-1 targeting therapeutics.
Mcl-1 is one of the top 10 most frequently amplified genes in cancers and an important factor in resistance to chemotherapeutic agents.1−3 Despite this, no effective Mcl-1 inhibitors have yet been brought to the clinic. Mcl-1 is a member of the family of anti-apoptotic Bcl-2 homologues, which also includes Bcl-xL, Bcl-2, Bcl-w, Bfl-1, and Bcl-b in humans. These anti-apoptotic Bcl-2 proteins block apoptosis by preventing the pro-apoptotic proteins Bax and Bak from homo-oligomerizing and creating pores in the mitochondrial outer membrane. The anti-apoptotic proteins can either bind directly to Bax and Bak or bind the related pro-apoptotic BH3-only activator proteins (Bim, Bid, and Puma), preventing activation of Bax and Bak. Other BH3-only proteins, called sensitizers, antagonize anti-apoptotic function by binding competitively with Bax/Bak and activators.4
Synthetic reagents that mimic BH3 sensitizers have high therapeutic potential. For example, the small-molecule BH3 mimetic ABT-263, or Navitoclax, is being tested in humans following extremely promising results in mice.5−8 However, ABT-263 only binds Bcl-2, Bcl-xL, and Bcl-w, and cancers that also express Bfl-1 or Mcl-1 show resistance to the drug.9,10 Furthermore, ABT-263 exhibits platelet toxicity, attributed to an inhibitory effect on Bcl-xL.5,6 Identifying more selective molecules that can target the Bcl-2 family member(s) responsible for blocking apoptosis in a given cancer may be a better strategy for differentially killing cancer cells while avoiding off-target effects. In support of this, ABT-199, a selective Bcl-2 inhibitor, has recently exhibited high activity in chronic lymphocytic leukemia patients, with no off-target killing of platelets.11 Several small-molecule Mcl-1 inhibitors have been reported recently.12−17 These compounds, some of which display specificity for Mcl-1 over other Bcl-2 family proteins, have IC50 values in the nanomolar to micromolar range and represent progress toward achieving a tight, Mcl-1-specific inhibitor that can be considered for clinical development.
Another strategy for developing BH3-mimetic molecules is to more directly mimic natural sensitizers by modifying peptides corresponding to the α-helical BH3 regions of known Bcl-2 family members. Peptide inhibitors can have higher affinities and specificities than small molecules, due to their larger interaction interfaces, and it is possible to identify peptides with desired properties by screening large genetically encoded libraries. In the clinical diagnostic setting, BH3 peptides are useful in BH3 profiling assays, which assess similarities between the in vitro binding profiles and mitochondrial permeabilization patterns of different peptides to determine which anti-apoptotic family member a cancer is dependent upon for resistance to apoptosis.18,19 There is a need for peptides that have high affinity and specificity for individual Bcl-2 proteins to enhance the diagnostic power of these types of assays. Finally, there are promising precedents in which helical peptides have been modified to render them cell-penetrating and highly protease-resistant, indicating that peptides should also be considered as candidate therapeutics.20−23
Thus far, several Mcl-1-specific peptides have been reported in the literature. Peptides with higher affinity for Mcl-1 (Kd <100 nM) and lower affinities (Kd >1 μM) for the other human Bcl-2 proteins have been selected from Bim BH3-based libraries or generated by targeted mutagenesis.24,25 A promising Mcl-1-specific peptide was derived from the BH3 region of Mcl-1 itself. Mcl-1 BH3 has a Kd of 245 nM for binding to Mcl-1, but chemical modification by hydrocarbon stapling significantly improved affinity (Kd = 10 nM).21 The stapled peptide, called Mcl-1 SAHBD, showed no binding to Bcl-2, Bcl-xL, Bcl-w, or Bfl-1 up to 1 μM and sensitized cells to caspase-dependent killing by TRAIL in culture at concentrations of 10–20 μM. A more potent molecule similar to Mcl-1 SAHBD would have great potential.
We have engineered three peptides, MS1, MS2, and MS3, based on the BH3 region of pro-apoptotic Bim that show high specificity and affinity for Mcl-1. These peptides use novel specificity mechanisms compared to previously designed peptide ligands targeting Mcl-1. The higher affinity Mcl-1 binding exhibited by the peptides makes them better Mcl-1-targeting reagents than the previously engineered Mcl-1-specific peptides or the natural BH3 NoxaA. All three peptides also exhibit high potency and specificity in BH3 profiling assays, demonstrating their usefulness in cellular assays.
Results and Discussion
Selection of Mcl-1-Specific Peptides
Three Mcl-1-specific peptides were discovered while screening a yeast-surface display library of Bim-BH3 variants. The original purpose of the library screening was to identify peptides that bound selectively to the viral Bcl-2 homologue Kaposi sarcoma herpesvirus Bcl-2 (KSBcl-2). Of the human Bcl-2 proteins, Mcl-1 is most similar to KSBcl-2 (29% sequence identity in the BH3 binding groove, compared to 16% sequence identity in the groove for KSBcl-2 and Bcl-xL). These two proteins also exhibit similar binding profiles to native BH3-only proteins.26 Therefore, it was not unexpected that peptides that were selected for high-affinity binding to KSBcl-2 also bound to Mcl-1.
