

Abstract
Fragile X syndrome, a common cause of intellectual disability and a well-known cause of autism spectrum disorder, is the result of loss or dysfunction of fragile X mental retardation protein (FMRP), a highly selective RNA-binding protein and translation regulator. A major research priority has been the identification of the mRNA targets of FMRP, particularly as recent studies suggest an excess of FMRP targets among genes implicated in idiopathic autism and schizophrenia. Several large-scale studies have attempted to identify mRNAs bound by FMRP through several methods, each generating a list of putative target genes, leading to distinct hypotheses by which FMRP recognizes its targets; namely, by RNA structure or sequence. However, no in depth analyses have been performed to identify the level of consensus among the studies. Here, we analyze four large FMRP target datasets to generate high-confidence consensus lists, and examine all datasets for sequence elements within the target RNAs to validate reported FMRP binding motifs (GACR, ACUK and WGGA). We found GACR to be highly enriched in FMRP datasets, while ACUK was not. The WGGA pattern was modestly enriched in several, but not all datasets. The previous association between FMRP and G-quadruplexes prompted the analysis of the distribution of WGGA in the target genes. Consistent with the requirements for G-quadruplex formation, we observed highly clustered WGGA motifs in FMRP targets compared with other genes, implicating both RNA structure and sequence in the recognition motif of FMRP. In addition, we generate a list of the top 40 FMRP targets associated with FXS-related phenotypes.
INTRODUCTION
Fragile X syndrome [FXS (MIM 300624)] is the most common cause of inherited intellectual disability in males and is caused by the absence or dysfunction of the fragile X mental retardation protein (FMRP). In the vast majority of cases, the loss of FMRP is due to a CGG repeat expansion in the 5′UTR of FMR1, the gene encoding FMRP (1). When the allele expands to >200 repeats, known as the full mutation, an epigenetic event is triggered where the promoter of FMR1 and flanking regions become heavily methylated, silencing FMR1 expression (2). FXS affects ∼1 in 5000 males and patients display cognitive impairment with variable severity (3). In addition to intellectual disability, FXS patients often exhibit behaviors associated with autism spectrum disorders (ASDs), making FMR1 the most prevalent monogenic cause of ASD known to date.
The main function of FMRP appears to be the selective binding of mRNA transcripts via one of its two KH-type domains or arginine-glycine rich domain (RGG box) and to regulate their translation in an activity-dependent manner, playing a critical role in the modulation of synaptic plasticity by local protein synthesis. It has been estimated that FMRP recognizes perhaps 4% of the mRNAs in the mammalian brain (4,5) and substantial efforts have been made to determine their identities to help define pathways affected by the absence of the protein. Another major reason for classifying these target genes is the emerging correlation between FMRP targets genes and neuropsychiatric diseases such as autism and schizophrenia (6–8). Given this relationship, identification of authentic FMRP targets could be extremely useful in studying the molecular mechanisms underlying these diseases.
For over a decade, studies have attempted to characterize the targets of FMRP using several methods including RNA immunoprecipitation followed by microarray interrogation (RIP-Chip) (5,9), crosslinking immunoprecipitation (CLIP) followed by high-throughput sequencing (9,10), in vitro RNA selection (11), and antibody positioned RNA amplification in cultured neurons (12). Despite these many and varied efforts, only a handful of mRNA targets have a validated association with FMRP (13,14). All of these studies have generated a list of putative FMRP-associated transcripts, though a thorough investigation of the level of overlap between studies has not been performed.
In addition to identifying the mRNA targets, substantial effort has been put forth to determine the binding elements recognized by FMRP within these ligands. One of the most well documented RNA elements mediating FMRP interaction is the G-quadruplex (11,15–18), a secondary structure comprised of two to four stacked guanine tetrads in a planar conformation (19). With regard to linear sequence recognition motifs, FMRP has been reported to bind U-rich sequences by cDNA-SELEX and yeast three hybrid experiments (20,21). More recently, two studies have determined recognition sequences of FMRP by immunoprecipitation of the protein and analysis of bound RNAs (9,22). Ascano et al. discovered two sequences, ACUK (K = G or U) and WGGA (W = A or U), to be enriched in FMRP targets using an in vivo CLIP approach, whereas Ray et al., using the KH domains of FMRP in an in vitro approach, detected a seven nucleotide consensus sequence with a highly conserved GAC core that does not correspond to the findings of Ascano et al. The incongruous sequences derived by these two studies highlight the need for further investigation of FMRP recognition elements.
