

Abstract
Bladder exstrophy-epispadias complex (BEEC), the severe end of the urorectal malformation spectrum, has a profound impact on continence as well as sexual and renal functions. It is widely accepted that for the majority of cases the genetic basis appears to be multifactorial. Here, we report the first study which utilizes genome-wide association methods to analyze a cohort comprising patients presenting the most common BEEC form, classic bladder exstrophy (CBE), to identify common variation associated with risk for isolated CBE. We employed discovery and follow-up samples comprising 218 cases/865 controls and 78 trios in total, all of European descent. Our discovery sample identified a marker near SALL1, showing genome-wide significant association with CBE. However, analyses performed on follow-up samples did not add further support to these findings. We were also able to identify an association with CBE across our study samples (discovery: P = 8.88 × 10−5; follow-up: P = 0.0025; combined: 1.09 × 10−6) in a highly conserved 32 kb intergenic region containing regulatory elements between WNT3 and WNT9B. Subsequent analyses in mice revealed expression for both genes in the genital region during stages relevant to the development of CBE in humans. Unfortunately, we were not able to replicate the suggestive signal for WNT3 and WNT9B in a sample that was enriched for non-CBE BEEC cases (P = 0.51). Our suggestive findings support the hypothesis that larger samples are warranted to identify association of common variation with CBE.
INTRODUCTION
The Bladder exstrophy-epispadias complex (BEEC; OMIM %600057) represents the severe end of the urorectal malformation spectrum and has a profound impact on continence as well as sexual and renal functions. The BEEC comprises three levels of severity ranging from epispadias (E) (Supplementary Material, Fig. S1A and B) and classic bladder exstrophy (CBE) (Supplementary Material, Fig. S1C and D), to the most severe form, cloacal exstrophy (CE), often referred to within the OEIS complex (omphalocele, exstrophy, imperforate anus and spinal defects; Supplementary Material, Fig. S1E) (1,2). In approximately, one-third of all BEEC patients, there are also associated urological malformations (e.g. ectopic kidney, renal agenesis and hydronephrosis). Management of the BEEC is primarily surgical, and the main aims are the achievement of secure abdominal wall closure, urinary continence with preservation of renal function and adequate cosmetic and functional genital reconstruction (3). Following reconstructive surgery of the bladder during the neonatal period, continence rates of around 80% are expected during childhood. Additional surgery might be needed to optimize bladder storage and emptying function. Psychosocial and psychosexual outcome and adequate health-related quality of life depend on long-term multidisciplinary care (4–7). Among children of European descent, the overall birth prevalence for the entire spectrum has been estimated to be 1 in 10 000 (2). The birth prevalence for the specific subtypes, including terminated pregnancies, has also been estimated and established values for E of 1 in 117 000 for males and 1 in 484 000 for females (2), along with 1 in 37 000 for CBE (8) and 1 in 200 000 to 1 in 400 000 for CE (9).
Although the BEEC can occur as part of a complex malformation syndrome, the majority of cases (∼98.5%) are classified as isolated (10–12). Formal genetic studies have suggested the involvement of genetic factors in the etiology of BEEC. For CBE, the recurrence risk among siblings in families with non-consanguineous and non-affected parents ranges between 0.3 and 2.3%, and the recurrence risk for the offspring of affected patients was reported to be 1.4% (13,14). Hence, the recurrence risk for the offspring of affected CBE patients and the risk of having a second affected child for parents who are non-consanguineous and non-affected exhibits an ∼400-fold increase as compared with the general population. Further evidence for genetic factors underlying the BEEC comes from a classic twin study which clearly showed higher concordance rates among monozygotic (62%) as compared with dizygotic (11%) twin pairs (15).
Despite the overall rare occurrence of these congenital malformations, a total of 30 families with multiple affected individuals have been reported. In some of these, the pattern of BEEC incidences within single families resembles a Mendelian mode of inheritance (15–18); however, the general consensus in the field is that, in the majority of patients, the genetic basis appears to be multifactorial (11). To further support formal evidence for a genetic contribution to BEEC, recent studies have identified various molecular genetic factors including chromosomal aberrations such as trisomy 21 and micro-duplications on chromosome 22q11.21, as well as the association of polymorphisms within the ΔNp63 promoter with an increased incidence of BEEC phenotypes (19–22). However, these associated chromosomal risk factors account for only a small portion of the BEEC, whereas most patients represent idiopathic cases.
To the best of our knowledge, we report here the first study using genome-wide association methods to analyze a BEEC cohort sample, comprising of patients presenting the most common form, CBE. Our aim was to identify genetic susceptibility loci for isolated CBE presentation. We therefore conducted a genome-wide association study (GWAS), and subsequently followed up the most promising genomic regions in independent replication samples, with the goal of identifying common single-nucleotide polymorphisms (SNPs) in the genome that are associated with risk for isolated CBE in patients of European descent.