Briefly, a library designed to be enriched in KSBcl-2 binders was sorted for binding to KSBcl-2 for two rounds, followed by five rounds of competition sorts in which clones were selected that could bind to KSBcl-2 in the presence of 50- to 100-fold excess Mcl-1 and Bcl-xL (see Supplementary Methods). BH3 sequences from two clones (B3 and A12) were chosen for further study as soluble peptides. B3 was chosen because it had the highest frequency of recovery, and A12 was chosen based on the presence of I2dM, an untested substitution, and E2gG, which was shown to provide modest specificity against Bcl-w in previously published Bim substitution SPOT arrays (Supplementary Table 1).24,27,28 The notation used to refer to mutants lists the original residue, the peptide position, and then the substitution. Synthetic peptides of 23 amino acids with the sequences of B3 and A12 and an N-terminal fluorescein were made and tested in solution for binding to KSBcl-2 and five human Bcl-2 family proteins (Table 2; see Table 1 for all sequences). These experiments showed that, although we identified peptides that bound to KSBcl-2 and Mcl-1 in preference to Bcl-2, Bfl-1, Bcl-w, and to a lesser extent Bcl-xL, we could not discriminate KSBcl-2 from Mcl-1 binding. Because the peptides that we identified bound tightly to Mcl-1 and showed good specificity for Mcl-1 over other human Bcl-2 family members, we chose to develop them further as Mcl-1 binders.
Table 2. Affinities of Native and Designed BH3 Peptides for Six Bcl-2 Homologsa.
| receptor |
||||||
|---|---|---|---|---|---|---|
| peptide | Mcl-1 | Bfl-1 | Bcl-w | Bcl-xL | Bcl-2 | KSBcl-2 |
| MS1 | 1.9 ± 1.0 | 5000 ± 3200 | 1300 ± 230 | 1600 ± 1000 | 2300 ± 1500 | 2.9 ± 0.68 |
| MS2 | 1.5 ± 1.2 | 3100 ± 2300 | 250 ± 76 | 1400 ± 500 | 6200 ± 4100 | <1 |
| MS3 | 2.0 ± 1.2 | 790 ± 140 | 340 ± 69 | 2300 ± 1000 | >3000 | 3.3 ± 1.6 |
| A12 | 2.4 ± 2.3 | 22 ± 6.6 | 210 ± 110 | 9.8 ± 3.1 | 42 ± 8.9 | 4.0 ± 2.6 |
| B3 | <1 | 26 ± 7.7 | 16 ± 6.2 | 4.2 ± 2.1 | 35 ± 11 | 1.43 ± 1.39 |
| NoxaA | 46 ± 11 | ND | ND | ND | ND | ND |
| Bim 23-mer | <1 | <1 | 1.75 ± 1.0 | 2.6 ± 1.9 | 1.9 ± 1.3 | 1.2 ± 0.79 |
| Bim_ A2eT | <1 | 31 ± 6.8 | 39 ± 9.4 | 17 ± 4.8 | 43 ± 12 | 1.8 ± 0.81 |
| Bim_ A2eT_ I2dM | <1 | 150 ± 69 | 260 ± 52 | 83 ± 53 | 210 ± 71 | 0.75 ± 0.37 |
| Bim_ A2eT_E2gG | 1.6 ± 0.57 | 250 ± 63 | 94 ± 36 | 37 ± 7.0 | 150 ± 27 | 0.66 ± 0.31 |
| Bim_ A2eT_ I3dL | <1 | 120 ± 27 | 12 ± 2.6 | 7.6 ± 3.8 | 37 ± 14 | 1.0 ± 0.38 |
| Bim_ A2eT_F4aI | <1 | 7.9 ± 2.1 | 16 ± 4.1 | 110 ± 62 | 150 ± 43 | 1.7 ± 0.80 |
Dissociation constants for direct binding of fluoresceinated peptides to Bcl-2 proteins (in nM) with 95% confidence intervals. Values designated <1 were too tight to be accurately fit. Values designated >3000 were too weak to be accurately fit. ND, not determined. See Table 1 for sequences of all peptides used. Binding data and fits are shown in Supplementary Figure 1.
Table 1. Sequences of Peptides Used for Fluorescence Anisotropy and BH3 Profiling Assaysa.

The heptad convention used to refer to positions in the BH3 peptide is shown. Bim point mutant positions are underlined.
We sought to improve the Mcl-1 binding selectivity of peptides identified in screening using rational mutagenesis guided by prior studies. Wild-type Bim has an alanine at position 2e (see Table 1 for peptide position labels), and SPOT-array tests of Bim BH3 point mutants have shown that glycine at 2e, found in B3, A12, and G9 (a point mutant of B3 that was also identified in screening), is tolerated by or increases binding to all of the receptors. Threonine at 2e was identified using SPOT arrays as a mutation that could decrease binding to Bfl-1, Bcl-xL, Bcl-2, and Bcl-w, while maintaining strong binding to Mcl-1.24,27,28 The specificity of peptides corresponding to B3, A12, and G9 was greatly improved by replacing the glycine at the 2e position with a threonine, generating the MS1, MS2, and MS3 variants (for Mcl-1 specific), corresponding to the sequences of A12, B3, and G9 with a G2eT mutation, respectively.
