

Abstract
Duchenne muscular dystrophy (DMD) is a devastating muscle wasting disease caused by mutations in dystrophin. Several downstream consequences of dystrophin deficiency are triggers of endoplasmic reticulum (ER) stress, including loss of calcium homeostasis, hypoxia and oxidative stress. During ER stress, misfolded proteins accumulate in the ER lumen and the unfolded protein response (UPR) is triggered, leading to adaptation or apoptosis. We hypothesized that ER stress is heightened in dystrophic muscles and contributes to the pathology of DMD. We observed increases in the ER stress markers BiP and cleaved caspase-4 in DMD patient biopsies, compared with controls, and an increase in multiple UPR pathways in muscles of the dystrophin-deficient mdx mouse. We then crossed mdx mice with mice null for caspase-12, the murine equivalent of human caspase-4, which are resistant to ER stress. We found that deleting caspase-12 preserved mdx muscle function, resulting in a 75% recovery of both specific force generation and resistance to eccentric contractions. The compensatory hypertrophy normally found in mdx muscles was normalized in the absence of caspase-12; this was found to be due to decreased fibre sizes, and not to a fibre type shift or a decrease in fibrosis. Fibre central nucleation was not significantly altered in the absence of caspase-12, but muscle fibre degeneration found in the mdx mouse was reduced almost to wild-type levels. In conclusion, we have identified heightened ER stress and abnormal UPR signalling as novel contributors to the dystrophic phenotype. Caspase-4 is therefore a potential therapeutic target for DMD.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is a devastating muscle wasting disease, affecting 1:5000 males (1,2). It is caused by mutations in dystrophin, a component of the dystrophin glycoprotein complex (DGC) found at the sarcolemma of muscle fibres. The DGC links the extracellular matrix to the intracellular cytoskeleton, providing structural integrity to the myofibre and the sarcolemma. In the absence of dystrophin, the DGC is lost from the sarcolemma, rendering the muscle more susceptible to damage during contraction. The sarcolemma becomes leaky and calcium homeostasis is impaired (reviewed in 3). Cycles of degeneration and regeneration ensue, eventually leading to the replacement of muscle fibres with fibrotic and adipose tissue (4). Other downstream consequences of dystrophin deficiency include hypoxia, due to respiratory muscle weakness and loss of regulation of vasoconstriction/dilation during exercise by the DGC-associated neuronal nitric oxide synthesis (5,6), and oxidative stress, arising from increased NADPH oxidase activity (reviewed in 7). Patients experience progressive loss of muscle strength, dependence on a wheelchair, cardiomyopathy, respiratory insufficiency and a shortened lifespan (2,4,8). There is currently no effective treatment available.
Several secondary consequences of dystrophin deficiency, namely calcium dysregulation, hypoxia/ischaemia and oxidative stress, are causes of endoplasmic reticulum (ER) stress, a condition in which normal ER function is disrupted (9–15). During ER stress, unfolded and misfolded proteins accumulate in the ER lumen, triggering a set of signalling pathways termed the unfolded protein response (UPR). In the short term, the UPR inhibits protein synthesis. In the longer term, the UPR leads to adaptive changes, such as an increase in ER protein folding chaperones, or, if this is not sufficient to alleviate the stress, to apoptosis (reviewed in 16). A low level of basal UPR signalling occurs in normal skeletal muscle (17). Interestingly, in mice null for hexose-6-phosphate dehydrogenase, an enzyme that helps maintain the ER redox environment, ER stress markers are upregulated in skeletal muscle and a severe myopathy develops (18), suggesting that excessive ER stress leads to muscle dysfunction. ER stress has also been proposed to be a primary cause of the pathology of dystroglycanopathies, based on a zebrafish model (19).
Three main pathways comprise the apoptotic arm of the UPR (Fig. 1). The first involves upregulation of CCAAT/enhancer binding protein homologous protein (CHOP) downstream of protein kinase RNA-like endoplasmic reticulum kinase (PERK) activation, the second involves IRE-1α-dependent activation of c-jun N-terminal kinase (JNK) and the third involves murine caspase-12 or its human equivalent caspase-4 (20) (human caspase-12 is an inactive pseudogene in most individuals (21,22); here caspase-12 will refer to the murine gene). Caspase-12 and -4 are initiator caspases localized to the cytoplasmic face of the ER membrane (23,24) and are expressed in skeletal and cardiac muscle (25–28). Caspase-12 and -4 are activated specifically by ER stress, but not by other apoptotic stimuli (20,29); in turn they cleave and activate caspase-9, which activates caspase-3, leading to apoptosis (30,31). In addition to initiating apoptosis via caspase-3, activated caspase-12 translocates to the nucleus, and may carry out apoptotic events directly (32). Caspase-12/4 is activated via cleavage by caspase-7 (33) or m-calpain (23,34), and/or via release from TNF receptor-associated factor 2 (TRAF2), homodimerization and autoprocessing (35). Caspase-12-null mice exhibit no overt abnormalities (29), but are resistant to ER stress-induced apoptosis, as are caspase-12/4-deficient cultured cells (20,29,36). Therefore, caspase-12/4 may represent an attractive target for blocking deleterious effects of ER stress in vivo.
Figure 1.

The unfolded protein response. Under ER stress, the chaperone BiP binds to misfolded proteins in the ER lumen, sequestering it away from ER membrane proteins IRE1α, PERK and ATF6α and causing their activation. IRE1α and PERK are activated by phosphorylation, ATF6α by translocation to the Golgi and proteolytic cleavage. The three arms of UPR signalling collectively bring about the acute response (protein synthesis inhibition and ER-associated mRNA degradation), the adaptive response (upregulation of adaptive genes such as BiP) and, if ER stress cannot be overcome, the apoptotic response (mediated by caspase-12, CHOP and JNK). BiP, binding protein; IRE1α, inositol requiring enzyme 1α; PERK, protein kinase RNA-like endoplasmic reticulum kinase; ATF6α, activating transcription factor 6α; eIF2α, eukaryotic initiation factor 2α; casp-12, caspase-12; JNK, c-jun N-terminal kinase; CHOP, CCAAT/enhancer binding protein homologous protein. Dashed arrows indicate pathways proposed but not known conclusively to occur.
We hypothesized that heightened ER stress occurs in dystrophin-deficient muscle, and that subsequent UPR activation contributes to the pathology of DMD. We crossed caspase-12-null (casp) mice with dystrophin-deficient (mdx) mice, a model for DMD, to generate dystrophin-deficient, caspase-12-null mice (mdx-casp), and used measures of muscle function, morphology and UPR activation to determine the contribution of ER stress to dystrophic pathology.
RESULTS
ER stress is heightened in dystrophic muscle
Several triggers for ER stress occur as a result of dystrophin deficiency in DMD and in mdx mice. To determine whether dystrophin loss led to ER stress and UPR activation, muscle samples were obtained from biopsies of DMD patients and healthy controls and levels of UPR proteins were measured by immunoblotting. The ER chaperone BiP was increased in all five DMD patients, compared with the three healthy controls, with increases ranging from 2-fold to over 40-fold (Fig. 2A and C). On measuring caspase-4 it was found that levels of procaspase-4 were not different between control and DMD muscles. However, cleaved caspase-4 was observed in all five DMD patient samples, whereas it was not detected in any of the healthy controls (Fig. 2A and B). Therefore, in DMD muscle, ER stress is heightened and UPR signalling is increased, including cleavage of caspase-4.
Figure 2.

Heightened UPR activation in DMD patient muscles. Immunoblot (A) and quantification for pro- and cleaved caspase-4 (B) and BiP (C) in quadriceps muscles from three healthy control and five DMD patient biopsies. Bars represent mean ± standard error. *Significantly different from healthy controls by one sample T test (for caspase-4) or Mann–Whitney test (for BiP). AU, arbitrary units. n.d., not detected.
Having found that ER stress is elevated in DMD, ER stress was evaluated in multiple muscles from mdx mice. UPR signalling proteins were examined by immunoblotting of the tibialis anterior (TA), extensor digitorum longus (EDL) and diaphragm muscles from 5-month-old C57 and mdx mice. Similar to DMD muscles, BiP was expressed at higher levels in mdx TA, EDL and diaphragm muscles, compared with C57 (Figs 3A and 4B and Supplementary Material, Fig. S1B). In terms of the apoptotic UPR, mdx muscles had an increase in both the pro- and cleaved forms of the ER stress-specific caspase-12 (Figs 3B and 4A and Supplementary Material, Fig. S1A). Mdx TA muscles also had increased levels of the ER stress-activated, pro-apoptotic protein CHOP and increased activation of JNK, which responds to ER stress (Fig. 3C and D; see also Fig. 1). These results validated the hypothesis that ER stress is heightened, and that both the adaptive and the apoptotic arms of the UPR are more active, in different dystrophin-deficient muscles.
Figure 3.

UPR activation in C57, casp, mdx and mdx-casp TA muscles. Representative immunoblots and quantification for BiP (A), pro- and active (cleaved) caspase-12 (B), CHOP (C) and phosphorylated (P)- and total (T)-JNK (D) in C57, mdx, casp and mdx-casp TA muscles at 5 months of age. Bars represent mean ± standard error. *Significantly different from C57 and ‡significantly different from casp by one-way ANOVA with Tukey's Multiple Comparison Test for BiP and P-JNK and by unpaired T test for caspase-12. n = 4–8 mice per genotype. AU, arbitrary units.
Figure 4.