Here, we describe an analysis of the four largest FMRP target studies available (5,9,10,12) in an effort to identify a set of consensus transcripts most highly and reproducibly associated with FMRP. We found that most datasets overlap very significantly with each other, but not perfectly, indicating that although each method used in these studies is likely effective in identifying FMRP targets to a certain extent, variation exists due to the assays, downstream analyses and the biological material utilized. We discovered that the consensus genes of all studies are highly enriched for autism susceptibility and intellectual disability genes, supporting them as high-confidence targets of FMRP. We also explored each dataset for information about potential FMRP recognition sites within the RNA targets. We found no evidence of enrichment for the ACUK pattern in the FMRP targets of any dataset except that of the data from which it was derived. Conversely, we found a high level of enrichment of GACR and GACARG in FMRP targets, supporting the in vitro consensus generated by Ray et al. and implicating these sequences as KH domain specific recognition motifs. We also detected a modest excess of WGGA sequences in several gene sets, suggesting the sequence may be part of a recognition motif of FMRP. We demonstrate that the WGGA sequences in FMRP targets are highly clustered and propose that the formation of G-quadruplex structures by this sequence serve as FMRP binding sites, resolving the mechanism of mRNA recognition by FMRP as being governed by structure and sequence.
RESULTS
Inter-study concordance of genes identified as FMRP targets
We analyzed five FMRP target sets from four published studies (Ascano et al. performed RIP-Chip in addition to PAR-CLIP to validate and rank FMRP targets) to evaluate the concordance of putative FMRP targets between each study and identify genes that are most reproducibly associated with FMRP. First, we examined these five datasets (Ascano-PAR, Ascano-RIP, Brown, Darnell and Miyashiro; experimental summaries are outlined in Supplementary Material, Table S1) to determine the genes in common between each in a pairwise manner. We found that four of the five overlapped to a very high degree, with Fisher's Exact P-values ranging from 6.2 × 10−23 to 1.3 × 10−137. The Miyashiro gene set did not overlap significantly with any set except for Ascano-PAR, where the Fisher's Exact P-value was much less impressive than any of the other comparisons (P = 0.01). Due to the lack of concordance and the comparatively fewer genes identified, we excluded the Miyashiro et al. dataset from the rest of the text, though the analysis of this dataset was performed and results are provided in Supplementary Material, Table S2.
Using the Brown, Darnell and Ascano-PAR datasets, we found 183 overlapping genes between Brown and Darnell (Fisher's Exact P-value = 1.34 × 10−137; permutation P-value <1 × 10−6), 251 between Brown and Ascano-PAR (Fisher's Exact test P-value = 6.21 × 10−23; permutation P-value <1 × 10−6) and 520 between Darnell and Ascano-PAR (Fisher's Exact P-value = 1.45 × 10−41; permutation P-value <1 × 10−6). To put these statistical results in context, a Fisher's Exact P-value of 10−137 indicates that if we assessed the level of overlap in two randomly generated gene sets, the likelihood that we would detect the amount of overlap observed in the FMRP datasets is 1 in 10137. The permutation P-value measures the number of times a permuted dataset, comprised of many randomly selected gene sets, exceeds the amount of overlap in the FMRP datasets. Thus, a permutation P-value of <1 × 10−6 indicates that of 1 000 000 random gene set comparisons, not once were there more overlapping genes than the FMRP target sets, revealing a very high level of correlation far beyond that of random chance.
Performing the same overlap analysis with the Ascano-RIP set yielded 87 genes common between Brown and Ascano-RIP (Fisher's Exact P-value = 1.35 × 10−30) and 178 genes common between Darnell and Ascano-RIP (Fisher's Exact P-value = 1.92 × 10−60; Supplementary Material, Table S3A). Though all comparisons are very highly correlated, in each case the degree of overlap improved by several orders of magnitude with all comparisons using the Ascano-RIP validated set of genes compared with the PAR-CLIP data.
Next, we examined the level of three-way overlap between the Brown, Darnell and Ascano-PAR datasets and found 135 genes that were present in all three studies (permutation P-value <1 × 10−6; Fig. 1). By comparing the Ascano-RIP, Brown and Darnell FMRP targets, we discovered 53 common genes between all three datasets (permutation P-value <1 × 10−6; Fig. 1). To emphasize the high level of concordance between these datasets, we also found that each pairing was still significantly overlapping even while excluding the genes from the 3-way overlaps, with the Brown and Darnell sets being the most concordant (Supplementary Material, Table S3B).
Figure 1.

Overlap of FMRP targets identified by three studies. Venn diagrams depicting the overlap of FMRP target genes identified by Brown et al. (green), Darnell et al. (purple) and Ascano et al. (brown) by PAR-CLIP assay (left) and RIP-Chip assay (right). P-values represent the results of 1 000 000 permutations of the data; the number of FMRP target genes discovered by each study is in parentheses.
Altogether, these analyses reveal an extremely high level of FMRP target concordance between each study, albeit to a lower degree in the Ascano datasets, and identifies 53 mRNA targets as having the most reproducible association with FMRP as determined by multiple laboratories and methods. The consensus lists from each comparison are available in the online Supplementary Materials.