MS1, MS2, and MS3 labeled with an N-terminal fluorescein were tested for binding to the five human Bcl-2 receptors in fluorescence anisotropy assays. As shown in Table 2 and Supplementary Figure 1, all three peptides bound Mcl-1 with Kd ≤ 2 nM. MS1 bound with Kd > 1 μM to the other four receptors. MS2 bound with micromolar affinity to Bcl-xL, Bcl-2, and Bfl-1 and bound in the hundred-nanomolar range to Bcl-w. MS3 also displayed micromolar affinity for Bcl-xL and Bcl-2 and several-hundred nanomolar affinity to Bcl-w and Bfl-1. In contrast, the NoxaA BH3 peptide from murine Noxa, for which no binding up to 2500 nM for Bcl-xL, Bcl-2, Bcl-w, or Bfl-1 is reported in the literature, bound Mcl-1 more weakly than the three designed peptides, with a Kd of 46 nM (Table 2).18 NoxaA is the most Mcl-1-selective natural BH3, and a NoxaA BH3 peptide is routinely used in BH3 profiling assays to detect apoptotic resistance dependent upon Mcl-1.19,29,30 Compared to NoxaA, the three designed peptides MS1, MS2 and MS3 have high affinity for Mcl-1 and also show high specificity against Bcl-xL, Bcl-2, Bcl-w, and Bfl-1.
To assess the influence of the N-terminal fluorescein dye on binding, we tested a subset of unlabeled peptides, MS1, MS2, Bim, and NoxaA in competition with a fluorescently labeled Bim variant (Supplementary Figure 2). The Ki values for Mcl-1 binding to MS1, MS2, and NoxaA were weaker than the Kd values determined using labeled peptides. The Ki for MS1 binding to Mcl-1 was between 8 and 24 nM, depending on the fitting protocol (see Supplementary Methods and Supplementary Table 2). The competition experiments indicated that MS1 is between ∼40- and 190-fold specific for Mcl-1 over Bcl-w, the next-tightest binding family member. Competition experiments also confirmed that MS1 and MS2 are considerably tighter binders to Mcl-1 than is NoxaA; NoxaA binding to Mcl-1 was very weak and thus difficult to quantify with the competition assay.
Specificity Mechanisms
A peptide corresponding to the BH3 region of Bim binds very tightly to all receptors. To better understand the determinants of binding specificity for MS1, MS2, and MS3, we sought to identify residues in these peptides that differ from Bim and destabilize interactions with receptors other than Mcl-1. The 2eT mutation was vital in generating highly Mcl-1-specific peptides. This single point mutation in Bim (giving Bim_A2eT, Table 1) provides a 6-fold reduction in Bcl-xL binding and over 20-fold reduction in Bcl-2, Bcl-w, and Bfl-1 binding in a peptide with the wild-type Bim background (Table 2). Likewise, introducing 2eT into library peptides A12 and B3 reduced binding to Bfl-1, Bcl-2, and Bcl-xL by ∼100-fold and gave a more moderate ∼10-fold reduction in Bcl-w binding affinity. Thus, it is clear that threonine at position 2e is highly destabilizing for all human Bcl-2 receptors other than Mcl-1, in several different peptide contexts.
Position 2e is conserved as small (alanine, glycine, serine) in natural BH3 sequences. Mcl-1 can bind BH3 peptides with larger residues at position 2e, including Bim _A2eT and a peptide corresponding to the Mcl-1 BH3 region, which has a leucine at position 2e.21 To look for possible reasons that the other Bcl-2 paralogs cannot accommodate threonine at position 2e, we compared structures of Bcl-xL, Bfl-1, and Mcl-1 bound to the BH3 region of Bim (3FDL, 2VM6, and 2PQK, respectively).31−33 As shown in Figure 1a, helix 4 of Bfl-1 is closer to the peptide near the 2e position than is helix 4 in Mcl-1. Simple modeling of preferred threonine rotamers at 2e on static Mcl-1:Bim BH3 and Bfl-1:Bim BH3 structures illustrates that threonine is easily accommodated in the Mcl-1 structure in a helix-preferred rotomer but clashes with helix-4 residues of Bfl-1 (Figure 1b).33,34 In the Bcl-xL:Bim BH3 structure, the BH3 peptide is positioned slightly differently in the groove, resulting in Ala2e being oriented further into the groove than in the Mcl-1:Bim BH3 structure (Figure 1c). Rearrangement of Bcl-xL helix 4 would likely be required to accommodate threonine, and such rearrangement could be disfavored if it led to disruption of a three-residue salt-bridge network that forms in the Bcl-xL:Bim BH3 structure between Glu129 and Arg132 of Bcl-xL (on helix 4) and Arg3b of Bim (Figure 1d). This network cannot be formed in a structure like that of Mcl-1:Bim BH3 or Mcl-1:Mcl-1 BH3 (in which position 2e is leucine), because the equivalent of Bcl-xL residue 129 is farther away from peptide position 3b in this complex. The charged residues in Bcl-xL that participate in salt-bridge formation are also conserved in Bcl-2, suggesting that a similar mechanism might operate to disfavor threonine or larger residues at 2e for that protein.
Figure 1.