UPR activation in C57, casp, mdx and mdx-casp EDL muscles. Representative immunoblots and quantification for pro- and active (cleaved) caspase-12 (A) and BiP (B) in C57, mdx, casp and mdx-casp EDL muscles at 5 months of age. Bars represent mean ± standard error. *Significantly different from C57, ‡significantly different from casp and †significantly different from mdx by one-way ANOVA with Tukey's Multiple Comparison Test. n = 3–5 mice per genotype. AU, arbitrary units.
Compensatory changes in the UPR in the absence of caspase-12
To examine the contribution of ER stress to the dystrophic phenotype, mdx mice were crossed with caspase-12-null (casp) mice, which are resistant to ER stress, generating mdx-casp mice. To determine the effect on the UPR of deleting caspase-12, immunoblotting was used to measure levels and activation of UPR proteins in TA, EDL and diaphragm muscles from 5-month-old C57, casp, mdx and mdx-casp mice. In the TA muscle, levels of BiP were unchanged between C57 and casp mice, but were increased further in mdx-casp mice, compared with mdx mice (Fig. 3A). In the EDL and diaphragm muscles, BiP was not increased in casp mice compared with C57, but was decreased in mdx-casp mice, compared with mdx, although it remained elevated compared with C57 (Figs 4B and S1B). In the TA muscle, activation (phosphorylation) of JNK was decreased in mdx-casp muscles compared with mdx, while levels of CHOP were not different between mdx and mdx-casp mice. Neither phospho (P)-JNK or CHOP levels were increased in casp mice compared with C57 (Fig. 3C and D). Taken together, these data show that while deletion of caspase-12 has no effect on basal UPR signalling found in C57 mice, deletion of caspase-12 in mdx mice leads to changes in other UPR pathways, in a muscle-specific manner.
Lack of caspase-12 preserves mdx muscle function
To assess the effect of caspase-12 deletion on muscle function, EDL muscles were dissected from C57, casp, mdx and mdx-casp mice aged 5 months or 1 year, and subjected to a series of ex vivo muscle mechanics measurements. Consistent with previous studies, EDL muscles from 5-month-old mdx mice had a 17% deficit in specific force production, compared with C57 mice. Mdx-casp mice showed a 74% recovery of this deficit, having specific force production only 6% less than C57 mice (Fig. 5A). Five-month-old mdx mice also showed a characteristic susceptibility to loss of force production following a series of eccentric contractions (ECCs), having a 4-fold greater loss of force than did C57 muscles subjected to the same protocol. Again, this deficit was corrected by 75% in mdx-casp muscles, which had only a 1.8-fold greater force loss than did C57 muscles (Fig. 5B). These effects persisted to some extent even in 1-year-old mice. One-year-old mdx mice had a 29% deficit in EDL specific force, compared with C57, which was 30% corrected in mdx-casp mice. Similarly, 1-year-old mdx EDL muscles had an 11-fold greater loss of force on ECC than C57 muscles, and this was recovered by 27% in mdx-casp muscles, although neither result was statistically significant (Fig. 5C and D). Casp mice were similar to C57 mice in all cases (Fig. 5A–D). Measurement of twitch and absolute tetanic forces of the EDL did not reveal differences between mdx and mdx-casp mice, nor was there any recovery of diaphragm muscle function (Table 1). These results demonstrate that deletion of caspase-12 preserves muscle function in mdx mice at 5 months of age, and that some beneficial effects persist up to 1 year of age. These findings implicate caspase-12 and ER stress in the pathology of dystrophin-deficient muscles.
Figure 5.

Muscle function in C57, casp, mdx and mdx-casp mice. Specific EDL force production by the EDL muscle (A and C) and loss of force production after a series of ECCs (B and D) in C57, casp, mdx and mdx-casp aged 5 months (A and B) and 1 year (C and D). Bars represent mean ± standard error. *Significantly different from C57, ‡significantly different from casp and †significantly different from mdx by one-way ANOVA with Tukey's Multiple Comparison Test. n = 6–11 mice per genotype.
Table 1.
Additional functional and mass data
| 5 months |
1 year |
|||||||
|---|---|---|---|---|---|---|---|---|
| C57 | casp | mdx | mdx-casp | C57 | casp | mdx | mdx-casp | |
| EDL twitch force (mN) | 112 ± 7.2 | 89.9 ± 10.3 | 103 ± 5.9 | 119 ± 4.8b | 120 ± 5.4 | 123 ± 4.7 | 105 ± 6.6 | 98.3 ± 4.6a,b |
| EDL tetanic force (mN) | 406 ± 20.1 | 410 ± 33.4 | 415 ± 14.6 | 396 ± 13.0 | 428 ± 16.6 | 405 ± 15.5 | 403 ± 20.3 | 408 ± 8.4 |
| Diaphragm specific force (N/cm2) | 13.8 ± 1.8 | 13.1 ± 2.7 | 7.9 ± 0.7a | 9.3 ± 1.2 | 17.1 ± 1.1 | 16.5 ± 1.4 | 5.8 ± 0.6a,b | 6.0 ± 0.6a,b |
| Diaphragm force drop after ECCs (%)c | 5.6 ± 1.3 | 6.6 ± 1.9 | 10.5 ± 1.9 | 12.3 ± 2.1 | 0.7 ± 0.9 | 5.6 ± 1.1 | 5.3 ± 0.8 | 6.1 ± 1.5a |
| Body weight (g)c | 29.2 ± 0.8 | 24.0 ± 0.2 | 32.2 ± 0.5 | 31.8 ± 1.1 | 35.7 ± 1.5 | 30.3 ± 1.1a | 33.4 ± 0.7 | 35.0 ± 0.6 |
| Tibialis anterior weight (mg) | 60.6 ± 1.6 | 58.7 ± 1.2 | 75.3 ± 3.3a,b | 67.6 ± 4.3 | 57.9 ± 1.7 | 52.5 ± 3.8 | 66.5 ± 1.9b | 68.2 ± 2.9a,b |
| Soleus weight (mg)c | 10.9 ± 0.6 | 9.5 ± 0.3 | 12.7 ± 0.6 | 12.6 ± 0.6 | 10.7 ± 0.6 | 10.0 ± 0.4 | 13.4 ± 0.6a,b | 13.6 ± 0.7a,b |
| Heart weight (mg) | 134 ± 7.5 | 135 ± 8.2 | 140 ± 3.4 | 136 ± 6.3 | 150 ± 5.7 | 163 ± 0.9 | 154 ± 0.6 | 154 ± 0.3 |
aDifferent to C57 by one-way ANOVA with Tukey's Multiple Comparison Test.
bDifferent to casp by one-way ANOVA with Tukey's Multiple Comparison Test.
cMeans significantly different by one-way ANOVA at 5 months.
Susceptibility to ECCs is an indicator of sarcolemmal fragility. Therefore, serum creatine kinase (CK) was assayed in 5-month-old C57, casp, mdx and mdx-casp mice. Although mdx-casp mice showed a trend towards decreased serum CK compared with mdx mice, the magnitude was relatively small (a 29% recovery) and thus is unlikely to explain the improvement in muscle function (Fig. 6).
Figure 6.

Serum creatine kinase in C57, casp, mdx and mdx-casp mice. CK activity in serum drawn from C57, casp, mdx and mdx-casp mice aged 5 months. Boxes represent 25–75th percentile, lines within boxes represent median and whiskers represent minimum and maximum. *Significantly different from C57 and ‡significantly different from casp by one-way ANOVA with Tukey's Multiple Comparison Test. n = 6–10 animals per genotype.
Caspase-12 null mice have a C57Bl6 background, while mdx mice have a C57Bl10 background, so that mdx-casp mice have a mixed background. We have previously demonstrated that there is no difference in the mechanical properties of these strains (37). To confirm that the improvements in muscle function, hypertrophy and degeneration were not due to the background strain, mdx-casp mice were backcrossed with mdx mice and F2 mdx and mdx-casp littermates were used to key mechanical measurements. F2 mdx mice had a more severe deficit in muscle function than previous mdx mice, which suggested that the introduction of the C57Bl6 background alone did not improve muscle function. F2 mdx-casp mice still displayed an improvement in specific force, resistance to ECCs and reduced muscle mass and CSA, compared with their mdx littermates. However, based on a limited number of animals, the size of the effect was somewhat reduced (data not shown), suggesting there could be genetic modifiers of the phenotype.
Lack of caspase-12 corrects mdx muscle hypertrophy
Muscles from mdx mice have an increased mass and cross-sectional area (CSA), due to compensatory hypertrophy and increased fibrosis. Measurement of EDL muscles from 5-month-old mdx mice showed a 23% increase in mass, compared with C57 EDL muscles, which was completely corrected in mdx-casp mice (Fig. 7A). Furthermore, there was a 28% increase in the calculated CSA of mdx EDL muscles, which was 84% corrected in mdx-casp mice (Fig. 7B). Again, these effects partially persisted in 1-year-old mdx mice, where EDL muscles had a 26% increase in mass and a 36% increase in CSA, and these parameters were corrected in mdx-casp mice by 72 and 53%, respectively (Fig. 7C and D). Casp mice were similar to C57 mice in all cases (Fig. 7A–D). The mass of the TA showed a similar pattern to the mass of the EDL at 5 months of age, but this pattern was lost at 1 year of age. In contrast, the mass of the soleus was increased in mdx mice at 5 months and 1 year of age, but was not affected by deletion of caspase-12. Finally, the mass of the heart was not different between any of the four genotypes (Table 1).