Analysis of putative FMRP recognition elements
Two sequence motifs, ACUK and WGGA, have recently been proposed as FMRP recognition elements in mRNA targets (9). We sought to validate this finding by analyzing the Brown and Darnell datasets for these sequence patterns, in addition to the Ascano datasets from which these sequences were derived. First, to establish a baseline distribution of the patterns in the expressed regions of the genome, we examined the frequency of ACUK and WGGA in all protein coding genes. Overall, there is a greater frequency of both sequence motifs in exons compared with introns, and a greater frequency of each pattern in both exons and introns when compared with the a priori value normalized for length (Fig. 2). This indicates that, in general, the expressed portions of the genome have a higher level of these sequences per kilobase than expected, especially in exons.
Figure 2.
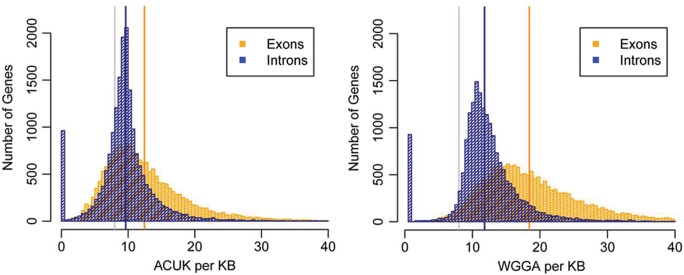
Frequency of putative FMRP recognition sequences. The mean frequency and position within each gene in the genome of ACUK (left) and WGGA (right) was evaluated and normalized by length. Cross-hatched vertical bars represent the count of genes containing each pattern as a function of the frequency per kilobase. The pattern frequency is categorized by location within a gene (i.e.—intronic or exonic). The a priori mean was calculated assuming equal probability of each nucleotide genome-wide. Solid vertical lines show the mean frequency of each pattern in each group. Grey, a priori; blue, introns; orange, exons.
We next examined the frequency of ACUK and WGGA in putative targets of FMRP compared with the rest of the genes in the genome. We first investigated the ∼6900 genes identified as FMRP targets by Ascano-PAR and found that both sequence patterns are more frequent compared with other genes, supporting the results described by Ascano et al. using our alternative analysis methods (Fig. 3A, Table 1). Next, we analyzed the genes identified in the Ascano-RIP dataset and discovered the WGGA motif to be enriched in the FMRP targets, while there was a significant deficit of ACUK sequences compared with the rest of the genes in the genome (Fig. 3B, Table 1). We performed the same analysis using the Brown and Darnell datasets; in both cases, we found a significant lack of ACUK in the FMRP targets. For the WGGA motif, we observed no detectable enrichment in the targets identified by Brown, whereas we found a modest overrepresentation of the pattern in the Darnell dataset compared with the rest of the genome (Fig. 3C and D, respectively; Table 1). The lack of enrichment of WGGA in the Brown dataset, coupled with relatively minor enrichment in the other datasets suggests that the pattern is enriched to some degree in FMRP targets compared with other genes in the genome, but not exceptionally so, leaving its role as an FMRP recognition motif unclear.
Figure 3.

Frequency of ACUK and WGGA in FMRP target datasets. Sequence motif frequency evaluation of the Ascano PAR-CLIP data (A), Ascano RIP-Chip data (B), Brown data (C) and Darnell data (D). The box plots show the median, quartiles, 1.5× interquartile range, and outliers in the number of ACUK (left) and WGGA (right) patterns per kilobase compared with the rest of the genes in the genome (Others). The difference in mean number of patterns per kilobase between the two groupings and the permutation P-value are given at the top of each graph; the number of genes in each grouping is given below each graph; number of permutations = 1 000 000.
Table 1.