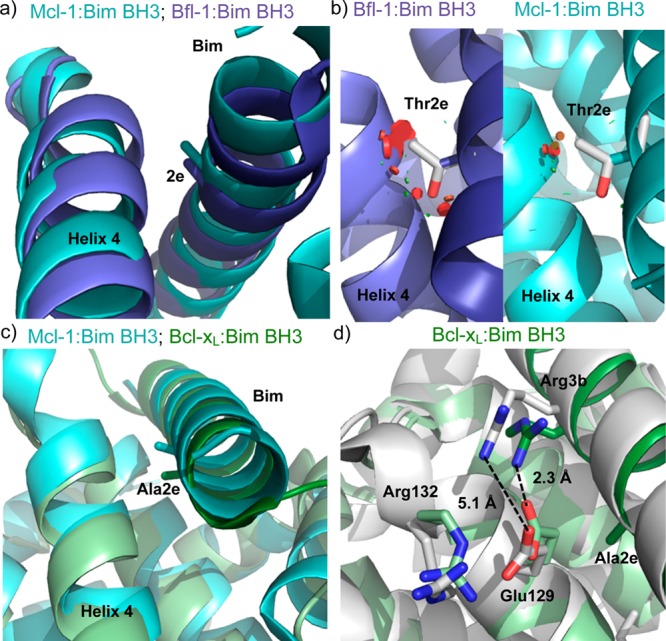
Comparison of Bim BH3 position 2e in structures of Bfl-1, Bcl-xL, and Mcl-1. (a) Helix 4 is closer to Ala2e in the Bfl-1:Bim BH3 structure (light:dark purple, 2VM6) than in the Mcl-1:Bim BH3 structure (light:dark cyan, 2PQK), as shown by aligning the structures in PyMOL.32,33 (b) Threonine modeled at position 2e clashes (red disks) with helix 4 in the Bfl-1 complex (left) but does not clash significantly in the Mcl-1 complex (right). Mutations were modeled in PyMOL by selecting the most preferred backbone-dependent rotamer for Thr; all rotamers are predicted to clash on the Bfl-1 structure backbone. (c) Ala2e is angled further into the BH3 binding groove in the Bcl-xL:Bim BH3 structure (light:dark green, 3FDL) than in the Mcl-1:Bim BH3 structure (light:dark cyan, 2PQK).31 (d) A salt bridge network that exists in the Bcl-xL:Bim BH3 structure (green) between Glu129, Arg132, and Arg3b would be disrupted at the equivalent sites in Mcl-1:Bim BH3 structure 2PQK (white). Glutamate and arginine are shown modeled in the place of His252 and Ser255 in the 2PQK structure, with rotamers chosen to position the side chains as close as possible to the orientations in the 3FDL structure.
MS1, MS2, and MS3 all have different substitutions at 2g, which is a glutamate in wild-type Bim and is typically a medium-to-large residue in other known BH3 regions. MS1, our most selective peptide, has a glycine at this position, and mutating glutamate to glycine at position 2g in Bim_A2eT decreased binding to all receptors. The change in affinity for Mcl-1 could not be quantified, but affinities for Bfl-1, Bcl-2, Bcl-w, and Bcl-xL were reduced an additional 2–8 fold-compared to Bim_A2eT. Thus, glycine at 2g provides some of the negative design disfavoring interactions with off-target receptors, although at the cost of weakening binding to the Mcl-1 target (Table 2).
Three mutations in peptides MS1, MS2, and MS3 occur in positions that are usually conserved as hydrophobic in known BH3 motifs (positions 2d, 3d, and 4a). When tested in the Bim_A2eT context, I2dM (found in MS1) provided a roughly 4-fold reduction in binding to Bcl-xL, Bcl-2, Bcl-w, and Bfl-1 (Table 2). Notably, significant decreases in Bcl-w binding for Bim_A2eT_E2gG and Bim_A2eT_I2dM may explain why MS1 is more selective for Mcl-1 vs Bcl-w than are MS2 and MS3, which have different mutations at 2g and 2d. Mutation I3dL reduced binding of Bim_A2eT to Bfl-1 by 4-fold, while this mutation increased binding slightly to Bcl-xL, Bcl-2, and Bcl-w (Table 2). The F4aI mutation increased Bim_A2eT binding slightly to Bfl-1 and Bcl-w but decreased binding by 6- and 3-fold to Bcl-xL and Bcl-2, respectively (Table 2). Position 4a is a well-documented source of specificity for Mcl-1 binding.21,24,33,35 Mutagenesis studies and peptide library screens have demonstrated that Bcl-xL binds preferentially to peptides that include a phenylalanine or tyrosine to fill the enclosed hydrophobic pocket near 4a, whereas Mcl-1 tolerates a wide variety of substitutions at this position.21,24,35
Cellular BH3 Profiling Assays
A whole-cell BH3 profiling assay was used to test the specificity of our Mcl-1-binding peptides in several cell lines with differing dependencies on Bcl-2, Mcl-1, Bcl-xL, or Bfl-1.29,36 In this assay, permeabilized cells were treated with increasing doses of BH3 peptides, and mitochondrial outer membrane permeabilization (MOMP) was monitored using the dye JC-1 (see Methods). EC50 values for BH3 profiling experiments involving peptides from this study are given in Figure 2 and Supplementary Table 3, with full titration curves shown in Supplementary Figure 3.
Figure 2.