RESULTS
For both GWASs, we followed the same stringent quality control (QC) protocol that has been successfully applied to other datasets (23,24). Details on the analytical pipeline can be found in Supplementary Material, Figure S2. The post-QC datasets (comprising 43 cases and 259 controls for GWAS1 and 55 cases and 267 controls for GWAS2, respectively) were subjected to imputation based on 1000 Genomes Project and HapMap 3 reference panels (25,26). After applying postimputation QC, single marker analyses were conducted for 4 510 680 markers each using a logistic regression (additive model) as implemented into SNPTEST (27). The subsequent meta-analysis of both datasets was performed using a fixed-effects model as implemented into YAMAS (28). The most significant results were obtained for two markers in (i) an intergenic region flanked by CYLD and SALL1 for rs4785484 on chromosome 16q12.1, P = 4.55 × 10−8 and (ii) in an intergenic region flanked by EEF1E1 and SLC35B3 for rs73374907 on chromosome 6p24.3, P = 6.13 × 10−8 (more information in the Supplementary Material, Table S1; Figs 1 and 2). Follow-up on the most promising genomic regions based on the results of the meta-analysis was performed for a total of 75 SNPs that were successfully genotyped and (meta) analyzed after pruning for linkage disequilibrium in the initially obtained SNP list. The post-QC dataset for our first follow-up sample comprised 27 index cases with their parents and 120 cases/339 controls (Supplementary Material, Table S1 and Fig. S2; for more details on the follow-up samples, see the Materials and Methods). Based on the results from our first follow-up sample, a second independent sample (Follow-up 2, 51 index cases and their parents; Supplementary Material, Table S1 and Fig. S2) was analyzed for four markers that showed evidence of association. Evidence for replication (nominal significant association with same directionality based on effect allele across GWAS, Follow-up 1 and Follow-up 2) was found for two SNPs (Table 1) located in an intergenic region on chromosome 17 flanked by WNT3 and WNT9B (rs9890413, PUSA = 0.0095, PEU + USA= 0.0025) and intronically to MECOM (rs56273700, PUSA = 0.0423, PEU + USA= 0.0045). The result for rs9890413 remained significant after correcting for the number of tests in the second follow-up sample. Subsequent meta-analysis of all samples tested in the discovery and follow-up steps showed P-values of 1.09 × 10−6 (rs9890413) and 1.24 × 10−6 (rs56273700), respectively. It is of note that for rs9890413 all samples (i.e. both GWAS samples and all four follow-up samples) shared the effect allele and the direction of the effect. Unfortunately, we were not able to replicate our findings for the WNT3-WNT9B locus in a final sample of 78 cases and 336 controls that was enriched for non-CBE BEEC cases (P = 0.51; more information in the Supplementary Material). However, it is of note that the MAF in the control cohort was higher than expected from observations in the GWAS samples (0.36 versus 0.29 and 0.30, respectively), and that a test for deviation from Hardy–Weinberg equilibrium (HWE) in controls did show a trend towards significance (P = 0.067). Expression of Wnt3 and Wnt9b was detected in the genital region of mouse embryos at all stages examined (Fig. 3). While Wnt3 was found to be widely expressed within the epithelia, including that of the cloacal membrane and genital tubercle, Wnt9b was found to be strongly expressed in the cloacal membrane between E9.5 and E10.5, and the epithelia of the genital tubercle at later stages.
Figure 1.
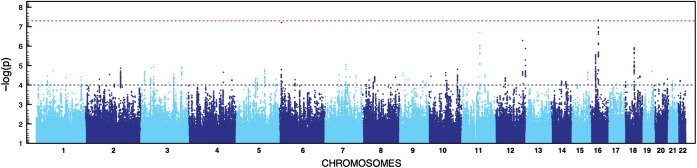
Manhattan plot for meta-analysis. Results based on fixed effect meta-analysis of GWAS1 and GWAS2 signals. Dashed red line shows threshold for genome-wide significance (P< 5 × 10−8) and dashed gray line shows cut off for the selection of SNPs for follow-up genotyping (P < 1 × 10−4).
Figure 2.

Regional association plots for regions on chromosome 6 (EEF1E1/SLC35B3) and chromosome 16 (CYLD/SALL1). The P-values from all imputed SNPs in the regions that passed post-imputation QC procedures are plotted against positions from the February 2009 human reference sequence, annotated by Ref Seq genes. The most associated marker (chromosome 6: rs73374907, Pmeta = 6.13 × 10−8; chromosome 16: rs4785484, Pmeta = 2.65 × 10−8) from the meta-analysis is indicated by a purple dot which is centered in a genomic window of ∼1 Mb. The strength of LD (in r2) between the top SNP and its adjacent markers is demonstrated by the red (high) to dark blue (low) color bar (top right corner). The recombination rate (second y-axis) is plotted in light blue, according to 1000 genomes project data. Plots were generated using Locuszoom (http://csg.sph.umich.edu/locuszoom/) (53).
Table 1.