Figure 7.

Mass and cross-sectional area of EDL in C57, casp, mdx and mdx-casp mice. Mass (A and C) and calculated CSA (B and D; see Methods for details) of EDL muscles from C57, casp, mdx and mdx-casp mice aged 5 months (A and C) and 1 year (C and D). Bars represent mean ± standard error. *Significantly different from C57, ‡significantly different from casp and †significantly different from mdx by one-way ANOVA with Tukey's Multiple Comparison Test. n = 6–11 mice per genotype.
Lack of caspase-12 affects fibre size and number but not type
To investigate these differences further, the distribution of fibre sizes was measured in EDL muscles from 5-month-old C57, casp, mdx and mdx-casp mice. In accordance with the known phenotype, mdx EDL muscles had an increased number of smaller fibres (under 700 µm2), representing newly regenerated fibres, and an increased number of very large fibres (over 2100 µm2), representing degenerating or hypertrophied fibres, compared with C57 (Fig. 8A and B). Surprisingly, given their completely normal muscle function (Fig. 5 and Table 1), the distribution of fibre sizes in casp EDL muscles was shifted towards smaller sizes, compared with C57 (Fig. 8A). Mdx-casp mice showed an additive phenotype, with the distribution of fibre sizes being further shifted towards smaller sizes, compared with mdx mice (Fig. 8B). The average fibre size was not different between C57 and mdx EDL muscles, but was decreased by ∼20% in casp and mdx-casp (Fig. 8C).
Figure 8.

Distribution of fibre sizes in C57, casp, mdx and mdx-casp EDL muscles. (A and B) Comparison of fibre size distributions between C57 and casp (A) and mdx and mdx-casp (B) EDL muscles at 5 months of age. (C and D). Average fibre size (C) and total number of fibres (D) in C57, casp, mdx and mdx-casp EDL muscles at 5 months of age. Bars represent means and error bars represent standard error. n = 4–10 animals per genotype.
Mdx mice also showed a 31% increase in the number of fibres in the EDL muscle. This was normalized by 50% in mdx-casp mice (Fig. 8D).
To determine whether the changes in fibre size in the absence of caspase-12 were due to a change in fibre type, the proportion and size of fibres positive for myosin heavy chain (MyHC) IIb were measured in 5-month-old C57, casp, mdx and mdx-casp EDL muscles. The percentage of IIb fibres was not different between the four genotypes, demonstrating that absence of caspase-12 does not result in the loss of the IIb fibres (Fig. 9A). However, the size of IIb fibres was reduced in the absence of caspase-12, reflecting the size reduction observed for total fibres. Specifically, the average size of IIb fibres was reduced by ∼25% in casp and 20% in mdx-casp EDL muscles, compared with C57. (Fig. 9B). As was the case for total fibres, deletion of caspase-12 shifted the IIb fibre size distribution towards smaller fibres in both C57 and mdx mice (Fig. 9C and D). Taken together, these results demonstrate that deletion of caspase-12 does not cause a fibre type shift, at least in the predominant IIb fibre population of the EDL muscle, but rather causes a decrease in the size of the IIb fibres.
Figure 9.

Percentage and size of MyHC IIb positive fibres in C57, casp, mdx and mdx-casp muscles. (A) Percentage of fibres positive for MyHC IIb in EDL muscles of C57, casp, mdx and mdx-casp mice at 5 months of age. (B–D) Average size (B) and size distribution (C and D) of IIb positive fibres in EDL muscles of C57, casp, mdx and mdx-casp mice at 5 months of age. Bars represent means and error bars represent standard error. n = 3–5 animals per genotype.
Neither muscle morphology nor fibrosis is altered in the absence of caspase-12
The increased size of dystrophin-deficient muscles reflects both true hypertrophy and the replacement of muscle with fibrous and adipose tissue. To determine whether a reduction in fibrosis contributed to the correction of mass and CSA in mdx-casp EDL muscles, the collagen content of 5-month-old C57, casp, mdx and mdx-casp EDL muscles was assessed. Collagen content was moderately increased (1.5-fold) in mdx EDL muscles compared with C57 muscles, although this was not statistically significant. Mdx-casp EDL collagen content was not different to mdx (Fig. 10A and C). In order to see any differences in fibrosis more clearly, collagen content was also assessed in 5-month-old diaphragm muscles, where mdx fibrosis is more extensive. Collagen content was increased 2-fold in mdx diaphragm muscles compared with C57, but no reduction in fibrosis was observed in mdx-casp diaphragm muscles (Fig. 10B and D). These findings exclude a decrease in fibrosis as the mechanism of improved muscle function in mdx-casp mice.
Figure 10.

Collagen staining in C57, casp, mdx and mdx-casp EDL and diaphragm. Representative images (A and B) and quantification (C and D) of 5-month-old C57, casp, mdx and mdx-casp EDL (A and C) and diaphragm (B and D) muscles stained for collagen using Sirius Red. Bars represent mean ± standard error. *Significantly different from C57 and ‡significantly different from casp by one-way ANOVA with Tukey's Multiple Comparison Test. n = 3–6 animals per genotype and muscle.
To examine the general morphology of C57 and dystrophic muscle in the presence and absence of caspase-12, TA muscles from 5-month-old C57, casp, mdx and mdx-casp mice were stained with haematoxylin and eosin. Without quantitation of specific features, the general appearance of casp muscles was similar to C57 muscles, while the general appearance of mdx-casp muscles was similar to mdx muscles (Fig. 11).
Figure 11.

General morphology of C57, casp, mdx and mdx-casp muscles. Representative images of TA muscles from 5-month-old C57, casp, mdx and mdx-casp mice, stained with haematoxylin and eosin.
Muscle fibre degeneration is reduced in the absence of caspase-12
Dystrophic muscles undergo continuous cycles of degeneration and regeneration, and centrally nucleated fibres (CNFs) serve as an index of this process. To determine whether the absence of caspase-12 altered the number of regenerating fibres, the percentage of CNFs was measured in 5-month-old C57, casp, mdx and mdx-casp EDL muscles. As has been previously shown, CNFs were greatly increased in mdx EDL muscles (12-fold more than C57). Mdx-casp EDL muscles showed a trend to a slightly lower percentage of CNFs, but this was not statistically significant (Fig. 12). Therefore, a change in the number of regenerating fibres is unlikely to explain the changes in fibre size observed between mdx and mdx-casp mice.
Figure 12.

Fibre central nucleation in C57, casp, mdx and mdx-casp muscles. Percentage of CNFs in EDL muscles of C57, casp, mdx and mdx-casp mice at 5 months of age. Bars represent mean ± standard error. *Significantly different from C57 and ‡significantly different from casp by one-way ANOVA with Tukey's Multiple Comparison Test. n = 4–10 animals per genotype.
Fibres remain centrally nucleated for an extended period after regeneration, and therefore CNFs may not capture all aspects of the degeneration/regeneration process. To measure degeneration in the absence of caspase-12, 5-month-old C57, casp, mdx and mdx-casp EDL muscles were immunostained for IgG to label degenerating fibres. Fibres in two stages of degeneration could be observed. Fibres in late stages of degeneration were small, misshapen and intensely stained with anti-IgG. These fibres were only observed in mdx EDL muscles (Fig. 13, arrow), but not in muscles from C57, casp or, notably, mdx-casp mice. Fibres in earlier stages of degeneration were large, rounded, and had less intense staining for IgG. (Fig. 13, arrowheads). These fibres occurred frequently in mdx muscles but their appearance was reduced in mdx-casp muscles. The total number of degenerating fibres in mdx-casp mice was decreased compared with mdx mice (Fig. 13). Taken together, these data show that the excessive degeneration of muscle fibres in mdx mice is substantially diminished in the absence of caspase-12.
Figure 13.
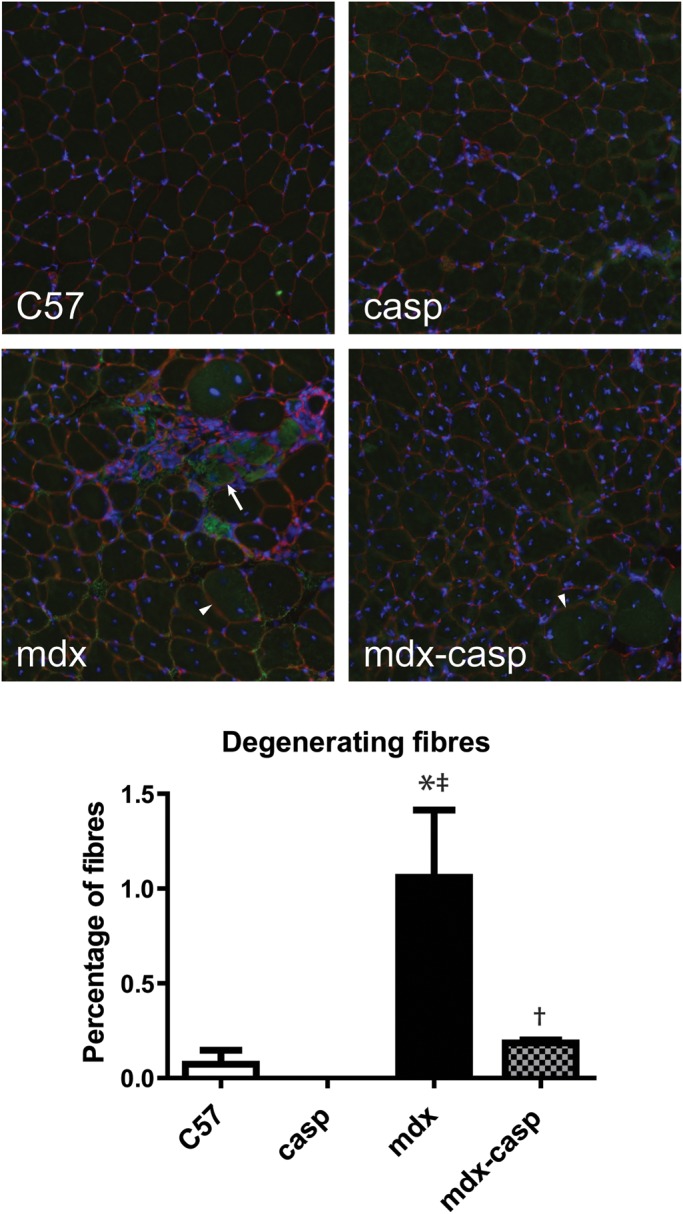
Muscle fibre degeneration in C57, casp, mdx and mdx-casp muscles. Representative images (upper panel) and quantification (lower panel) of IgG immunostaining in 5-month-old C57, casp, mdx and mdx-casp EDL. Arrow shows small, misshapen, intensely stained fibre; arrowheads show large, round, less intensely stained fibres. Green, IgG; red, laminin; blue, DAPI. Bars represent mean ± standard error. *Significantly different from C57, ‡significantly different from casp and †significantly different from mdx by one-way ANOVA with Tukey's Multiple Comparison Test. n = 3–4 mice per genotype.