Heat map of the EC50 values (peptide concentration in nM) for mitochondrial depolarization induced by engineered and native BH3 peptides in four cell lines. Engineered peptide names are shaded in gray. Each cell line is dependent upon a single Bcl-2 family member: Mcl-1, Bcl-2, Bcl-xL, or Bfl-1 as determined by BH3 profiling with native BH3-only peptides (Supplementary Figure 3e–h). All experiments were performed at least three times. The inset shows an example titration curve; see Supplementary Figure 3 and Supplementary Table 3 for other curves and all EC50 values with 95% confidence intervals.
Mcl-1/Myc 2640 is an engineered murine leukemia cell line overexpressing murine Mcl-1 and Myc, and Bcl-2/Myc 2924 is a similarly engineered cell line overexpressing human Bcl-2.30 By Western blot and BH3 profiling, these cells exhibit Mcl-1 and Bcl-2 dependencies, respectively.30 MS1, MS2, and MS3 elicited potent mitochondrial depolarization responses in Mcl-1/Myc 2640, with EC50 values of 70 nM, 700 nM, and 860 nM, respectively. These peptides were much more potent than NoxaA in this assay (EC50 = 20 μM). Human and murine Mcl-1 are over 90% identical in the Bcl-2 domain and 94% identical in the BH3 binding groove. Human multiple myeloma cell lines dependent upon Mcl-1 (as indicated by response to NoxaA and Bad) gave EC50 values of 2.5–3.3 μM for MS1, compared to EC50 values >100 μM for NoxaA (Supplementary Figure 3i–k and Supplementary Table 3). Thus, multiple Mcl-1-dependent cell lines were much more sensitive to MS1 than to NoxaA.
MS1 and MS2 were highly selective in BH3 profiling. In a Bcl-2-dependent line, EC50 values were >100 μM for MS1, MS2, MS3, and NoxaA. MDA-MB-231 is a human breast cancer cell line that has been shown to have a Bcl-xL-dependent profile.29 EC50 values for MS1, MS2, MS3, and NoxaA were over 100 μM for MDA-MB-231 cells. MS1 and MS2 showed EC50 values >100 μM in Pfeiffer, a lymphoma line with high Bfl-1 mRNA expression that has previously been shown to exhibit a Bfl-1-dependent BH3 profile.19 MS3 gave a stronger response in Pfeiffer than MS1 or MS2, but MS3 also exhibited tighter Bfl-1 binding by fluorescence anisotropy (Table 2). Finally, Bim_A2eT_E2gG, which showed modest specificity for Mcl-1 by fluorescence anisotropy (Table 2), exhibited a strong depolarization response in Mcl-1/Myc 2640 and a depolarization response intermediate to that of Bim and MS1 in Bcl-2/Myc 2924, MDA-MB-231, and Pfeiffer. Thus, in vitro binding specificities are replicated in BH3 profiling assays in cell lines showing all of the currently identified dependencies on Bcl-xL, Bcl-2, Mcl-1, and Bfl-1, as a Bcl-w-dependent cell line has not yet been identified or constructed.
The engineered peptides tested here were derived from Bim BH3, which is an activator BH3 peptide. Nevertheless, these peptides did not cause strong depolarization in cell lines that are not dependent upon Mcl-1, indicating that they act as sensitizers rather than activators in these assays. Depolarization activity was specific to the Bcl-2 pathway, because the peptides did not depolarize mitochondria in the Bax/Bak-deficient cell line Su-DHL10 (Supplementary Figure 4).19
The three peptides presented here exhibit high affinity for Mcl-1 in solution binding assays. MS1, the most specific peptide, was at least 40-fold specific over Bcl-xL, Bcl-2, Bcl-w, and Bfl-1 as assessed using biochemical competition binding experiments. We demonstrated the potency and specificity of these peptides as Mcl-1-specific reagents in BH3 profiling assays. Given that Mcl-1 overexpression is a key source of resistance to ABT-263, the ability to detect Mcl-1 activity will be important in designing treatment strategies.9,10 The high affinity of the peptides for Mcl-1 also presents a good starting point for developing peptide-based therapeutics.
Methods
Library Construction and Sorting
Fluorescence Anisotropy Assays
The Bim variant peptides were synthesized by the MIT Biopolymers Laboratory and were 23-mers as shown in Table 1 with N-terminal 5/6-fluorescein amidite and C-terminal amidation. The crude synthesis product was validated to contain primarily the peptide of interest by mass spectrometry and purified by HPLC on a C18 column using a linear gradient of acetonitrile. FITC-AHA-NOXAA peptide was produced by the Tufts University Physiology core facility, purified by HPLC, and validated by mass spectrometry. Bcl-2 receptors were c-myc-tagged variants without the C-terminal trans-membrane region and without the N-terminal domain of Mcl-1, expressed, and purified as in Dutta et al.24 The KSBcl-2 construct is described in the Supplementary Methods. Direct fluorescence anisotropy assays were performed in a buffer of 25 mM Tris pH 7.8, 50 mM NaCl, 1 mM EDTA, 0.001% Triton-X-100, 5% v/v DMSO. A 12-point dilution of receptor protein was added to the wells of a Corning 96-well, black, polystyrene, nonbinding surface plate, followed by the fluoresceinated peptide for a total volume of 120 μL with a final peptide concentration of 10 nM. Plates were incubated for 2 h before being read on a SpectraMax M5 plate reader (Molecular Devices) at 25 °C. Equilibration was checked by comparing curves read at 2 and 24 h. Most curves exhibited no change, so the 2-h data were used. The exceptions were the Bfl-1 curves for peptides MS3 and MS2 for which the 24-h curves were used. See Supplementary Methods for the curve-fitting protocols and competition fluorescence anisotropy methods.