Association results for rs9890413 (WNT3/WNT9b) and rs56273700 (MECOM) at all stages of the analysis: discovery, Follow-up 1 and Follow-up 2
| SNP | Sample |
N |
Test, P | OR | EA | MAF |
||
|---|---|---|---|---|---|---|---|---|
| Cases | Controls | Cases | Controls | |||||
| rs9890413 | Meta GWAS | 98 | 526 | Z-score, 8.88 × 10−5 | ||||
| GWAS1 | 43 | 259 | LOGISTIC, 0.0239 | 1.86 | G | 0.40 | 0.29 | |
| GWAS2 | 55 | 267 | LOGISTIC, 0.0013 | 2.11 | G | 0.45 | 0.30 | |
| Follow-up 1 | 147 | 339 | Z-score, 0.0390 | |||||
| Germany | 94 | 278 | LOGISTIC, 0.0995 | 1.32 | G | 0.39 | 0.32 | |
| Sweden | 26 | 61 | LOGISTIC, 0.4915 | 1.24 | G | 0.40 | 0.34 | |
| Spain–Italy | 27 | N/A | TDT, 0.2752 | 1.63 | G | N/A | N/A | |
| Follow-up 2 | 51 | N/A | TDT, 0.0095 | 2.31 | G | N/A | N/A | |
| rs56273700 | Meta GWAS | 98 | 526 | Z-score, 5.15 × 10−5 | ||||
| GWAS1 | 43 | 259 | LOGISTIC, 0.0074 | 0.48 | G | 0.15 | 0.28 | |
| GWAS2 | 55 | 267 | LOGISTIC, 0.0013 | 0.49 | G | 0.15 | 0.29 | |
| Follow-up 1 | 147 | 339 | Z-score, 0.0317 | |||||
| Germany | 94 | 278 | LOGISTIC, 0.0171 | 0.62 | G | 0.22 | 0.31 | |
| Sweden | 26 | 61 | LOGISTIC, 0.2897 | 0.66 | G | 0.23 | 0.31 | |
| Spain–Italy | 27 | N/A | TDT, 0.4913 | 1.38 | G | N/A | N/A | |
| Follow-up 2 | 51 | N/A | TDT, 0.0423 | 0.52 | G | N/A | N/A | |
N, number of cases and controls in study sample (for trio samples, the number of index patients is given in the case column); test, P, test statistic (LOGISTIC, logistic regression; TDT, transmission disequilibrium test) used and P-value for association; EA, effect allele; OR, odds ratio (based on effect allele); MAF, minor allele frequency (same as effect allele for both SNPs, no MAF given for trio samples).
Figure 3.

Analysis of Wnt3 and Wnt9b expression in mid-gestational mouse embryos by in situ hybridization. (A) Expression of Wnt3 was widely detected from E9.5 to E15.5, with stronger expression observed in the skin epithelia, limb buds and neural tissues. Wnt3 is also present in the genital tubercle at these stages. Expression at E14.5 and E15.5 is not shown. gt, genital tubercle; tl, tail. (B) Expression was detectable between E9.5 and E13.5 in the epithelia of the branchial arches, frontonasal process, limb buds and genital tubercle (including the cloacal membrane between E9.5 and E10.5). Strong expression was also found in the nephric ducts. The expression pattern of E13.5 (not shown) was identical to that at E12.5. ba, branchial arch; cm, cloacal membrane; flb, forelimb bud; fnp, frontonasal process; gt, genital tubercle; hlb, hindlimb bud; nd, nephric duct; nt, neural tube; tl, tail.
DISCUSSION
Presumably our constrained sample size did not allow us to find association of genetic variation with CBE (or BEEC) beyond reasonable doubt. Our results are therefore mainly to be regarded as suggestive evidence for association and larger studies are needed to confirm our findings. In the following, we would like to discuss these findings in light of the statistical power for GWAS of rare disorders such as BEEC (birth prevalence is 0.0001).
As outlined above, we found suggestive evidence for association with CBE across our study samples (rs9890413; discovery: P = 8.88 × 10−5; follow-up: P = 0.0025) in a highly conserved 32 kb intergenic region containing regulatory elements between WNT3 and WNT9B. However, we were not able to replicate this finding in an independent sample that was enriched for non-CBE BEEC cases. It is reasonable to assume that (at least in part) the inability to find stronger evidence for association in the intergenic region between WNT3 and WNT9b is based on a lack in statistical power: the risk allele frequency in our discovery samples was ∼30% in controls and the effect size was estimated with an OR of ∼2. Under the assumption of an additive model and genome-wide significance (5 × 10−8) power of 80% to see association at this significance level would require a sample of 357 cases (and equal number of controls). While our combined study samples (comprising 218 cases/865 controls and 78 trios in total) comes reasonably close to this number it has to be taken into consideration that the estimated effect size is presumably inaccurate and the true effect size is likely to be smaller (‘winner's curse’). The lower boundary of the 95% confidence interval (CI) for rs9890413 is 1.57 (which is still higher than the OR of the European replication sample) and results in a requirement of 866 cases and an equal number of controls to obtain genome-wide significance. Considering a recessive mode of inheritance even further increases sample size requirements (1702 cases). On top of the above outlined limitations our final replication sample, comprising a patient sample with a broader defined BEEC phenotype (including patients with CE and E), did show evidence for deviation from the expected allele frequencies in the controls (36 versus 31% in the independent German controls from discovery and replication; test for deviation from HWE: P = 0.067).
Similar observations with regards to the power considerations can be made for our initial genome-wide significant hit at the SALL1 locus. Under the assumption of valid effect size estimation (OR = 2.89), a power of 80% for observing this locus with genome-wide significance (additive model, minor allele frequency 20%) is reached with ∼100 cases (which is the size of our discovery sample). However, at the lower boundary of the 95% CI (OR = 2.20), an estimate that is still likely to be higher than the true effect size, a sample size of 200 cases (additive model) and 410 cases (recessive model) are required. For our replication study, we attempted replication for 75 SNPs, i.e. a P < 6.7 × 10−4 was required for experiment-wide significance. Hence, a sample of 150 cases (equal number of controls) would have been sufficient under the assumption of an additive model and requiring 80% power to see this experiment-wide significant at SALL1 (369 cases for a recessive model). In sum, our datasets could have provided enough power to detect association of SALL1 with CBE at the level of genome- or experiment-wide significant. However, it remains to be seen whether overestimation of the effect size did hinder a reasonable assessment of power and therefore could have hindered a valid interpretation of the results.