Summary of pattern enrichment results for each dataset (WGGA/ACUK)
| Dataset | Pattern | Total genes | Total genes (others) | Pattern per KB | Pattern per KB (others) | Difference of means | Permutation P-value | T-test P-value | Wilcoxon P-value | Effect size |
|---|---|---|---|---|---|---|---|---|---|---|
| Ascano (PAR-CLIP) | ACUK | 6658 | 11 751 | 14.09 | 11.77 | 2.32 | <1 × 10−6 | 1.82 × 10−113 | 2.62 × 10−140 | 0.358 |
| WGGA | 6658 | 11 751 | 19.68 | 18.65 | 1.03 | <1 × 10−6 | 6.09 × 10−17 | 4.22 × 10−21 | 0.128 | |
| Ascano (RIP-CHIP) | ACUK | 921 | 17 488 | 11.75 | 12.66 | −0.91 | 4.4 × 10−5 | 1.24 × 10−7 | 0.0024 | 0.140 |
| WGGA | 921 | 17 488 | 20.45 | 18.95 | 1.50 | <1 × 10−6 | 5.01 × 10−10 | 3.23 × 10−15 | 0.187 | |
| Brown | ACUK | 354 | 18 055 | 11.22 | 12.64 | −1.41 | 6.5 × 10−5 | 1.29 × 10−6 | 1.25 × 10−5 | 0.219 |
| WGGA | 354 | 18 055 | 19.08 | 19.02 | 0.06 | 0.8903 | 0.8730 | 0.4273 | 0.007 | |
| Darnell | ACUK | 807 | 17 602 | 11.83 | 12.65 | −0.81 | 0.0005 | 0.0002 | 5.68 × 10−5 | 0.126 |
| WGGA | 807 | 17 602 | 19.76 | 18.99 | 0.77 | 0.0077 | 0.0094 | 0.0062 | 0.096 | |
| Common B_D_A-PAR | ACUK | 135 | 18 274 | 11.97 | 12.62 | −0.65 | 0.2443 | 0.2003 | 0.2166 | 0.100 |
| WGGA | 135 | 18 274 | 20.05 | 19.02 | 1.04 | 0.1349 | 0.1170 | 0.0959 | 0.129 | |
| Common D_A-PAR | ACUK | 516 | 17 893 | 12.40 | 12.62 | −0.22 | 0.4464 | 0.4111 | 0.6597 | 0.034 |
| WGGA | 516 | 17 893 | 19.73 | 19.00 | 0.73 | 0.0431 | 0.0421 | 0.0134 | 0.090 | |
| Common B_D | ACUK | 180 | 18 229 | 11.34 | 12.62 | −1.28 | 0.0084 | 0.0027 | 0.0025 | 0.198 |
| WGGA | 180 | 18 229 | 19.59 | 19.02 | 0.57 | 0.3432 | 0.3040 | 0.2264 | 0.071 | |
| Common B_D_A-RIP | ACUK | 53 | 18 356 | 11.82 | 12.61 | −0.79 | 0.3721 | 0.2810 | 0.4739 | 0.122 |
| WGGA | 53 | 18 356 | 21.64 | 19.02 | 2.63 | 0.0177 | 0.0082 | 0.0023 | 0.327 | |
| SFARI | ACUK | 517 | 17 892 | 13.72 | 12.58 | 1.14 | 0.0001 | 0.0001 | 6.91 × 10−6 | 0.177 |
| WGGA | 517 | 17 892 | 20.06 | 18.99 | 1.06 | 0.0031 | 0.0030 | 0.0007 | 0.132 | |
| MR_OMIM | ACUK | 803 | 17 606 | 13.45 | 12.57 | 0.88 | 0.0002 | 0.0002 | 7.92 × 10−5 | 0.137 |
| WGGA | 803 | 17 606 | 20.00 | 18.98 | 1.02 | 0.0004 | 0.0007 | 0.0011 | 0.127 |
Each dataset was evaluated for the ACUK and WGGA sequences by several statistical metrics.
Common B_D_A-PAR reflects the list of 135 overlapping genes in the Brown, Darnell and Ascano PAR-CLIP data; Common D_A-PAR reflects the list of 516 overlapping genes in the Darnell and Ascano PAR-CLIP data; Common B_D reflects the list of 180 overlapping genes in the Brown and Darnell data; Common B_D_A-RIP reflects the list of 53 overlapping genes in the Brown, Darnell and Ascano RIP-Chip data.
Although the ACUK pattern does not appear to be overrepresented in any dataset other than Ascano-PAR, and WGGA only modestly so, we reasoned that if either motif were substantially enriched in the genes identified by all three studies, which confers a higher confidence in the authenticity of the interaction, the likelihood of these sequences being bona fide recognition motifs would greatly increase. To investigate this, we examined four different consensus target sets derived from comparison of the individual target lists. First, we analyzed the 135 genes common to each dataset (Ascano-PAR, Darnell and Brown) for the two sequence patterns. As shown in Figure 4A, neither ACUK nor WGGA are significantly enriched in this FMRP target population. In the second set of genes, we compared the two most recent and similarly assayed datasets, Darnell and Ascano-PAR, which both used a form of CLIP to identify FMRP targets. The overlapping genes between these datasets showed no enrichment of the ACUK pattern, but a small enrichment of the WGGA pattern in the FMRP targets was detected (difference in means = 0.725; Fig. 4B, Table 1). A third comparison assessed the overlapping genes between Brown and Darnell, which were generated by dissimilar experimental approaches, but both of which used brain tissue to evaluate FMRP interactions. Here, we found no enrichment of ACUK or WGGA patterns in the overlapping targets (Fig. 4C, Table 1). Lastly, we analyzed the 53 genes in common among the Brown, Darnell and Ascano-RIP datasets for each of the recognition motifs. In this case, while we did not detect an enrichment of ACUK sequences, we discovered a significant increase in frequency of the WGGA pattern in these 53 genes (difference in means = 2.63, effect size = 0.327; Fig. 4D and Table 1). Altogether, the ACUK motif is not present at a higher frequency in any of the consensus FMRP target lists. The WGGA pattern is statistically enriched in several datasets, though to a relatively low degree; in most datasets, both individual and consensus, there is only ∼1 more WGGA pattern per kilobase in the FMRP targets compared with other genes on average. It should also be noted that in most of the individual and consensus gene list analyses, the effect size is relatively small. This indicates that the enrichment of either pattern, even if statistically significant, likely does not represent an exceptionally strong relationship. The two exceptions to this, the Ascano-PAR dataset for ACUK (effect size = 0.358) and the overlapping genes of Ascano-RIP, Brown and Darnell for WGGA (effect size = 0.327), imply a robust association. Since ACUK was not enriched in any other dataset including the consensus sets, and actually deficient in most datasets, assay bias may have contributed to this result in the Ascano-PAR data. In contrast, the enrichment of WGGA in the 53 genes common to Ascano-RIP, Brown and Darnell datasets is supported by the same finding in several other datasets, suggesting that WGGA may indeed be a recognition sequence of FMRP.