Cellular BH3 Profiling Assays
Assay plates were produced by serial dilution of each peptide from 200 μM to 0.2 nM using 10-fold dilutions in DTEB (Derived from Trehalose Experimental Buffer: 135 mM trehalose, 50 mM KCl, 20 μM EDTA, 20 μM EGTA, 0.1% BSA, 5 mM succinate, 10 mM HEPES-KOH pH 7.5) containing 0.005% w/v digitonin, 10 mM 2-mercaptoethanol, 2 μM JC-1, and 20 mg mL–1 oligomycin. Triplicate wells for each peptide were made for each cell line by adding 15 μL of the peptide dilutions to each well of a black, untreated 384-well plate. Control wells containing no peptide or 20 μM FCCP (carbonyl cyanide-4(trifluoromethoxy) phenylhydrazone, a chemical uncoupler of oxidative phosphorylation) were included for zero and complete depolarization, respectively. Multiple plates were produced from the same stock and frozen at −80 °C for later use. Frozen plates were brought to RT prior to use, cells were suspended in DTEB at a density of 1.34 × 106 cells/mL, and 15 μL of cell suspension was added to each well of the dilution series to yield wells ranging from 0.1 nM to 100 μM peptide and 20000 cells/well. Fluorescence of JC-1 aggregates was measured at 590 nm with 545 nm excitation on a Tecan Safire2 at 5 min intervals for 3 h. The area under each signal-vs-time curve was calculated and normalized to the untreated and FCCP values to produce the percent depolarization. Curves were plotted as the log [peptide] vs percent depolarization, with sigmoidal dose–response curves fitted using Graphpad PRISM 6. For curves without an upper baseline, an upper limit on the EC50 was estimated by fitting the curve with the upper baseline fixed at 100% depolarization, as this was the upper limit reached by most curves with a complete upper baseline.
Acknowledgments
We thank the Koch Institute Biopolymers and Proteomics Facility for peptide synthesis and the Koch Institute Flow Cytometry Core at MIT for assistance with cell sorting and analysis. This work was supported by NIGMS awards R01-GM084181, P50-GM068762, and P01-CA129980, and G.W.F. was supported by an NSF Graduate Research Fellowship. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supporting Information Available
This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
∥ Center for Therapeutic Innovation Boston, Pfizer Inc., 3 Blackfan Cir., Room 1840, Boston, MA 02148.
The authors declare the following competing financial interest(s): A.L. is a Leukemia and Lymphoma Society Scholar. A.L. was a cofounder and formerly served on the scientific advisory board of Eutropics Pharmaceuticals, which has a license for BH3 profiling.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Beroukhim R.; Mermel C. H.; Porter D.; Wei G.; Raychaudhuri S.; Donovan J.; Barretina J.; Boehm J. S.; Dobson J.; Urashima M.; Mc Henry K. T.; Pinchback R. M.; Ligon A. H.; Cho Y.-J.; Haery L.; Greulich H.; Reich M.; Winckler W.; Lawrence M. S.; Weir B. A.; Tanaka K. E.; Chiang D. Y.; Bass A. J.; Loo A.; Hoffman C.; Prensner J.; Liefeld T.; Gao Q.; Yecies D.; Signoretti S.; Maher E.; Kaye F. J.; Sasaki H.; Tepper J. E.; Fletcher J. A.; Tabernero J.; Baselga J.; Tsao M.-S.; Demichelis F.; Rubin M. A.; Janne P. A.; Daly M. J.; Nucera C.; Levine R. L.; Ebert B. L.; Gabriel S.; Rustgi A. K.; Antonescu C. R.; Ladanyi M.; Letai A.; Garraway L. A.; Loda M.; Beer D. G.; True L. D.; Okamoto A.; Pomeroy S. L.; Singer S.; Golub T. R.; Lander E. S.; Getz G.; Sellers W. R.; Meyerson M. (2010) The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertz I. E.; Kusam S.; Lam C.; Okamoto T.; Sandoval W.; Anderson D. J.; Helgason E.; Ernst J. A.; Eby M.; Liu J.; Belmont L. D.; Kaminker J. S.; O’Rourke K. M.; Pujara K.; Kohli P. B.; Johnson A. R.; Chiu M. L.; Lill J. R.; Jackson P. K.; Fairbrother W. J.; Seshagiri S.; Ludlam M. J. C.; Leong K. G.; Dueber E. C.; Maecker H.; Huang D. C. S.; Dixit V. M. (2011) Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 471, 110–114. [DOI] [PubMed] [Google Scholar]
- Wei S.-H.; Dong K.; Lin F.; Wang X.; Li B.; Shen J.-J.; Zhang Q.; Wang R.; Zhang H.-Z. (2008) Inducing apoptosis and enhancing chemosensitivity to gemcitabine via RNA interference targeting Mcl-1 gene in pancreatic carcinoma cell. Cancer Chemother. Pharmacol. 62, 1055–1064. [DOI] [PubMed] [Google Scholar]
- Letai A.; Bassik M. C.; Walensky L. D.; Sorcinelli M. D.; Weiler S.; Korsmeyer S. J. (2002) Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2, 183–192. [DOI] [PubMed] [Google Scholar]