The effect size for common variation associated with CBE/BEEC seems to be not higher than those observed with other (more common) congenital disorders in human (e.g. orofacial clefts). Association of markers at 8q24.21 with non-syndromic cleft lip with or without cleft palate was found with an effect size similar to the one observed with our SALL1 association (OR = 2.57, frequency of effect allele 20% in controls, 462 cases and 954 controls (29)). Accounting for winners curse (and taking into account effect size estimates of replication studies for 8q24.21), it is therefore reasonable to assume that future successful GWAS in CBE/BEEC will (at least) require a (combined) sample size of ∼400 cases and equal number of controls (additive model, OR = 2, risk allele frequency 20%, power = 80%). Including more controls (e.g. three times the number of cases) potentially reduces the required number of cases (∼250).
Although we consider our findings as suggestive evidence for association, we would like to briefly put them into context with earlier studies. We hope that insights from this discussion might help to guide future studies. A homozygous nonsense mutation in WNT3 has been associated with tetra-amelia and urorectal malformations, which include persistent cloaca (30), and loss of Wnt9b causes urogenital defects (31). Our most associated SNP in this region (rs9890413) resides ∼4 kb next to the WNT3 promoter (http://promoter.cdb.riken.jp/), a region highly conserved among amniotes. Moreover, this transcriptional regulatory region is CpG enriched (ObsCpG/ExpCpG:0.718) and likely to be regulated by methylation status. Nakamura et al. identified several potential transcription factor-binding motifs to exist within the WNT3 promoter region (32), several of which were previously found to be differentially expressed in human newborn bladder exstrophy tissue and known to be important for promotion of the embryonic urorectal septation process (33,34). Furthermore, this region has been shown to contain regulatory elements which regulate Wnt signaling via p63 (35). Thus, it is tempting to speculate that there are also regulatory domains within this intergenic region able to modulate Wnt signaling via a conserved WNT3-WNT9B-p63 regulatory module in the context of urorectal and urogenital development. Indeed, expression profiling of human urinary bladder exstrophy tissue showed dysregulation of both the WNT and p63 pathways (34,36,37). Furthermore, the only known BEEC-associated animal knockout model to date is the ΔNp63−/− mouse described by Cheng et al. (38), who were recently able to show association of a 12 bp deletion within the TP63 promoter in BEEC patients of Canadian, U.S. American, Spanish origins and a 4 bp insertion in BEEC patients of Indian, Bangladeshi and Chinese origins (19).
We also found suggestive evidence for association of CBE with genetic variation at the MECOM gene locus. Data from the GenitoUrinary Development Molecular Anatomy Project (http://www.gudmap.org) (39,40) and the GenePaint.org project (41) suggest that Mecom is strongly expressed at E14.5 in the metanephros, the ureter, the urinary bladder and the urethra (39,40), and at E15.5 in the female reproductive system, the renal interstitium, the ureteric trunk and female urethra (41). Although E14.5–15.5 represents a developmental timeframe later than the critical period for the urorectal septation process, a modifier role for Mecom in the manifestation of the BEEC cannot be excluded, as a recent study revealed zebrafish mecom expression in the cloacal chamber, a space proximal to the urogenital pore where the pronephric duct, intestine and the oviduct or sperm duct empties (42).
The initial genome-wide significant marker rs4785484 (P = 4.55 × 10−8) resides close to SALL1. Patients with SALL1 mutations (Townes–Brocks syndrome, OMIM #107480) frequently present with urogenital anomalies (43) and Sall1 mutant mice exhibit neural tube defects, which can be affiliated with the OEIS complex (44). A recent study proposed heterochromatin localization of SALL1 as a new mechanism for the activation of Wnt signaling (45) which was also shown to be affected by SALL1-dependent signals in the context of ureter tip fate to initiate kidney development (46). Future studies are warranted to shed light on the potential interplay of SALL1 and the conserved WNT3-WNT9B-p63 regulatory module in the etiology of BEEC.
MATERIALS AND METHODS
Ethics statement and subjects
Written informed consent was obtained from all subjects or their proxies, in case of minors. Demographic information was collected from both patients and controls through a structured questionnaire. This study was approved by each participating center's Institutional Ethics Committee and was conducted according to Declaration of Helsinki principles. Experienced physicians trained in the diagnosis of the BEEC personally recruited all BEEC patients included in this study. More details about the recruitment process (for discovery and follow-up samples) can be found elsewhere (20,47) and in the Supplementary Materials, Methods.