Apoptosis is minimal in 5-month-old C57 and mdx muscles
Muscle fibre death may occur by necrosis or apoptosis, and one of the functions of caspase-12/4 is to initiate apoptosis. However, apoptosis is not found at high levels in adult muscle, and its contribution to the loss of muscle fibres in dystrophy is unclear. To determine whether the mechanism of recovery in the absence of caspase-12 is likely to involve apoptosis, we used TUNEL to identify nuclei with fragmented DNA in sections from 5-month-old C57 and mdx EDL muscle. We found that the percentage of nuclei with fragmented DNA was low (2.6 ± 1.7% in C57 and 1.7 ± 1.0% in mdx, n = 3–4 animals per genotype) and that there was no significant difference between C57 and mdx by unpaired T test. Thus, the functional improvements imparted by caspase-12 ablation cannot be attributed to a reduction in apoptosis at this age.
DISCUSSION
In this study, we discovered that heightened ER stress and UPR activation occurs in the skeletal muscles of dystrophin-deficient DMD patients and mdx mice. Furthermore, we found that blocking one pathway of the UPR by deletion of the caspase-12 gene in mdx mice preserved muscle function, restoring both specific force production and resistance to ECCs. In addition to these functional improvements, compensatory hypertrophy was abrogated, due to a decrease in fibre size, and the muscle fibre degeneration associated with loss of dystrophin was reduced almost to wild-type levels.
We found heightened UPR activation in mdx TA, EDL and diaphragm muscles at 5 months of age, including increased levels of BiP, pro- and active caspase-12, CHOP and P-JNK. Importantly, BiP and cleaved caspase-4 were also elevated in muscle samples from DMD patients. The increase in caspase-12 is consistent with a previous study that found increased caspase-12 expression in the masseter of young mdx mice (38). Increased P-JNK, which is not specific to ER stress, has also been previously observed in 8-week-old mdx TA muscles (39) and 5-month-old mdx hearts (40). However, to our knowledge, this is the first demonstration that the UPR components BiP and CHOP are upregulated in dystrophic muscle.
We found that deletion of caspase-12 in mdx mice led to a 75% recovery in EDL specific force production, and a 75% recovery in resistance to ECCs. These results compare favourably with other studies using transgenesis or gene targeting to modulate gene expression in the mdx mouse (Table 2). For example, expression of a minidystrophin gene completely preserved EDL specific force, but only led to a 20% recovery in resistance to ECCs (41), while overexpression of the dystrophin homologue utrophin completely preserved specific force in EDL and diaphragm muscles, and resistance to ECCs in EDL (42). Overexpression of γ-actin reduced susceptibility of the EDL to ECCs by ∼40% compared with mdx, but did not improve specific force (43). C57 controls were not included in this study, so percentage recovery could not be calculated. For comparison, deletion of caspase-12 in our study reduced ECC susceptibility by 55% compared to mdx mice. IGF-I overexpression led to a 45% recovery of EDL susceptibility to ECCs with no difference in specific force (44). Overexpression of the TRPV2 ion channel in mdx mice led to a 55% recovery of ECC susceptibility in the EDL (45). Knockout of the MAPK phosphatase MKP5 led to an 80% recovery of the specific force of the EDL (46). Therefore, with the exception of studies using utrophin or dystrophin itself, the recovery in muscle function that we obtained was comparable to, or better than, other studies that used similar protocols and ages of mice. It is worth noting that many studies on the mdx mouse rely on biochemical or histological assessment of the phenotype, and do not assess muscle function directly.
Table 2.
Comparison with other studies
| Study | mdx mouse intervention | Age of mice | EDL specific force recovery, % | EDL ECC resistance recovery, % | Reduction in ECC susceptibility compared with mdx, % |
|---|---|---|---|---|---|
| Moorwood and Barton 2014 | Caspase-12 deletion | 5 months | 75 | 75 | 55 |
| Dunant et al. 2013 | Minidystrophin expression in muscle | 7 months | 100 | 20 | |
| Tinsley et al. 1998 | Utrophin overexpression in muscle | 3 months | 100 | 100 | |
| Baltgalvis et al. 2011 | γ-actin overexpression in muscle | 4 months | No recovery | Not determined (no C57 controls) | 40 |
| Barton et al. 2002 | IGF-I overexpression in muscle | 3 months | No recovery | 45 | |
| Zanou et al. 2009 | TRPV2 overexpression in muscle | 3–4 months | No recovery | 55 | |
| Shi et al. 2013 | MKP5 deletion | Not given | 80 | Not measured |
Our mdx-casp mouse strain had a mixed C57Bl10 and C57Bl6 background, while mdx mice have a C57Bl10 background. To ensure that the recovery in function in mdx-casp mice was not due to background differences, we backcrossed mdx-casp mice with mdx mice and compared mdx-casp and mdx littermates. We found that mdx littermates with a mixed background did not have improved muscle function compared with normal mdx mice, implying that introduction of the C57Bl6 background alone does not result in improvements in muscle function. This is consistent with our previous work demonstrating that there is no difference in the mechanical properties of C57Bl6 and C57Bl10 muscles (37). Furthermore, the same differences in muscle function and size were found when comparing mdx and mdx-casp mice with identical backgrounds. However, the magnitude of the differences was different, suggesting that there may be genetic modifiers that influence the effects of caspase-12 ablation in mdx mice.
When we compared 1-year-old mice to 5-month-old mice, we found that the beneficial effects of caspase-12 deletion were partially preserved, but less pronounced than at 5 months. It may simply be that as other pathological processes not affected by ER stress become more prevalent, this outweighs the benefits provided by caspase-12 ablation. Indeed, in the mdx diaphragm, where pathology is more severe, there was no improvement in muscle function at 5 months of age, although we found that ER stress was also heightened in this muscle. An important future direction will be to investigate whether caspase-12 ablation preserves diaphragm function at earlier ages. Similarly, backcrossing mdx-casp mice onto the mdx background and comparing littermates produced more severely affected mdx mice and a less pronounced improvement in muscle function and hypertrophy, again suggesting that the degree of pathology affects the degree of recovery brought about by caspase-12 ablation.
Caspase-12 deletion reduced myofibre size, and counteracted the hypertrophy present in mdx muscles. However, there were some differences in the degree of this effect between different muscles. Whereas muscle mass was completely normalized in the EDL, the mass of the TA was only normalized by 50%, and the mass of the soleus was not affected. The lack of effect on the soleus could be due to its different fibre type composition, having mostly type I and IIa fibres with little or no IIb fibres, compared with the EDL, which has predominantly type IIb fibres (47). The fact that changes in the mass of the TA were more moderate supports this, because the TA is composed of all three fibre types.
Our data show that caspase-12 deletion prevented much of the susceptibility to ECC-induced damage in mdx mice. Susceptibility to ECCs is considered an indicator of sarcolemmal fragility, as is increased serum CK. However, we did not find a significant decrease in CK in mdx-casp mice compared with mdx mice. This indicates that baseline sarcolemmal permeability and/or membrane resealing was not improved in the absence of caspase-12. During ECCs, however, mdx-casp muscles are protected from further sarcolemmal damage or from damage to the contractile apparatus, possibly because of their smaller size, as discussed below. The fact that muscle fibre degeneration is reduced in mdx-casp muscles is consistent with their protection from contractile damage.
The mechanism by which caspase-12 deletion preserves mdx muscle function is not known. Possible mechanisms include a reduction in apoptosis, improvement in regeneration, protection of contractile proteins from proteolytic degradation or a greater robustness conferred by smaller fibre size. These are discussed in more detail in what follows.