Figure 4.

Frequency of ACUK and WGGA among the Consensus FMRP targets. Sequence motif frequency evaluation of the genes common to the Brown, Darnell, Ascano PAR-CLIP or RIP-Chip data. The box plots show the median, quartiles, 1.5× interquartile range, and outliers in the number of ACUK (left) and WGGA (right) patterns per kilobase compared with the rest of the genes in the genome (Others). The difference in the mean number of patterns per kilobase between the two groupings and the permutation P-value are given at the top of each graph; the number of genes in each grouping is given below each graph; number of permutations = 1 000 000. The different dataset combinations are as follows: (A) Ascano PAR-CLIP, Brown and Darnell datasets; (B) Ascano PAR-CLIP and Darnell; (C) Brown and Darnell; and (D) Ascano RIP-Chip, Brown and Darnell datasets. B, Brown; D, Darnell; Asc-P, Ascano PAR-CLIP; Asc-R, Ascano RIP-Chip.
Another recent study reported the binding motifs for hundreds of RNA-binding proteins (RBPs), including FMRP, by an in vitro technique called RNAcompete whereby hundreds of thousands of short RNA oligos (30–41 nt) were incubated with affinity-tagged RBPs, then co-purified and analyzed by microarrays (22). The FMRP binding sequence by this method was found to be a seven nucleotide consensus with a conserved GAC core. We explored each FMRP target set for three motifs derived from this data: GAC, GACR and GACARG. If we consider the data with a relatively large effect size (≥0.2), GACR is significantly enriched in every FMRP target dataset except for Ascano-PAR, while GAC is enriched in both three-way consensus sets. The motif GACARG is significantly enriched only in the Brown, Darnell and Ascano-RIP consensus genes, though the effect size approaches significance in the Brown, Darnell and Ascano-PAR consensus genes as well (effect size = 0.179; Table 2). Overall, the GACR motif is enriched in most FMRP target datasets, while the GAC/GACARG motifs are enriched in the three-way consensus sets. This data indicate that these motifs are indeed more prevalent in FMRP targets than other genes, particularly in the consensus sets where the genes are more likely to be authentic targets of FMRP, supporting them as true recognition sequences.
Table 2.
Summary of pattern enrichment results for each dataset (GAC/GACR/GACARG)
| Dataset | Pattern | Total genes | Total genes (others) | Pattern per KB | Pattern per KB (others) | Difference of means | Permutation P-value | T-test P-value | Wilcoxon P-value | Effect size |
|---|---|---|---|---|---|---|---|---|---|---|
| Ascano (PAR-CLIP) | GAC | 6658 | 11 751 | 25.37 | 25.19 | 0.18 | 0.5019 | 0.5053 | 0.8149 | 0.01 |
| GACARG | 6658 | 11 751 | 0.93 | 0.93 | 0.01 | 0.6300 | 0.6151 | 5.52 × 10−6 | 0.008 | |
| GACR | 6658 | 11 751 | 8.35 | 8.22 | 0.14 | 0.0286 | 0.0259 | 0.0021 | 0.033 | |
| Ascano (RIP-CHIP) | GAC | 921 | 17 488 | 26.50 | 25.19 | 1.30 | 0.0247 | 0.0198 | 4.79 × 10−6 | 0.076 |