Genetic analyses (GWAS and follow-up)
For the purposes of phenotypic homogeneity, the GWAS sample consisted only of isolated, non-syndromic CBE, the most common form based on the definition described in detail elsewhere (2). Patients with additional malformations or congenital anomalies, not associated with the CBE or the BEEC, respectively, were excluded from the analysis. DNA was extracted from blood or saliva samples and genotyping of 107 isolated CBE patients of Central European ancestry was performed in two batches (51 and 56, respectively). Due to discontinuation of the genotyping array utilized for the first batch, different arrays were used for Batches 1 and 2. For case–control comparison, we also obtained genotypes of 538 (270 and 268, respectively) ethnically matched population-based controls that have been described elsewhere (48). All QC and subsequent procedures were applied to both batches separately (due to insufficient overlap in the SNP content of the utilized genotyping arrays). For both GWASs, we followed the same stringent QC protocol that has been successfully applied to other datasets (23,24). A detailed description of the protocol can be found in the Supplementary Materials, Methods. The post-QC datasets (comprising 43 cases and 259 controls for GWAS1 and 55 cases and 267 controls for GWAS2, respectively) were subjected to imputation based on 1000 Genomes Project and HapMap 3 reference panels (25,26). After applying postimputation QC, single marker analyses were conducted for 4 510 680 markers each using a logistic regression (additive model) as implemented into SNPTEST (27). The subsequent meta-analysis of both datasets was performed using a fixed-effects model as implemented into YAMAS (28). A detailed overview on the analytical pipeline is provided in Supplementary Material, Figure S2. Results from our meta-analysis (for all 4 510 680 markers) are publicly available and can be downloaded from http://www.sharing.biostats.info after registration with the website. Follow-up on the most promising genomic regions was performed in a first independent sample (Follow-up 1) comprising three samples of European ancestry from Germany (94 cases and 278 controls post-QC), Sweden (26 cases and 61 controls post-QC), Spain and Italy (a total of 27 trios post-QC). All analyses for the downstream steps were performed using PLINK (49). In order to be selected for downstream analyses, SNPs were required to show P < 0.0001 in the meta-analysis of the two GWAS samples and P < 0.05 in the individual GWASs (with same effect allele and direction of effect; see Supplementary Material, Table S1). A total of 75 SNPs were successfully genotyped and (meta) analyzed after pruning for linkage disequilibrium in the initially obtained SNP list. Based on the results from our first follow-up sample, a second independent sample (Follow-up 2) was analyzed for four markers that showed evidence for association (see Supplementary Material, Table S2 for results of all 75 markers). The second follow-up sample comprised of 49 trios of European ancestry from North America with an isolated non-syndromic CBE patient as index (post-QC number). Finally, we attempted to replicate the most associated marker in the discovery and follow-up samples in a final sample of European ancestry. Due to a limited number of samples available for genetic studies on CBE, and in order to study the potential impact of rs9890413 on a broader defined phenotype of BEEC (including also patients with CE and E), we decided to study a sample comprising 78 samples (32 with CBE, 19 with CE and 27 with E; along with 336 controls from a cohort of ethnically matched blood donors).
Mouse expression data
Mouse embryos were fixed overnight in 4% paraformaldehyde (PFA)/PBS and processed for in situ hybridization as described elsewhere (50). For hybridization on sections (E12.5–E15.5), embryos were processed into paraffin wax and sections (5 µm) were made using a microtome. Antisense RNA probes were transcribed from PCR products generated either from our in-house MAMEP collection (Wnt9b) (51) or as previously described for Wnt3 (52), which was kindly made available by Andy McMahon. Riboprobes were synthesized using the appropriate RNA polymerases and a nucleotide mix containing digoxigenin-11-UTP (Roche Diagnostics, Mannheim, Germany) and were purified using G-50 sephadex columns (GE-Healthcare, Solingen, Germany). Following probe hybridization and washes, an anti-DIG antibody conjugated to alkaline phosphatase (AP) (Roche Diagnostics, Mannheim, Germany) was incubated with embryos overnight at 4°C, and detection of AP activity was carried out using BM Purple (Roche Diagnostics, Mannheim, Germany). For each probe, embryos were processed concurrently and staining reaction times were maintained between embryos in order to limit variations in signal intensity. For whole mounts, at least three embryos were examined for each gene and stage. For slides, three sections from at least two different embryos were analyzed for each stage shown and figures depict representative staining. Images were captured using AxioVision software (Zeiss, Jena, Germany) with a Zeiss AxioCam and SteREO Discovery.V12 microscope.