Caspase-12 and its human equivalent caspase-4 are activated specifically by ER stress, leading to the activation of caspase-9, which activates caspase-3, leading to apoptosis. Therefore, one possible mechanism by which caspase-12 deletion preserves the function of mdx muscles is through a reduction in apoptosis. Muscle fibre death by apoptosis occurs in DMD and in the mdx mouse, although its contribution to the dystrophic phenotype remains unclear (48,49). We conducted our studies in 5-month- and 1-year-old mice, and consistent with previous studies (50,51), we found that levels of apoptosis are minimal by 5 months of age in mdx muscle, and are not greater than in C57 muscle. However, prevention of apoptosis at an earlier age could have led to a preservation of muscle function in the mdx-casp mouse, delaying loss of function. This could explain the partial loss of the recovery in 1-year-old mice, because the deterioration is delayed, rather than prevented completely. As discussed earlier, the progression of other pathological processes that do not involve ER stress and the UPR may gradually overcome the benefit gained through caspase-12 deletion. Studies in young or exercised mice would help to elucidate any differences in muscle fibre apoptosis between mdx and mdx-casp mice.
Myofibre degeneration and death by necrosis, and subsequent regeneration, persists into adulthood in mdx mice. We found a reduction in the number of degenerating fibres in mdx-casp mice compared with mdx, indicating that caspase-12 deletion promotes muscle fibre survival. We did not find a significant decrease in CNFs in mdx-casp mice, suggesting that the degree of regeneration was not different. Hence, a shift in the balance between degeneration and regeneration may underlie the improvements in muscle function. Interestingly, caspases have been implicated in the process of myogenesis, in some cases in an apoptosis-independent manner (52–55). Specifically, the UPR is activated during differentiation of cultured myoblasts and during developmental myogenesis, and caspase-12, -9 and -3 activation occurs in apoptotic cells during in vitro differentiation and in vivo myogenesis. However, active caspase-12 is also present in non-apoptotic myogenic cells during development, indicating that it has additional functions beyond initiating apoptosis (56). Several studies suggest a role for caspase-12, -9 and -3 in promoting myoblast differentiation (52,53,55). In contrast, Moresi et al. (54) found that increased caspase activity delayed muscle regeneration, by a mechanism independent of caspase-3 and apoptosis, while caspase inhibition improved muscle regeneration.
Little is known about the substrates of caspase-12/4, other than caspase-9 and autoprocessing. Because both the total amount of caspase-12 and the extent of its cleavage are greater in mdx muscle, its protein targets would be more susceptible to degradation. Although the identification of caspase-12 substrates was not part of the current study, it is possible that caspase-12 may target a protein or proteins of the muscle contractile apparatus, leading to loss of contractile capacity when caspase-12 is chronically over-activated. This would then lead to the preservation of force generating capacity and resistance to damage observed when caspase-12 is deleted in mdx mice.
It is not clear whether the lack of hypertrophy in the absence of caspase-12 in mdx mice is a cause or a consequence of the recovery of muscle function. It may be the case that in the absence of a large deficit in function, hypertrophy is not triggered as a compensatory measure. On the other hand, it is possible that hypertrophy in the context of dystrophin deficiency is actually maladaptive, and that blocking this response by deleting caspase-12 results in more robust muscle fibres. The latter possibility is supported by the fact that casp mice also had smaller fibres, suggesting that fibre size reduction is a primary outcome of caspase-12 deletion. Hypertrophy might be maladaptive in mdx mice if the blood supply did not adapt to the increased size of the muscles, leading to less efficient oxygenation, if nuclear number was not increased, resulting in failure to maintain the contractile apparatus (57), or if larger fibres resulted in a greater mechanical impact on the sarcolemma. Interestingly, myostatin-null mice exhibit muscle hypertrophy coupled with a decrease in force production, while food restriction normalizes muscle mass and preserves specific force in these animals, suggesting that fibre size economy may be beneficial (58).
A key question in light of our findings is whether caspase-4, the human equivalent of caspase-12, could be targeted pharmaceutically as a potential therapy for DMD. We observed increased levels of BiP and cleaved caspase-4 in muscles of patients with DMD, demonstrating that ER stress is heightened and the UPR is activated in dystrophic human muscle. This strongly suggests that, similar to the mdx mouse, aberrant UPR activation may contribute to pathogenesis in humans with DMD. It is encouraging to note that there is already an FDA-approved, orally administered drug, ursodeoxycholic acid (Ursodiol), that is known to alleviate ER stress, including caspase-12/4 activation (http://www.accessdata.fda.gov/scripts/cder/drugsatfda/ (last accessed on 27 May 2014)), and that the closely related drug, tauro-UDCA (TUDCA), is in clinical trials (http://clinicaltrials.gov/ct2/results?term=TUDCA (last accessed on 27 May 2014)). These compounds are broad inhibitors of the UPR, and have been shown to be beneficial in conditions where ER stress occurs, such as diabetes and obesity (59–62). On the other hand, a more specific drug may be desirable. Caspase-12 peptide inhibitors have been developed, such as Z-ATAD-FMK, which has been used widely in vitro, and Q-ATAD-OPH, which was designed for in vivo use. Future studies are warranted to evaluate pharmacological inhibition of this pathway for its therapeutic potential.
It is important to make clear that targeting ER stress and caspase-12/4 in DMD will not correct the primary defect, i.e. lack of dystrophin. However, there is currently no effective treatment for this devastating disease, and any drug that can substantially alleviate the decline in muscle function, especially one that could be brought rapidly to clinical trials, would be of enormous benefit to the patient community and their families. Moreover, a drug treatment that targets one of the secondary pathological consequences of dystrophin loss, i.e. ER stress, could be used in combination with other treatments currently being developed to address the primary deficit, such as exon-skipping, gene therapy or utrophin upregulation, which show great promise, but are not by any means 100% efficacious (63–69).
In summary, our findings reveal ER stress and caspase-12/4 as a novel pathway contributing to the pathology of dystrophinopathies, and identify human caspase-4 as a potential therapeutic target for DMD. Further studies will be important to fully understand the mechanism by which caspase-12 deletion preserves dystrophic muscle function, and to determine whether pharmaceutical targeting of caspase-12/4 produces similar results. Encouragingly, drugs targeting this pathway are already FDA-approved or in clinical trials, and could be rapidly brought to clinical use if found to be effective in animal models.
MATERIALS AND METHODS
Animals
All animal experiments were approved by the University of Pennsylvania Institutional Animal Care and Use Committee. Caspase-12-null (casp) mice were a generous gift of the Yuan lab (Harvard University) (29). These were maintained on a C57Bl/6 background strain. C57Bl/6 mice were used as wild-type controls, and mdx (C57Bl/10 background strain) were used as dystrophic controls. Male casp mice were bred with female mdx mice, resulting in a hemizygous F1 generation. Males and females from this generation were bred, resulting in a group of mice that were homozygous null for Caspase-12 and also homozygous for the X-linked mdx mutation. Screening to confirm the mdx dystrophin gene mutation was done by PCR using primers spanning the site of mutation, and subsequent sequencing of the product. Screening to confirm caspase-12 ablation was done by PCR using pairs of primers specific to the wild-type and ablated caspase-12 gene, respectively. The F2 generation had the expected proportions of homozygosity according to Mendelian genetics. Mice exhibiting the desired genotype were bred to establish homozygotic lines of mdx-casp mice. Mice from the F4 and subsequent generations were used for analysis. For all animal groups, experiments were performed at 5 months and 1 year of age for functional analysis and at 5 months for all other analyses.
Immunoblotting
TA, EDL and diaphragm muscles were dissected from 5-month-old C57, casp, mdx and mdx-casp mice, snap-frozen in liquid nitrogen and stored in liquid nitrogen. Lysates were prepared using RIPA buffer (50 mm HEPES pH7.5, 150 mm NaCl, 5 mm EDTA, 1 mm EGTA, 15 mm p-nitrophenyl phosphate disodium hexahydral, 1% NP-40, 0.1% SDS, 1% deoxycholate, 0.025% sodium azide) with protease and phosphatase inhibitor cocktails added (Sigma). Protein concentration was assayed using a Bradford method protein assay kit (Bio-Rad). 30–90 μg total protein were separated by SDS–PAGE using precast Tris–HCl gels (Bio-Rad) and transferred to PVDF membranes, which were blocked in 5% non-fat milk in TBST (10 mm Tris, 150 mm NaCl, 0.1% v/v Tween-20, pH8), then probed with antibodies to BiP (Becton Dickinson 60979), CHOP (Santa Cruz sc-7351), caspase-12, caspase-4 (Abcam ab10455 and ab22687), P-JNK, total (T)-JNK (Cell Signaling 9255 and 9252), GAPDH (Santa Cruz sc-32233) and tubulin (Sigma T5168) diluted in blocking solution. Membranes were washed with three changes of blocking solution for 10 min each, incubated with HRP-conjugated secondary antibodies (Cell Signaling), washed with three changes of TBST for 10 min each and labelled bands visualized using ECL Plus reagent (Pearce) and autoradiography film (Kodak/GeneMate). Band densities were quantified using ImageJ (NIH). BiP, CHOP and caspase-12 were normalized to tubulin and P-JNK was normalized to T-JNK.
For human samples, 25 pooled, 10 µm frozen sections of quadriceps biopsies from DMD patients and healthy male controls were lysed in RIPA buffer, and 5% of the lysate was used for immunoblotting, as described earlier. Samples were obtained from the Muscular Dystrophy Tissue and Cell Repository at the University of Iowa. DMD patients were aged between 5 and 8 years, and controls were aged between 3 and 13 years.