| GACARG | 921 | 17 488 | 1.00 | 0.93 | 0.07 | 0.0265 | 0.0044 | 3.94 × 10−9 | 0.075 | |
| GACR | 921 | 17 488 | 9.04 | 8.23 | 0.81 | <1 × 10−6 | 4.17 × 10−7 | 1.29 × 10−16 | 0.201 | |
| Brown | GAC | 354 | 18 055 | 27.91 | 25.20 | 2.71 | 0.0036 | 0.0046 | 1.50 × 10−5 | 0.158 |
| GACARG | 354 | 18 055 | 0.99 | 0.93 | 0.07 | 0.1893 | 0.0784 | 9.39 × 10−5 | 0.07 | |
| GACR | 354 | 18 055 | 9.32 | 8.25 | 1.07 | <1 × 10−6 | 2.02 × 10−7 | 2.20 × 10−10 | 0.265 | |
| Darnell | GAC | 807 | 17 602 | 27.90 | 25.14 | 2.77 | 1.6 × 10−5 | 2.91 × 10−5 | 4.75 × 10−10 | 0.161 |
| GACARG | 807 | 17 602 | 1.00 | 0.93 | 0.08 | 0.0222 | 0.0037 | 9.46 × 10−9 | 0.083 | |
| GACR | 807 | 17 602 | 9.38 | 8.22 | 1.16 | <1 × 10−6 | 4.34 × 10−14 | 6.61 × 10−20 | 0.288 | |
| Common B_D_A-PAR | GAC | 135 | 18 274 | 29.22 | 25.23 | 3.99 | 0.0075 | 0.0165 | 0.0002 | 0.233 |
| GACARG | 135 | 18 274 | 1.09 | 0.93 | 0.17 | 0.0377 | 0.0135 | 0.0003 | 0.179 | |
| GACR | 135 | 18 274 | 9.70 | 8.26 | 1.44 | 7.4 × 10−5 | 0.0001 | 5.31 × 10−6 | 0.358 | |
| Common D_A-PAR | GAC | 516 | 17 893 | 27.86 | 25.18 | 2.68 | 0.0006 | 0.0015 | 3.58 × 10−6 | 0.156 |
| GACARG | 516 | 17 893 | 0.97 | 0.93 | 0.04 | 0.3278 | 0.1884 | 0.0001 | 0.044 | |
| GACR | 516 | 17 893 | 9.17 | 8.24 | 0.93 | <1 × 10−6 | 1.77 × 10−7 | 6.43 × 10−11 | 0.23 | |
| Common B_D | GAC | 180 | 18 229 | 28.50 | 25.22 | 3.27 | 0.0114 | 0.0199 | 0.0002 | 0.191 |
| GACARG | 180 | 18 229 | 1.06 | 0.93 | 0.13 | 0.0574 | 0.0192 | 0.0002 | 0.142 | |
| GACR | 180 | 18 229 | 9.63 | 8.25 | 1.38 | 1.6 × 10−5 | 1.28 × 10−5 | 2.38 × 10−7 | 0.342 | |
| Common B_D_A-RIP | GAC | 53 | 18 356 | 32.24 | 25.24 | 7.01 | 0.0046 | 0.0045 | 9.09 × 10−6 | 0.408 |
| GACARG | 53 | 18 356 | 1.23 | 0.93 | 0.30 | 0.0221 | 0.0138 | 0.0006 | 0.32 | |
| GACR | 53 | 18 356 | 10.61 | 8.26 | 2.35 | 0.0001 | 0.0002 | 2.16 × 10−6 | 0.582 | |
| SFARI | GAC | 517 | 17 892 | 25.16 | 25.26 | −0.10 | 0.9002 | 0.8913 | 0.2556 | 0.006 |
| GACARG | 517 | 17 892 | 0.98 | 0.93 | 0.05 | 0.2278 | 0.1463 | 0.0007 | 0.054 | |
| GACR | 517 | 17 892 | 9.15 | 8.24 | 0.91 | <1 × 10−6 | 1.37 × 10−6 | 2.33 × 10−8 | 0.225 | |
| MR_OMIM | GAC | 803 | 17 606 | 26.60 | 25.20 | 1.41 | 0.0234 | 0.0336 | 0.0178 | 0.082 |
| GACARG | 803 | 17 606 | 0.96 | 0.93 | 0.04 | 0.2649 | 0.2313 | 0.0121 | 0.04 | |
| GACR | 803 | 17 606 | 8.70 | 8.25 | 0.45 | 0.0019 | 0.0029 | 0.0043 | 0.112 |
Each dataset was evaluated for the GAC, GACR and GACARG sequences by several statistical metrics.
Common B_D_Asc-PAR reflects the list of 135 overlapping genes in the Brown, Darnell and Ascano PAR-CLIP data; Common D_Asc-PAR reflects the list of 516 overlapping genes in the Darnell and Ascano PAR-CLIP data; Common B_D reflects the list of 180 overlapping genes in the Brown and Darnell data; Common B_D_Asc-RIP reflects the list of 53 overlapping genes in the Brown, Darnell and Ascano RIP-Chip data.