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by research grant 01GM08107 from the German Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung, BMBF). Furthermore this work was funded by grant RE 1723/1-1 from the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) to H.R. S.A.B. is partially supported by Children's Miracle Network (CMN) endowed chair in pediatric genetics. This project has been partially supported through NIH grant R01 DE016886 from NIDCD/NIH and M01-RR00052 from NCRR/NIH and by CMN grant CMNSB06 to S.A.B. E.B. is supported by the BONFOR program of the University of Bonn (grant no. O-149.0099). M.M.N. also received support from the Alfried Krupp von Bohlen und Halbach-Stiftung, and he is a member of the DFG-funded Excellence Cluster ImmunoSensation.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank the many families who have participated in the study. M.D., E.B., T.P., L.W., A.K.E., W.R., R.S., E.S., E.J., N.Z., M.M.N., B.G.H., M.L. and H.R. are members of the ‘Network for the Systematic Investigation of the Molecular Causes, Clinical Implications, and Psychosocial Outcome of Congenital Uro-Rectal Malformations (CURE-Net)’. Controls for our GWAS studies were drawn from the Heinz Nixdorf Recall Study (HNR) cohort, which was established with the support of the Heinz Nixdorf Foundation. We thank Jessica Becker, Per Hoffmann, Peter Teßmann and Stefan Herms for their excellent technical support.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Carey J.C. Exstrophy of the cloaca and the OEIS complex: one and the same. Am. J. Med. Genet. 2001;99:270. doi: 10.1002/ajmg.1211. [DOI] [PubMed] [Google Scholar]
- 2.Gearhart J. In: Campbell's Urology. Campbell M., Walsh P., Retik A., editors. Philadelphia, PA: Saunders; 2002. pp. 2136–2196. [Google Scholar]
- 3.Ebert A.K., Reutter H., Ludwig M., Rosch W.H. The exstrophy-epispadias complex. Orphanet. J. Rare Dis. 2009;4:23. doi: 10.1186/1750-1172-4-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reutter H., Lee C., Grasser M.F., Noeker M. Subjective developmental outcome in bladder exstrophy and epispadias. A pilot study. Urologe A. 2005;44:57–63. doi: 10.1007/s00120-004-0739-7. [DOI] [PubMed] [Google Scholar]
- 5.Lee C., Reutter H.M., Grasser M.F., Fisch M., Noeker M. Gender-associated differences in the psychosocial and developmental outcome in patients affected with the bladder exstrophy-epispadias complex. BJU Int. 2006;97:349–353. doi: 10.1111/j.1464-410X.2005.05910.x. [DOI] [PubMed] [Google Scholar]
- 6.Ebert A.K., Bals-Pratsch M., Seifert B., Reutter H., Rosch W.H. Genital and reproductive function in males after functional reconstruction of the exstrophy-epispadias complex – long-term results. Urology. 2008;72:566–569. doi: 10.1016/j.urology.2007.11.166. discussion 569–570. [DOI] [PubMed] [Google Scholar]
- 7.Ebert A.K., Falkert A., Hofstadter A., Reutter H., Rosch W.H. Pregnancy management in women within the bladder-exstrophy-epispadias complex (BEEC) after continent urinary diversion. Arch. Gynecol. Obstet. 2011;284:1043–1046. doi: 10.1007/s00404-011-1945-3. [DOI] [PubMed] [Google Scholar]
- 8.Wiesel A., Queisser-Luft A., Clementi M., Bianca S., Stoll C. EUROSCAN Study Group. Prenatal detection of congenital renal malformations by fetal ultrasonographic examination: an analysis of 709,030 births in 12 European countries. Eur. J. Med. Genet. 2005;48:131–144. doi: 10.1016/j.ejmg.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 9.Hurwitz R.S., Manzoni G.A., Ransley P.G., Stephens F.D. Cloacal exstrophy: a report of 34 cases. J. Urol. 1987;138:1060–1064. doi: 10.1016/s0022-5347(17)43502-6. [DOI] [PubMed] [Google Scholar]
- 10.Anonymous. Epidemiology of bladder exstrophy and epispadias: a communication from the International Clearinghouse for Birth Defects Monitoring Systems. Teratology. 1987;36:221–227. doi: 10.1002/tera.1420360210. [DOI] [PubMed] [Google Scholar]
- 11.Boyadjiev S.A., Dodson J.L., Radford C.L., Ashrafi G.H., Beaty T.H., Mathews R.I., Broman K.W., Gearhart J.P. Clinical and molecular characterization of the bladder exstrophy-epispadias complex: analysis of 232 families. BJU Int. 2004;94:1337–1343. doi: 10.1111/j.1464-410X.2004.05170.x. [DOI] [PubMed] [Google Scholar]
- 12.Gambhir L., Holler T., Muller M., Schott G., Vogt H., Detlefsen B., Ebert A.K., Fisch M., Beaudoin S., Stein R., et al. Epidemiological survey of 214 families with bladder exstrophy-epispadias complex. J. Urol. 2008;179:1539–1543. doi: 10.1016/j.juro.2007.11.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shapiro E., Lepor H., Jeffs R.D. The inheritance of the exstrophy-epispadias complex. J. Urol. 1984;132:308–310. doi: 10.1016/s0022-5347(17)49605-4. [DOI] [PubMed] [Google Scholar]
- 14.Messelink E.J., Aronson D.C., Knuist M., Heij H.A., Vos A. Four cases of bladder exstrophy in two families. J. Med. Genet. 1994;31:490–492. doi: 10.1136/jmg.31.6.490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reutter H., Qi L., Gearhart J.P., Boemers T., Ebert A.K., Rosch W., Ludwig M., Boyadjiev S.A. Concordance analyses of twins with bladder exstrophy-epispadias complex suggest genetic etiology. Am. J. Med. Genet. A. 2007;143A:2751–2756. doi: 10.1002/ajmg.a.31975. [DOI] [PubMed] [Google Scholar]