Muscle functional testing
Five-month and 1-year-old male C57, casp, mdx and mdx-casp mice were anaesthetized using ketamine and xylazine, before EDL and diaphragm muscles were dissected. For the diaphragm, a strip was cut, following the line of the muscle fibres. EDL muscles and diaphragm strips were secured in an organ bath filled with oxygenated Ringer's solution (119 mm NaCl, 4.74 mm KCl, 2.54 mm CaCl2, 1.18 mm KH2PO4, 1.18 mm MgSO4, 25 mm HEPES, 2.75 mm glucose, 25 mm HEPES, 2.75 mm glucose) and adjusted to their optimum length (Lo) based on maximum twitch force. Lo was measured with Vernier callipers before the muscle was subjected to functional testing. Twitch force was measured by stimulating with a single 0.5 ms pulse at supramaximal stimulation. Tetanic force was measured by stimulating for 500 ms at 120 Hz for EDL and 800 ms at 100 Hz for diaphragm, at supramaximal stimulation. ECCs consisted of an isometric tetanic contraction lasting 700 ms with a stretch of 10% Lo superimposed during the last 200 ms. ECCs were repeated five times with a 5 min rest in between. For further details on muscle functional testing refer to Moorwood et al. (70). Following functional testing, EDL muscles and diaphragm strips were pinned at approximately physiological length, embedded in OCT (Thermo Scientific) and frozen in liquid nitrogen-cooled isopentane. Samples were stored at −80°C for subsequent morphological analysis.
CSAs of the EDL muscles and diaphragm strips were calculated using the formula: CSA (mm2) = mass (mg)/[Lo (mm)×(L/Lo)×1.06 mg/mm3], where Lo is the optimum muscle length, L is the fibre length and 1.06 mg/mm3 is the density of muscle. L/Lo is 0.45 for the EDL and 1 for the diaphragm. Specific force was calculated by the formula: specific force (N/cm2) = [absolute force (mN) /CSA (mm2)]×0.1.
Serum CK assay
Blood was drawn from anaesthetized mice by cardiac puncture prior to beginning dissection, and allowed to clot at room temperature before centrifuging for 20 min at 2000 g. Supernatants (serum) were retained and stored in a liquid nitrogen freezer. Creatine kinase activity was assayed using a creatine kinase-SL assay kit from Sekisui Diagnostics, using the manufacturer's protocol adapted for a 96-well plate reader.
Fibre size distribution, fibre type, central nucleation and degeneration measurement
Frozen 10 µm sections were cut from EDL muscles of 5-month-old C57, casp, mdx and mdx-casp mice. Sections were washed in PBS, blocked in 5% bovine serum albumin in PBS, and immunostained using antibodies to laminin (Thermo Scientific), to label the sarcolemma, and MyHC IIb (Developmental Studies Hybridoma Bank). For degeneration, anti-mouse IgG was added without any mouse primary antibody to label degenerating fibres (71), which were co-stained with laminin. Nuclei were labelled with DAPI incorporated into the mounting media (Vectashield). Images were taken using a Leica DM RBE microscope and Leica DFC350FX camera, and image analysis to measure the CSA of the fibres, the percentage of fibres positive for MyHC IIb and the percentage of fibres with central nuclei was done using Matlab (Mathworks). Degenerating fibres were counted manually and expressed as a percentage of total fibres counted on serial sections.
Haematoxylin and eosin staining
Frozen 10 µm sections of TA muscle from 5-month-old C57, casp, mdx and mdx-casp were incubated in two changes of PBS for 10 min each, then incubated in Haematoxylin Stain 1, Gill Method solution (Fisher) for 5 min. After washing in distilled water, they were incubated for 1 min in Eosin Y solution alcoholic (Sigma), washed twice in 90% ethanol, twice in 100% ethanol and twice in Citrisolv (Fisher) before mounting with Cytoseal 60 (Thermo Scientific). Images were taken using a Nikon Eclipse 80i microscope and Nikon Digital Sight DS-Fi1 camera.
Measurement of apoptotic index
Detection of DNA fragmentation in 10 µm frozen sections of EDL from 5-month-old C57 and mdx mice was done using the FragEL DNA Fragmentation Detection Kit, Colorimetric – TdT enzyme (EMD Millipore). The manufacturer's protocol was used, with the following modifications: the H2O2 incubation was omitted because it was found to increase background, the TdT enzyme was used at a dilution of 1:80 and the TdT incubation time was 30 min. Images were taken using a Nikon Eclipse 80i microscope and Nikon Digital Sight DS-Fi1 camera. One field from the centre of each EDL muscle was analysed. Labelled nuclei were counted and expressed as a percentage of total nuclei (counterstained with methyl green).
Measurement of fibrosis
Frozen 10 μm sections were cut from EDL muscles and diaphragm strips of 5-month-old C57, casp, mdx and mdx-casp mice. Sections were fixed in 4% formaldehyde for 10 min, rinsed in distilled water, air dried and stained in Pico-Sirius Red (1% w/v Direct Red 80 in saturated picric acid) for 1 h. Sections were washed in two changes of acidified water (5% v/v glacial acetic acid in tap water), dehydrated in three changes of 100% ethanol, cleared with Citrisolv (Fisher) and mounted with Cytoseal (Thermo Scientific). Images were taken using a Nikon Eclipse 80i microscope and Nikon Digital Sight DS-Fi1 camera, and image analysis to measure the percentage area positive for collagen staining was done using Matlab.
Statistics
Statistical significance was determined using one-way ANOVA with Tukey's Multiple Comparison Test for all comparisons between C57, casp, mdx and mdx-casp mice. For comparison of caspase-12 levels between C57 and mdx mice a two-tailed, unpaired T test was used, assuming equal variance. For analysis of BiP levels between control and DMD biopsies, a Mann–Whitney test was used. For analysis of cleaved caspase-4, a one sample T test was used, comparing to 0.
Percentage recovery was defined as 100×[(mdx-casp value−mdx value)/(C57 value−mdx value)], as described in Treat-NMD Protocol DMD_M.1.1.001.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by National Institutes of Health grant number AR052646 to E.R.B. C.M. was supported by a National Institutes of Health Wellstone Muscular Dystrophy Cooperative Research Center Training Grant.
Supplementary Material
ACKNOWLEDGEMENTS
Caspase-12-null mice were a generous gift of Dr Junying Yuan, Harvard University. Control and DMD human muscle samples were kindly provided by Steven Moore at the University of Iowa Muscular Dystrophy Tissue and Cell Repository. The authors thank Dr Kelly Jordan-Sciutto for helpful discussions and Ms Anya Gilroy and Ms Samantha Lincoln for technical assistance.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Mendell J.R., Shilling C., Leslie N.D., Flanigan K.M., al-Dahhak R., Gastier-Foster J., Kneile K., Dunn D.M., Duval B., Aoyagi A., et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann. Neurol. 2012;71:304–313. doi: 10.1002/ana.23528. [DOI] [PubMed] [Google Scholar]
- 2.Emery A.E., Muntoni F. Duchenne Muscular Dystrophy. Oxford: Oxford University Press; 2003. [Google Scholar]
- 3.Gailly P. New aspects of calcium signaling in skeletal muscle cells: implications in Duchenne muscular dystrophy. Biochim. Biophys. Acta. 2002;1600:38–44. doi: 10.1016/s1570-9639(02)00442-9. [DOI] [PubMed] [Google Scholar]
- 4.Engel A.G., Franzini-Armstrong C. Myology: Basic and Clinical. McGraw-Hill, Inc; 2004. Columbus, OH. [Google Scholar]
- 5.Smith P.E.M., Calverley P.M.A., Edwards R.H.T. Hypoxemia during Sleep in Duchenne Muscular Dystrophy. Am. J. Respir. Crit. Care Med. 1988;137:884–888. doi: 10.1164/ajrccm/137.4.884. [DOI] [PubMed] [Google Scholar]
- 6.Sander M., Chavoshan B., Harris S.A., Iannaccone S.T., Stull J.T., Thomas G.D., Victor R.G. Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. PNAS. 2000;97:13818–13823. doi: 10.1073/pnas.250379497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim J.H., Kwak H.B., Thompson L.V., Lawler J.M. Contribution of oxidative stress to pathology in diaphragm and limb muscles with Duchenne muscular dystrophy. J. Muscle Res. Cell Motil. 2013;34:1–13. doi: 10.1007/s10974-012-9330-9. [DOI] [PubMed] [Google Scholar]
- 8.Kohler M., Clarenbach C.F., Bahler C., Brack T., Russi E.W., Bloch K.E. Disability and survival in Duchenne muscular dystrophy. J. Neurol. Neurosurg. Psychiatry. 2009;80:320–325. doi: 10.1136/jnnp.2007.141721. [DOI] [PubMed] [Google Scholar]