- Roberts A. W.; Seymour J. F.; Brown J. R.; Wierda W. G.; Kipps T. J.; Khaw S. L.; Carney D. A.; He S. Z.; Huang D. C. S.; Xiong H.; Cui Y.; Busman T. A.; McKeegan E. M.; Krivoshik A. P.; Enschede S. H.; Humerickhouse R. (2012) Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J. Clin. Oncol. 30, 488–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin C. M.; Hann C. L.; Garon E. B.; Ribeiro de Oliveira M.; Bonomi P. D.; Camidge D. R.; Chu Q.; Giaccone G.; Khaira D.; Ramalingam S. S.; Ranson M. R.; Dive C.; McKeegan E. M.; Chyla B. J.; Dowell B. L.; Chakravartty A.; Nolan C. E.; Rudersdorf N.; Busman T. A.; Mabry M. H.; Krivoshik A. P.; Humerickhouse R. A.; Shapiro G. I.; Gandhi L. (2012) Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin. Cancer Res. 18, 3163–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse C.; Shoemaker A. R.; Adickes J.; Anderson M. G.; Chen J.; Jin S.; Johnson E. F.; Marsh K. C.; Mitten M. J.; Nimmer P.; Roberts L.; Tahir S. K.; Xiao Y.; Yang X.; Zhang H.; Fesik S.; Rosenberg S. H.; Elmore S. W. (2008) ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 68, 3421–3428. [DOI] [PubMed] [Google Scholar]
- Oltersdorf T.; Elmore S.; Shoemaker A. (2005) An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435, 677–681. [DOI] [PubMed] [Google Scholar]
- Konopleva M.; Contractor R.; Tsao T.; Samudio I.; Ruvolo P. P.; Kitada S.; Deng X.; Zhai D.; Shi Y.-X.; Sneed T.; Verhaegen M.; Soengas M.; Ruvolo V. R.; McQueen T.; Schober W. D.; Watt J. C.; Jiffar T.; Ling X.; Marini F. C.; Harris D.; Dietrich M.; Estrov Z.; McCubrey J.; May W. S.; Reed J. C.; Andreeff M. (2006) Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 10, 375–388. [DOI] [PubMed] [Google Scholar]
- Yecies D.; Carlson N.; Deng J.; Letai A. (2010) Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood 115, 3304–3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souers A. J.; Leverson J. D.; Boghaert E. R.; Ackler S. L.; Catron N. D.; Chen J.; Dayton B. D.; Ding H.; Enschede S. H.; Fairbrother W. J.; Huang D. C. S.; Hymowitz S. G.; Jin S.; Khaw S. L.; Kovar P. J.; Lam L. T.; Lee J.; Maecker H. L.; Marsh K. C.; Mason K. D.; Mitten M. J.; Nimmer P. M.; Oleksijew A.; Park C. H.; Park C.-M.; Phillips D. C.; Roberts A. W.; Sampath D.; Seymour J. F.; Smith M. L.; Sullivan G. M.; Tahir S. K.; Tse C.; Wendt M. D.; Xiao Y.; Xue J. C.; Zhang H.; Humerickhouse R. A.; Rosenberg S. H.; Elmore S. W. (2013) ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 19, 202–208. [DOI] [PubMed] [Google Scholar]
- Cohen N. A.; Stewart M. L.; Gavathiotis E.; Tepper J. L.; Bruekner S. R.; Koss B.; Opferman J. T.; Walensky L. D. (2012) A competitive stapled peptide screen identifies a selective small molecule that overcomes MCL-1-dependent leukemia cell survival. Chem. Biol. 19, 1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. B.; Balasis M. E.; Doi K.; Berndt N.; DuBoulay C.; Hu C.-C. A.; Guida W.; Wang H.-G.; Sebti S. M.; Del Valle J. R. (2012) Synthesis and evaluation of substituted hexahydronaphthalenes as novel inhibitors of the Mcl-1/BimBH3 interaction. Bioorg. Med. Chem. Lett. 22, 5961–5965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rega M. F.; Wu B.; Wei J.; Zhang Z.; Cellitti J. F.; Pellecchia M. (2011) SAR by interligand nuclear overhauser effects (ILOEs) based discovery of acylsulfonamide compounds active against Bcl-x(L) and Mcl-1. J. Med. Chem. 54, 6000–6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friberg A.; Vigil D.; Zhao B.; Daniels R. N.; Burke J. P.; Garcia-Barrantes P. M.; Camper D.; Chauder B. A.; Lee T.; Olejniczak E. T.; Fesik S. W. (2013) Discovery of potent myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods and structure-based design. J. Med. Chem. 56, 15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y.; Aikawa K.; Nishida G.; Homma M.; Sogabe S.; Igaki S.; Hayano Y.; Sameshima T.; Miyahisa I.; Kawamoto T.; Tawada M.; Imai Y.; Inazuka M.; Cho N.; Imaeda Y.; Ishikawa T. (2013) Discovery of potent Mcl-1/Bcl-xL dual inhibitors by using a hybridization strategy based on structural analysis of target proteins. J. Med. Chem. 56, 9635–9645. [DOI] [PubMed] [Google Scholar]
- Abulwerdi F. A.; Liao C.; Mady A. S.; Gavin J.; Shen C.; Cierpicki T.; Stuckey J. A.; Showalter H. D. H.; Nikolovska-Coleska Z. (2014) 3-Substituted-N-(4-hydroxynaphthalen-1-yl)arylsulfonamides as a novel class of selective Mcl-1 inhibitors: Structure-based design, synthesis, SAR, and biological evaluation. J. Med. Chem. 57, 4111–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Certo M.; Del Gaizo Moore V.; Nishino M.; Wei G.; Korsmeyer S.; Armstrong S. A.; Letai A. (2006) Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 9, 351–365. [DOI] [PubMed] [Google Scholar]