- 16.Ludwig M., Ching B., Reutter H., Boyadjiev S.A. Bladder exstrophy-epispadias complex. Birth Defects Res. A Clin. Mol. Teratol. 2009;85:509–522. doi: 10.1002/bdra.20557. [DOI] [PubMed] [Google Scholar]
- 17.Ludwig M., Utsch B., Reutter H. Genetic and molecular biological aspects of the bladder exstrophy-epispadias complex (BEEC) Urologe A. 2005;44:1037–1038. doi: 10.1007/s00120-005-0863-z. 1040–1034. [DOI] [PubMed] [Google Scholar]
- 18.Reutter H., Shapiro E., Gruen J.R. Seven new cases of familial isolated bladder exstrophy and epispadias complex (BEEC) and review of the literature. Am. J. Med. Genet. A. 2003;120A:215–221. doi: 10.1002/ajmg.a.20057. [DOI] [PubMed] [Google Scholar]
- 19.Wilkins S., Zhang K.W., Mahfuz I., Quantin R., D'Cruz N., Hutson J., Ee M., Bagli D., Aitken K., Fong F.N., et al. Insertion/deletion polymorphisms in the DeltaNp63 promoter are a risk factor for bladder exstrophy epispadias complex. PLoS Genet. 2012;8:e1003070. doi: 10.1371/journal.pgen.1003070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lundin J., Soderhall C., Lunden L., Hammarsjo A., White I., Schoumans J., Lackgren G., Kockum C.C., Nordenskjold A. 22q11.2 microduplication in two patients with bladder exstrophy and hearing impairment. Eur. J. Med. Genet. 2010;53:61–65. doi: 10.1016/j.ejmg.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 21.Draaken M., Reutter H., Schramm C., Bartels E., Boemers T.M., Ebert A.K., Rosch W., Schroder A., Stein R., Moebus S., et al. Microduplications at 22q11.21 are associated with non-syndromic classic bladder exstrophy. Eur. J. Med. Genet. 2010;53:55–60. doi: 10.1016/j.ejmg.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 22.Reutter H., Bokenkamp A., Ebert A.K., Rosch W., Boemers T.M., Nothen M.M., Ludwig M. Possible association of Down syndrome and exstrophy-epispadias complex: report of two new cases and review of the literature. Eur. J. Pediatr. 2009;168:881–883. doi: 10.1007/s00431-008-0852-5. [DOI] [PubMed] [Google Scholar]
- 23.Cichon S., Muhleisen T.W., Degenhardt F.A., Mattheisen M., Miro X., Strohmaier J., Steffens M., Meesters C., Herms S., Weingarten M., et al. Genome-wide association study identifies genetic variation in neurocan as a susceptibility factor for bipolar disorder. Am. J. Hum. Genet. 2011;88:372–381. doi: 10.1016/j.ajhg.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rietschel M., Mattheisen M., Degenhardt F. Genetic Risk and Outcome in Psychosis (GROUP Investigators) Mühleisen T.W., Kirsch P., Esslinger C., Herms S., Demontis D., Steffens M., et al., editors. Association between genetic variation in a region on chromosome 11 and schizophrenia in large samples from Europe. Mol. Psychiatry. 2012;17:906–917. doi: 10.1038/mp.2011.80. [DOI] [PubMed] [Google Scholar]
- 25.Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., McVean G.A. 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.International HapMap Consortium. The international HapMap project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 27.Marchini J., Howie B. Genotype imputation for genome-wide association studies. Nat. Rev. Genet. 2010;11:499–511. doi: 10.1038/nrg2796. [DOI] [PubMed] [Google Scholar]
- 28.Meesters C., Leber M., Herold C., Angisch M., Mattheisen M., Drichel D., Lacour A., Becker T. Quick, "imputation-free" meta-analysis with proxy-SNPs. BMC Bioinformatics. 2012;13:231. doi: 10.1186/1471-2105-13-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Birnbaum S., Ludwig K.U., Reutter H., Herms S., Steffens M., Rubini M., Baluardo C., Ferrian M., Almeida de Assis N., Alblas M.A., et al. Key susceptibility locus for nonsyndromic cleft lip with or without cleft palate on chromosome 8q24. Nat. Genet. 2009;41:473–477. doi: 10.1038/ng.333. [DOI] [PubMed] [Google Scholar]
- 30.Niemann S., Zhao C., Pascu F., Stahl U., Aulepp U., Niswander L., Weber J.L., Muller U. Homozygous WNT3 mutation causes tetra-amelia in a large consanguineous family. Am. J. Hum. Genet. 2004;74:558–563. doi: 10.1086/382196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carroll T.J., Park J.S., Hayashi S., Majumdar A., McMahon A.P. Wnt9b plays a central role in the regulation of mesenchymal to epithelial transitions underlying organogenesis of the mammalian urogenital system. Dev. Cell. 2005;9:283–292. doi: 10.1016/j.devcel.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 32.Nakamura Y., Tsiairis C.D., Ozbek S., Holstein T.W. Autoregulatory and repressive inputs localize Hydra Wnt3 to the head organizer. Proc. Natl. Acad. Sci. USA. 2011;108:9137–9142. doi: 10.1073/pnas.1018109108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kang S., Graham J.M., Jr., Olney A.H., Biesecker L.G. GLI3 frameshift mutations cause autosomal dominant Pallister-Hall syndrome. Nat. Genet. 1997;15:266–268. doi: 10.1038/ng0397-266. [DOI] [PubMed] [Google Scholar]
- 34.Qi L., Chen K., Hur D.J., Yagnik G., Lakshmanan Y., Kotch L.E., Ashrafi G.H., Martinez-Murillo F., Kowalski J., Naydenov C., et al. Genome-wide expression profiling of urinary bladder implicates desmosomal and cytoskeletal dysregulation in the bladder exstrophy-epispadias complex. Int. J. Mol. Med. 2011;27:755–765. doi: 10.3892/ijmm.2011.654. [DOI] [PubMed] [Google Scholar]