- 9.Brostrom M.A., Brostrom C.O. Calcium dynamics and endoplasmic reticular function in the regulation of protein synthesis: implications for cell growth and adaptability. Cell Calcium. 2003;34:345–363. doi: 10.1016/s0143-4160(03)00127-1. [DOI] [PubMed] [Google Scholar]
- 10.Hotokezaka Y., van Leyen K., Lo E.H., Beatrix B., Katayama I., Jin G., Nakamura T. [alpha]NAC depletion as an initiator of ER stress-induced apoptosis in hypoxia. Cell Death Differ. 2009;16:1505–1514. doi: 10.1038/cdd.2009.90. [DOI] [PubMed] [Google Scholar]
- 11.Pan C., Prentice H., Price A.L., Wu J.Y. Beneficial effect of taurine on hypoxia- and glutamate-induced endoplasmic reticulum stress pathways in primary neuronal culture. Amino Acids. 2011;43:845–855. doi: 10.1007/s00726-011-1141-6. [DOI] [PubMed] [Google Scholar]
- 12.Sawada N., Yao J., Hiramatsu N., Hayakawa K., Araki I., Takeda M., Kitamura M. Involvement of hypoxia-triggered endoplasmic reticulum stress in outlet obstruction-induced apoptosis in the urinary bladder. Lab Invest. 2008;88:553–563. doi: 10.1038/labinvest.2008.21. [DOI] [PubMed] [Google Scholar]
- 13.Xue X., Piao J.H., Nakajima A., Sakon-Komazawa S., Kojima Y., Mori K., Yagita H., Okumura K., Harding H., Nakano H. Tumor necrosis factor alpha (TNFalpha) induces the unfolded protein response (UPR) in a reactive oxygen species (ROS)-dependent fashion, and the UPR counteracts ROS accumulation by TNFalpha. J. Biol. Chem. 2005;280:33917–33925. doi: 10.1074/jbc.M505818200. [DOI] [PubMed] [Google Scholar]
- 14.Guo R., Ma H., Gao F., Zhong L., Ren J. Metallothionein alleviates oxidative stress-induced endoplasmic reticulum stress and myocardial dysfunction. J. Mol. Cell. Cardiol. 2009;47:228–237. doi: 10.1016/j.yjmcc.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang C., Koulajian K., Schuiki I., Zhang L., Desai T., Ivovic A., Wang P., Robson-Doucette C., Wheeler M.B., Minassian B., et al. Glucose-induced beta cell dysfunction in vivo in rats: link between oxidative stress and endoplasmic reticulum stress. Diabetologia. 2012;55:1366–1379. doi: 10.1007/s00125-012-2474-8. [DOI] [PubMed] [Google Scholar]
- 16.Rutkowski D.T., Kaufman R.J. That which does not kill me makes me stronger: adapting to chronic ER stress. Trends Biochem. Sci. 2007;32:469–476. doi: 10.1016/j.tibs.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 17.Iwawaki T., Akai R., Kohno K., Miura M. A transgenic mouse model for monitoring endoplasmic reticulum stress. Nat. Med. 2004;10:98–102. doi: 10.1038/nm970. [DOI] [PubMed] [Google Scholar]
- 18.Lavery G.G., Walker E.A., Turan N., Rogoff D., Ryder J.W., Shelton J.M., Richardson J.A., Falciani F., White P.C., Stewart P.M., et al. Deletion of hexose-6-phosphate dehydrogenase activates the unfolded protein response pathway and induces skeletal myopathy. J. Biol. Chem. 2008;283:8453–8461. doi: 10.1074/jbc.M710067200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin Y.-Y., White R.J., Torelli S., Cirak S., Muntoni F., Stemple D.L. Zebrafish Fukutin family proteins link the unfolded protein response with dystroglycanopathies. Hum. Mol. Genet. 2011;20:1763–1775. doi: 10.1093/hmg/ddr059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hitomi J., Katayama T., Eguchi Y., Kudo T., Taniguchi M., Koyama Y., Manabe T., Yamagishi S., Bando Y., Imaizumi K., et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Aβ-induced cell death. J. Cell Biol. 2004;165:347–356. doi: 10.1083/jcb.200310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fischer H., Koenig U., Eckhart L., Tschachler E. Human caspase 12 has acquired deleterious mutations. Biochem. Biophys. Res. Comm. 2002;293:722–726. doi: 10.1016/S0006-291X(02)00289-9. [DOI] [PubMed] [Google Scholar]
- 22.Saleh M., Vaillancourt J.P., Graham R.K., Huyck M., Srinivasula S.M., Alnemri E.S., Steinberg M.H., Nolan V., Baldwin C.T., Hotchkiss R.S., et al. Differential modulation of endotoxin responsiveness by human caspase-12 polymorphisms. Nature. 2004;429:75–79. doi: 10.1038/nature02451. [DOI] [PubMed] [Google Scholar]
- 23.Nakagawa T., Yuan J. Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J. Cell Biol. 2000;150:887–894. doi: 10.1083/jcb.150.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li C., Wei J., Li Y., He X., Zhou Q., Yan J., Zhang J., Liu Y., Liu Y., Shu H.-B. Transmembrane protein 214 (TMEM214) mediates endoplasmic reticulum stress-induced caspase 4 enzyme activation and apoptosis. J. Biol. Chem. 2013;288:17908–17917. doi: 10.1074/jbc.M113.458836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalai M., Lamkanfi M., Denecker G., Boogmans M., Lippens S., Meeus A., Declercq W., Vandenabeele P. Regulation of the expression and processing of caspase-12. J. Cell Biol. 2003;162:457–467. doi: 10.1083/jcb.200303157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Faucheu C., Diu A., Chan A.W., Blanchet A.M., Miossec C., Herve F., Collard-Dutilleul V., Gu Y., Aldape R.A., Lippke J.A., et al. A novel human protease similar to the interleukin-1 beta converting enzyme induces apoptosis in transfected cells. Embo J. 1995;14:1914–1922. doi: 10.1002/j.1460-2075.1995.tb07183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Munday N.A., Vaillancourt J.P., Ali A., Casano F.J., Miller D.K., Molineaux S.M., Yamin T.T., Yu V.L., Nicholson D.W. Molecular cloning and pro-apoptotic activity of ICErelII and ICErelIII, members of the ICE/CED-3 family of cysteine proteases. J. Biol. Chem. 1995;270:15870–15876. doi: 10.1074/jbc.270.26.15870. [DOI] [PubMed] [Google Scholar]
- 28.Kamens J., Paskind M., Hugunin M., Talanian R.V., Allen H., Banach D., Bump N., Hackett M., Johnston C.G., Li P., et al. Identification and characterization of ICH-2, a novel member of the interleukin-1 beta-converting enzyme family of cysteine proteases. J. Biol. Chem. 1995;270:15250–15256. doi: 10.1074/jbc.270.25.15250. [DOI] [PubMed] [Google Scholar]
- 29.Nakagawa T., Zhu H., Morishima N., Li E., Xu J., Yankner B.A., Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 30.Morishima N., Nakanishi K., Takenouchi H., Shibata T., Yasuhiko Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J. Biol. Chem. 2002;277:34287–34294. doi: 10.1074/jbc.M204973200. [DOI] [PubMed] [Google Scholar]
- 31.Yukioka F., Matsuzaki S., Kawamoto K., Koyama Y., Hitomi J., Katayama T., Tohyama M. Presenilin-1 mutation activates the signaling pathway of caspase-4 in endoplasmic reticulum stress-induced apoptosis. Neurochem. Int. 2008;52:683–687. doi: 10.1016/j.neuint.2007.08.017. [DOI] [PubMed] [Google Scholar]
- 32.Fujita E., Kouroku Y., Jimbo A., Isoai A., Maruyama K., Momoi T. Caspase-12 processing and fragment translocation into nuclei of tunicamycin-treated cells. Cell Death Differ. 2002;9:1108–1114. doi: 10.1038/sj.cdd.4401080. [DOI] [PubMed] [Google Scholar]
- 33.Rao R.V., Hermel E., Castro-Obregon S., del Rio G., Ellerby L.M., Ellerby H.M., Bredesen D.E. Coupling endoplasmic reticulum stress to the cell death program. J. Biol. Chem. 2001;276:33869–33874. doi: 10.1074/jbc.M102225200. [DOI] [PubMed] [Google Scholar]
- 34.Matsuzaki S., Hiratsuka T., Kuwahara R., Katayama T., Tohyama M. Caspase-4 is partially cleaved by calpain via the impairment of Ca2+ homeostasis under the ER stress. Neurochem. Int. 2009;56:352–356. doi: 10.1016/j.neuint.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 35.Yoneda T., Imaizumi K., Oono K., Yui D., Gomi F., Katayama T., Tohyama M. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem. 2001;276:13935–13940. doi: 10.1074/jbc.M010677200. [DOI] [PubMed] [Google Scholar]
- 36.Kim S.-J., Zhang Z., Hitomi E., Lee Y.-C., Mukherjee A.B. Endoplasmic reticulum stress-induced caspase-4 activation mediates apoptosis and neurodegeneration in INCL. Hum. Mol. Genet. 2006;15: 1826–1834. doi: 10.1093/hmg/ddl105. [DOI] [PubMed] [Google Scholar]
- 37.Barton E.R. Impact of sarcoglycan complex on mechanical signal transduction in murine skeletal muscle. Am. J. Physiol. Cell Physiol. 2006;290:C411–C419. doi: 10.1152/ajpcell.00192.2005. [DOI] [PubMed] [Google Scholar]
- 38.Honda A., Abe S., Hiroki E., Honda H., Iwanuma O., Yanagisawa N., Ide Y. Activation of caspase 3, 9, 12, and Bax in masseter muscle of mdx mice during necrosis. J. Muscle Res. Cell Motil. 2007;28:243–247. doi: 10.1007/s10974-007-9122-9. [DOI] [PubMed] [Google Scholar]