- Deng J.; Carlson N.; Takeyama K.; Dal Cin P.; Shipp M.; Letai A. (2007) BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell 12, 171–185. [DOI] [PubMed] [Google Scholar]
- Walensky L. D.; Kung A. L.; Escher I.; Malia T. J.; Barbuto S.; Wright R. D.; Wagner G.; Verdine G. L.; Korsmeyer S. J. (2004) Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science 305, 1466–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart M.; Fire E.; Keating A. (2010) The MCL-1 BH3 helix is an exclusive MCL-1 inhibitor and apoptosis sensitizer. Nat. Chem. Biol. 6, 595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muppidi A.; Doi K.; Edwardraja S.; Drake E. J.; Gulick A. M.; Wang H.-G.; Lin Q. (2012) Rational design of proteolytically stable, cell-permeable peptide-based selective Mcl-1 inhibitors. J. Am. Chem. Soc. 134, 14734–14737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B. J.; Lee E. F.; Checco J. W.; Evangelista M.; Gellman S. H.; Fairlie W. D. (2013) Structure-guided rational design of α/β-peptide foldamers with high affinity for BCL-2 family prosurvival proteins. ChemBioChem 14, 1564–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta S.; Gullá S.; Chen T. S.; Fire E.; Grant R. A.; Keating A. E. (2010) Determinants of BH3 binding specificity for Mcl-1 versus Bcl-xL. J. Mol. Biol. 398, 747–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E. F.; Fedorova A.; Zobel K.; Boyle M. J.; Yang H.; Perugini M. A.; Colman P. M.; Huang D. C. S.; Deshayes K.; Fairlie W. D. (2009) Novel Bcl-2 homology-3 domain-like sequences identified from screening randomized peptide libraries for inhibitors of the pro-survival Bcl-2 proteins. J. Biol. Chem. 284, 31315–31326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan A. M.; Letai A. (2008) BH3 domains define selective inhibitory interactions with BHRF-1 and KSHV BCL-2. Cell Death Differ. 15, 580–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBartolo J.; Dutta S.; Reich L.; Keating A. E. (2012) Predictive Bcl-2 family binding models rooted in experiment or structure. J. Mol. Biol. 422, 124–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London N.; Gullá S.; Keating A. E.; Schueler-Furman O. (2012) In silico and in vitro elucidation of BH3 binding specificity toward Bcl-2. Biochemistry 51, 5841–5850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan J. A.; Brunelle J. K.; Letai A. (2010) Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4+ CD8+ thymocytes. Proc. Natl. Acad. Sci. U.S.A. 107, 12895–12900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunelle J. K.; Ryan J.; Yecies D.; Opferman J. T.; Letai A. (2009) MCL-1-dependent leukemia cells are more sensitive to chemotherapy than BCL-2-dependent counterparts. J. Cell Biol. 187, 429–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E. F.; Sadowsky J. D.; Smith B. J.; Czabotar P. E.; Peterson-Kaufman K. J.; Colman P. M.; Gellman S. H.; Fairlie W. D. (2009) High-resolution structural characterization of a helical alpha/beta-peptide foldamer bound to the anti-apoptotic protein Bcl-xL. Angew. Chem., Int. Ed. 48, 4318–4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman M. D.; Nyman T.; Welin M.; Lehtiö L.; Flodin S.; Trésaugues L.; Kotenyova T.; Flores A.; Nordlund P. (2008) Completing the family portrait of the anti-apoptotic Bcl-2 proteins: crystal structure of human Bfl-1 in complex with Bim. FEBS Lett. 582, 3590–3594. [DOI] [PubMed] [Google Scholar]
- Fire E.; Gullá S.; Grant R.; Keating A. (2010) Mcl-1-Bim complexes accommodate surprising point mutations via minor structural changes. Protein Sci. 19, 507–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbrack R. L. Jr.; Cohen F. E. (1997) Bayesian statistical analysis of protein side-chain rotamer preferences. Protein Sci. 6, 1661–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E.; Czabotar P.; Van Delft M. (2008) A novel BH3 ligand that selectively targets Mcl-1 reveals that apoptosis can proceed without Mcl-1 degradation. J. Cell Biol. 180(2), 341–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan J.; Letai A. (2013) BH3 profiling in whole cells by fluorimeter or FACS. Methods (San Diego, CA, U.S.) 61, 156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.