- 35.Ferretti E., Li B., Zewdu R., Wells V., Hebert J.M., Karner C., Anderson M.J., Williams T., Dixon J., Dixon M.J., et al. A conserved Pbx-Wnt-p63-Irf6 regulatory module controls face morphogenesis by promoting epithelial apoptosis. Dev. Cell. 2011;21:627–641. doi: 10.1016/j.devcel.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ching B.J., Wittler L., Proske J., Yagnik G., Qi L., Draaken M., Reutter H., Gearhart J.P., Ludwig M., Boyadjiev S.A. p63 (TP73L) a key player in embryonic urogenital development with significant dysregulation in human bladder exstrophy tissue. Int. J. Mol. Med. 2010;26:861–867. doi: 10.3892/ijmm_00000535. [DOI] [PubMed] [Google Scholar]
- 37.Mahfuz I., Darling T., Wilkins S., White S., Cheng W. New insights into the pathogenesis of bladder exstrophy-epispadias complex. J. Pediatr. Urol. 2013;9:996–1005. doi: 10.1016/j.jpurol.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 38.Cheng W., Jacobs W.B., Zhang J.J., Moro A., Park J.H., Kushida M., Qiu W., Mills A.A., Kim P.C. DeltaNp63 plays an anti-apoptotic role in ventral bladder development. Development. 2006;133:4783–4792. doi: 10.1242/dev.02621. [DOI] [PubMed] [Google Scholar]
- 39.Harding S.D., Armit C., Armstrong J., Brennan J., Cheng Y., Haggarty B., Houghton D., Lloyd-MacGilp S., Pi X., Roochun Y., et al. The GUDMAP database – an online resource for genitourinary research. Development. 2011;138:2845–2853. doi: 10.1242/dev.063594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McMahon A.P., Aronow B.J., Davidson D.R., Davies J.A., Gaido K.W., Grimmond S., Lessard J.L., Little M.H., Potter S.S., Wilder E.L., et al. GUDMAP: the genitourinary developmental molecular anatomy project. J. Am. Soc. Nephrol. 2008;19:667–671. doi: 10.1681/ASN.2007101078. [DOI] [PubMed] [Google Scholar]
- 41.Visel A., Thaller C., Eichele G. GenePaint.org: an atlas of gene expression patterns in the mouse embryo. Nucleic Acids Res. 2004;32:D552–D556. doi: 10.1093/nar/gkh029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wingert R.A., Selleck R., Yu J., Song H.D., Chen Z., Song A., Zhou Y., Thisse B., Thisse C., McMahon A.P., et al. The cdx genes and retinoic acid control the positioning and segmentation of the zebrafish pronephros. PLoS Genet. 2007;3:1922–1938. doi: 10.1371/journal.pgen.0030189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Botzenhart E.M., Green A., Ilyina H., Konig R., Lowry R.B., Lo I.F., Shohat M., Burke L., McGaughran J., Chafai R., et al. SALL1 mutation analysis in Townes-Brocks syndrome: twelve novel mutations and expansion of the phenotype. Hum. Mutat. 2005;26:282. doi: 10.1002/humu.9362. [DOI] [PubMed] [Google Scholar]
- 44.Bohm J., Buck A., Borozdin W., Mannan A.U., Matysiak-Scholze U., Adham I., Schulz-Schaeffer W., Floss T., Wurst W., Kohlhase J., et al. Sall1, sall2, and sall4 are required for neural tube closure in mice. Am. J. Pathol. 2008;173:1455–1463. doi: 10.2353/ajpath.2008.071039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sato A., Kishida S., Tanaka T., Kikuchi A., Kodama T., Asashima M., Nishinakamura R. Sall1, a causative gene for Townes-Brocks syndrome, enhances the canonical Wnt signaling by localizing to heterochromatin. Biochem. Biophys. Res. Commun. 2004;319:103–113. doi: 10.1016/j.bbrc.2004.04.156. [DOI] [PubMed] [Google Scholar]
- 46.Kiefer S.M., Robbins L., Stumpff K.M., Lin C., Ma L., Rauchman M. Sall1-dependent signals affect Wnt signaling and ureter tip fate to initiate kidney development. Development. 2010;137:3099–3106. doi: 10.1242/dev.037812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reutter H., Boyadjiev S.A., Gambhir L., Ebert A.K., Rosch W.H., Stein R., Schroder A., Boemers T.M., Bartels E., Vogt H., et al. Phenotype severity in the bladder exstrophy-epispadias complex: analysis of genetic and nongenetic contributing factors in 441 families from North America and Europe. J. Pediatr. 2011;159:825–831 e821. doi: 10.1016/j.jpeds.2011.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schmermund A., Mohlenkamp S., Stang A., Gronemeyer D., Seibel R., Hirche H., Mann K., Siffert W., Lauterbach K., Siegrist J., et al. Assessment of clinically silent atherosclerotic disease and established and novel risk factors for predicting myocardial infarction and cardiac death in healthy middle-aged subjects: rationale and design of the Heinz Nixdorf RECALL Study. Risk factors, evaluation of coronary calcium and lifestyle. Am. Heart J. 2002;144:212–218. doi: 10.1067/mhj.2002.123579. [DOI] [PubMed] [Google Scholar]
- 49.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chotteau-Lelievre A., Dolle P., Gofflot F. Expression analysis of murine genes using in situ hybridization with radioactive and nonradioactively labeled RNA probes. Methods Mol. Biol. 2006;326:61–87. doi: 10.1385/1-59745-007-3:61. [DOI] [PubMed] [Google Scholar]
- 51.Herrmann B., Spörle R., Neidhardt L., Kietzmann S., Scholze M., Caparros-Rodriguez M., Werber M. 2013 http://mamep.molgen.mpg.de . [Google Scholar]
- 52.Parr B.A., Shea M.J., Vassileva G., McMahon A.P. Mouse Wnt genes exhibit discrete domains of expression in the early embryonic CNS and limb buds. Development. 1993;119:247–261. doi: 10.1242/dev.119.1.247. [DOI] [PubMed] [Google Scholar]
- 53.Prium R.J., Welch R.P., et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 26:2336–2337. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.