- 39.Smythe G.M., Forwood J.K. Altered mitogen-activated protein kinase signaling in dystrophic (mdx) muscle. Muscle Nerve. 2012;46:374–383. doi: 10.1002/mus.23312. [DOI] [PubMed] [Google Scholar]
- 40.Megeney L.A., Kablar B., Perry R.L.S., Ying C., May L., Rudnicki M.A. Severe cardiomyopathy in mice lacking dystrophin and MyoD. PNAS. 1999;96:220–225. doi: 10.1073/pnas.96.1.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dunant P., Larochelle N., Thirion C., Stucka R., Ursu D., Petrof B.J., Wolf E., Lochmuller H. Expression of dystrophin driven by the 1.35-kb MCK promoter ameliorates muscular dystrophy in fast, but not in slow muscles of transgenic MDX mice. Mol. Ther. 2003;8:80–89. doi: 10.1016/s1525-0016(03)00129-1. [DOI] [PubMed] [Google Scholar]
- 42.Tinsley J., Deconinck N., Fisher R., Kahn D., Phelps S., Gillis J.M., Davies K. Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat. Med. 1998;4:1441–1444. doi: 10.1038/4033. [DOI] [PubMed] [Google Scholar]
- 43.Baltgalvis K.A., Jaeger M.A., Fitzsimons D.P., Thayer S.A., Lowe D.A., Ervasti J.M. Transgenic overexpression of gamma-cytoplasmic actin protects against eccentric contraction-induced force loss in mdx mice. Skelet. Muscle. 2011;1:32. doi: 10.1186/2044-5040-1-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barton E.R., Morris L., Musaro A., Rosenthal N., Sweeney H.L. Muscle-specific expression of insulin-like growth factor I counters muscle decline in mdx mice. J. Cell Biol. 2002;157:137–148. doi: 10.1083/jcb.200108071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zanou N., Iwata Y., Schakman O., Lebacq J., Wakabayashi S., Gailly P. Essential role of TRPV2 ion channel in the sensitivity of dystrophic muscle to eccentric contractions. FEBS Lett. 2009;583:3600–3604. doi: 10.1016/j.febslet.2009.10.033. [DOI] [PubMed] [Google Scholar]
- 46.Shi H., Verma M., Zhang L., Dong C., Flavell R.A., Bennett A.M. Improved regenerative myogenesis and muscular dystrophy in mice lacking Mkp5. J. Clin. Invest. 2013;123:2064–2077. doi: 10.1172/JCI64375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Augusto V., Padovani C.R., Campos G.E.R. Skeletal muscle fiber types in C57BL6J mice. Braz. J. Morphol. Sci. 2004;21:89–94. [Google Scholar]
- 48.Miller J.B., Girgenrath M. The role of apoptosis in neuromuscular diseases and prospects for anti-apoptosis therapy. Trends Mol. Med. 2006;12:279–286. doi: 10.1016/j.molmed.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 49.Sandri M., Carraro U. Apoptosis of skeletal muscles during development and disease. Int. J. Biochem. Cell Biol. 1999;31:1373–1390. doi: 10.1016/s1357-2725(99)00063-1. [DOI] [PubMed] [Google Scholar]
- 50.Lim J.H., Kim D.Y., Bang M.S. Effects of exercise and steroid on skeletal muscle apoptosis in the mdx mouse. Muscle Nerve. 2004;30:456–462. doi: 10.1002/mus.20139. [DOI] [PubMed] [Google Scholar]
- 51.Tidball J.G., Albrecht D.E., Lokensgard B.E., Spencer M.J. Apoptosis precedes necrosis of dystrophin-deficient muscle. J. Cell Sci. 1995;108:2197–2204. doi: 10.1242/jcs.108.6.2197. [DOI] [PubMed] [Google Scholar]
- 52.Dee K., Freer M., Mei Y., Weyman C.M. Apoptosis coincident with the differentiation of skeletal myoblasts is delayed by caspase 3 inhibition and abrogated by MEK-independent constitutive Ras signaling. Cell Death Differ. 2002;9:209–218. doi: 10.1038/sj.cdd.4400930. [DOI] [PubMed] [Google Scholar]
- 53.Fernando P., Kelly J.F., Balazsi K., Slack R.S., Megeney L.A. Caspase 3 activity is required for skeletal muscle differentiation. PNAS USA. 2002;99:11025–11030. doi: 10.1073/pnas.162172899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moresi V., Pristera A., Scicchitano B.M., Molinaro M., Teodori L., Sassoon D., Adamo S., Coletti D. Tumor necrosis factor-alpha inhibition of skeletal muscle regeneration is mediated by a caspase-dependent stem cell response. Stem Cells. 2008;26:997–1008. doi: 10.1634/stemcells.2007-0493. [DOI] [PubMed] [Google Scholar]
- 55.Nakanishi K., Dohmae N., Morishima N. Endoplasmic reticulum stress increases myofiber formation in vitro. FASEB J. 2007;21:2994–3003. doi: 10.1096/fj.06-6408com. [DOI] [PubMed] [Google Scholar]
- 56.Nakanishi K., Sudo T., Morishima N. Endoplasmic reticulum stress signaling transmitted by ATF6 mediates apoptosis during muscle development. J. Cell Biol. 2005;169:555–560. doi: 10.1083/jcb.200412024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qaisar R., Renaud G., Morine K., Barton E.R., Sweeney H.L., Larsson L. Is functional hypertrophy and specific force coupled with the addition of myonuclei at the single muscle fiber level? FASEB J. 2012;26:1077–1085. doi: 10.1096/fj.11-192195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Matsakas A., Romanello V., Sartori R., Masiero E., Macharia R., Otto A., Elashry M., Sandri M., Patel K. Food restriction reverses the hyper-muscular phenotype and force generation capacity deficit of the myostatin null mouse. Int. J. Sports Med. 2013;34:223–231. doi: 10.1055/s-0032-1312605. [DOI] [PubMed] [Google Scholar]
- 59.Ben Mosbah I., Alfany-Fernandez I., Martel C., Zaouali M.A., Bintanel-Morcillo M., Rimola A., Rodes J., Brenner C., Rosello-Catafau J., Peralta C. Endoplasmic reticulum stress inhibition protects steatotic and non-steatotic livers in partial hepatectomy under ischemia-reperfusion. Cell Death Dis. 2010;1:e52. doi: 10.1038/cddis.2010.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miller S.D.W., Greene C.M., McLean C., Lawless M.W., Taggart C.C., O'Neill S.J., McElvaney N.G. Tauroursodeoxycholic acid inhibits apoptosis induced by Z alpha-1 antitrypsin via inhibition of bad. Hepatology. 2007;46:496–503. doi: 10.1002/hep.21689. [DOI] [PubMed] [Google Scholar]
- 61.Sohn J., Khaoustov V.I., Xie Q., Chung C.C., Krishnan B., Yoffe B. The effect of ursodeoxycholic acid on the survivin in thapsigargin-induced apoptosis. Cancer Lett. 2003;191:83–92. doi: 10.1016/s0304-3835(02)00624-9. [DOI] [PubMed] [Google Scholar]
- 62.Zhou L., Liu M., Zhang J., Chen H., Dong L.Q., Liu F. DsbA-L alleviates endoplasmic reticulum stress-induced adiponectin downregulation. Diabetes. 2010;59:2809–2816. doi: 10.2337/db10-0412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bowles D.E., McPhee S.W.J., Li C., Gray S.J., Samulski J.J., Camp A.S., Li J., Wang B., Monahan P.E., Rabinowitz J.E., et al. Phase 1 gene therapy for Duchenne muscular dystrophy using a translational optimized AAV vector. Mol. Ther. 2012;20:443–455. doi: 10.1038/mt.2011.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Goemans N.M., Tulinius M., van den Akker J.T., Burm B.E., Ekhart P.F., Heuvelmans N., Holling T., Janson A.A., Platenburg G.J., Sipkens J.A., et al. Systemic administration of PRO051 in Duchenne's muscular dystrophy. N. Engl. J. Med. 2011;364:1513–1522. doi: 10.1056/NEJMoa1011367. [DOI] [PubMed] [Google Scholar]
- 65. GSK (2013), Vol. 2013.
- 66.Mendell J.R., Rodino-Klapac L.R., Sahenk Z., Roush K., Bird L., Lowes L.P., Alfano L., Gomez A.M., Lewis S., Kota J., et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 2013;74:637–647. doi: 10.1002/ana.23982. [DOI] [PubMed] [Google Scholar]
- 67.Moorwood C., Khurana T.S. Duchenne muscular dystrophy drug discovery - the application of utrophin promoter activation screening. Expert Opin. Drug Discov. 2013;8:569–581. doi: 10.1517/17460441.2013.777040. [DOI] [PubMed] [Google Scholar]
- 68. Summit, p. (2013), Vol. 2013.
- 69.Tinsley J.M., Fairclough R.J., Storer R., Wilkes F.J., Potter A.C., Squire S.E., Powell D.S., Cozzoli A., Capogrosso R.F., Lambert A., et al. Daily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mouse. PLoS ONE. 2011;6:e19189. doi: 10.1371/journal.pone.0019189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Moorwood C., Liu M., Tian Z., Barton E.R. Isometric and eccentric force generation assessment of skeletal muscles isolated from murine models of muscular dystrophies. J. Vis. Exp. 2013;71:e50036. doi: 10.3791/50036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Janssen P.M., Murray J.D., Schill K.E., Rastogi N., Schultz E.J., Tran T., Raman S.V., Rafael-Fortney J.A. Prednisolone attenuates improvement of cardiac and skeletal contractile function and histopathology by lisinopril and spironolactone in the mdx mouse model of Duchenne muscular dystrophy. PLoS ONE. 2014;9:e88360. doi: 10.1371/journal.pone.0088360. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.