Lastly, we investigated the occurrence of ACUK or WGGA as a function of enrichment since it has been reported that more of these patterns in a gene increases enrichment by FMRP immunoprecipitation (9). We first plotted the total number of each pattern against the fold enrichment of all 940 genes in the Ascano-RIP data and found there was a weak positive correlation for both motifs (R2 = 0.010 for ACUK; R2 = 0.019 for WGGA), suggesting the more frequent the sequence is, the tighter FMRP binds the target (Supplementary Material, Fig. S1). However, when we examined the fold enrichment of each target gene as a function of its length, there was a similar positive relationship (R2 = 0.021), implicating gene length as a possible underlying cause of the apparent sequence correlation (Supplementary Material, Fig. S2). To assess the effect of gene length on the 940 RIP-Chip targets, we normalized each gene by length and plotted the number of sequence patterns per kilobase against the fold enrichment. In this analysis, we found almost no correlation between the number of times the patterns occurred and the enrichment (R2 = 0.001 with a negative slope for ACUK; R2 = 0.0001 for WGGA; Supplementary Material, Fig. S3), indicating that the length of the gene is responsible for the observed positive relationship between sequence element and target enrichment. Together these data indicate that when adjusted for the length of a gene, a greater number of these patterns do not increase the likelihood of FMRP binding.
Investigation of G-quadruplex formation by WGGA
Several previous studies have demonstrated that FMRP binds to a specific RNA secondary structure referred to as a G-quadruplex (11,16,17). A consensus sequence for the formation of G-quadruplexes bound by FMRP, DWGG was derived by RNA selection experiments (11). We noticed the similarity between this and WGGA, and hypothesized that the small enrichment of WGGA in the FMRP target gene sets could be due to the formation of G-quadruplexes by this sequence. Although the kinetics of G-quadruplex formation is not fully understood, there are a few generally accepted requirements in the formation of these structures (23–25). First, a core of 2–4 tandem guanines is necessary for the planar tetrad configuration. Second, there are typically 1–7 intervening nucleotides between these guanine cores known as loops, although loops of up to 21 nucleotides have been shown to be compatible with G-quadruplex formation (26). Therefore, we reasoned that if the WGGA patterns in the FMRP targets were situated in close proximity to one another, and the similar pattern DWGG is known to form G-quadruplexes targeted by FMRP, then the formation of these structures by WGGA would be possible and should be considered as a potential mechanism underlying the binding of FMRP to its targets. To evaluate this supposition, we analyzed the distribution of the WGGA patterns in the genes of each dataset compared with all other genes in the genome. Specifically, we filtered the data for sets of four WGGA patterns with a maximum of five intervening nucleotides, which we call a quadruplex forming motif (QFM; Fig. 5). We found an extraordinarily robust enrichment of QFMs in all FMRP target datasets, including the consensus datasets, compared with other genes (permutation P-value <1 × 107 in all cases), even in those that were not enriched for the WGGA pattern overall. This strong enrichment was detected in the total number of target genes with at least one QFM, as well as the total number of QFMs in all target genes (Tables 3A and B). Additionally, the percentage of genes with at least one QFM in both three-way common datasets was higher than any individual dataset, particularly in the 53 genes common to Ascano-RIP/Brown/Darnell, where over half of the genes contained at least one QFM. As a control for our method, we performed the same analysis using ACUK, another four-nucleotide sequence with a single ambiguous base. This motif cannot form G-quadruplexes, has no known biological reason to cluster, and therefore should not show enrichment as a QFM by our method. We found no enrichment of ACUK clusters in any of the datasets except for the Ascano-PAR data (Tables 3C and D). These results indicate that the WGGA pattern is specifically and highly clustered in FMRP targets, and potentially has the capacity to form a G-quadruplex structure that serves as a recognition element for FMRP.
Figure 5.
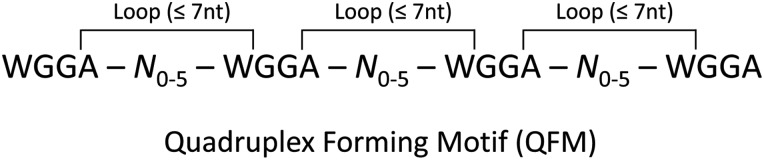
Schematic of a quadruplex forming motif. The QFM pattern used to query all datasets and evaluate the potential of G-quadruplex formation in FMRP targets. N is any nucleotide and is limited to a maximum of five since the ‘W’ and ‘A’ of the WGGA motif are considered part of the intervening loop sequence, which is typically seven nucleotides or less.
Table 3.
Quantification of quadruplex forming motifs in genes targeted by FMRP
| A | Genes with ≥1 WGGA QFM |
||
|---|---|---|---|
| Dataset | Genes | % of Total | Perm P-value |
| WGGA | |||
| Ascano-PAR | 1206 | 18.1 | <1 × 10−7 |
| Ascano-RIP | 281 | 30.5 | <1 × 10−7 |
| Brown | 112 | 31.6 | <1 × 10−7 |
| Darnell | 253 | 31.4 | <1 × 10−7 |
| B_D_A-PAR | 53 | 39.3 | <1 × 10−7 |
| B_D_A-RIP | 28 | 52.8 | <1 × 10−7 |
| Genome-wide | 2732 | 14.8 | N/A |
| B | Total number of WGGA QFMs | ||
| Dataset | QFMs | QFMs/Gene | Perm P-value |
| WGGA | |||
| Ascano-PAR | 1730 | 1.43 | <1 × 10−7 |
| Ascano-RIP | 443 | 1.58 | <1 × 10−7 |
| Brown | 166 | 1.48 | <1 × 10−7 |
| Darnell | 417 | 1.65 | <1 × 10−7 |
| B_D_A-PAR | 83 | 1.57 | <1 × 10−7 |
| B_D_A-RIP | 49 | 1.75 | 1 × 10−7 |
| Genome-wide | 3872 | 1.42 | N/A |
| C | Genes with ≥1 ACUK cluster | ||
| Dataset | Genes | % of Total | Perm P-value |
| ACUK | |||
| Ascano-PAR | 69 | 1.04 | <1 × 10−7 |
| Ascano-RIP | 8 | 0.87 | 0.118 |
| Brown | 2 | 0.56 | 0.376 |
| Darnell | 8 | 0.99 | 0.063 |
| B_D_A-PAR | 1 | 0.74 | 0.204 |
| B_D_A-RIP | 0 | 0.00 | 0.281 |
| Genome-wide | 114 | 0.62 | N/A |
| D | Total number of ACUK clusters | ||
| Dataset | Clusters | Clusters/gene | Perm P-value |
| ACUK | |||
| Ascano-PAR | 69 | 1.00 | <1 × 10−7 |
| Ascano-RIP | 8 | 1.00 | 0.118 |
| Brown | 2 | 1.00 | 0.376 |
| Darnell | 8 | 1.00 | 0.063 |
| B_D_A-PAR | 1 | 1.00 | 0.204 |
| B_D_A-RIP | 0 | – | 0.281 |
| Genome-wide | 114 | 1.00 | N/A |
Each dataset, including the overlapping genes from all three studies, were analyzed for clusters of each sequence motif. The total number of genes with at least one WGGA QFM/ACUK cluster (A and C, respectively) and the total number of WGGA QFMs/ACUK clusters (B and D, respectively) are shown, as well as the permutation P-values generated by comparison to random sets of genes; n = 10 000 000 permutations of the data. B_D_A-PAR, consensus genes from Brown, Darnell and Ascano PAR-CLIP; B_D_A-RIP, consensus genes from Brown, Darnell and Ascano RIP-Chip.
Assessment of kissing complexes in FMRP target sequences
A complex loop–loop pseudoknot structure known as a ‘kissing complex’ was identified by Darnell et al. as a target of the FMRP KH2 domain using in vitro selection methods (27). We assessed predicted kissing complexes (28) in consensus FMRP target genes identified in all three major datasets using the RNA sequence surrounding the dominant Ascano PAR-CLIP peak, as compared with randomly selected mRNA sequences of the same length. Although there were kissing complexes predicted in 44% of sequences (Supplementary Material, Fig. S4), this trend was not statistically significant (permutation P-value 0.27). This indicates that the kissing complex is not the primary determinant of FMRP targeting, but is consistent with KH2-bound kissing complexes contributing to target recognition either as a sub-dominant motif or as the dominant motif in only a subset of targets.
We also examined the enrichment and position of GAC motifs within the context of kissing complex RNAs. Notably, all the kissing complex RNAs identified by Darnell et al. contain the GAC sequence. In the Darnell kissing complex RNAs, the GAC is located with the GA in the 5′ single-stranded loop, while the C forms the first base-pair of the loop–loop interaction. Mutation of this 5′A was highly deleterious to KH2 binding (27), underlining the importance of this sequence. In the same set of FMRP target sequences used to assess kissing complexes, the GAC motif was enriched independently of whether the sequence was predicted to form a kissing complex. Within those sequences predicted to form kissing complexes, the location of the GAC sequence varied (Supplementary Material, Fig. S4), which suggests that there is not a strict sequence–structure relationship between the GAC motif and kissing complex pseudoknots.
Analysis of KH domain recognition sequence
Although the WGGA sequences are highly clustered in the FMRP targets compared with other genes, only 40–50% of the consensus genes contained at least one QFM, leaving the enrichment of the other targets unaccounted for in terms of recognition motif. This suggests that perhaps our QFM search parameters may be too stringent or, more likely, that additional RNA elements are also targeted by FMRP. Given that the RGG box has been shown to mediate FMRPs interaction with G-quadruplexes (11,16,17,29), the two KH-type domains, which function as mediators of RNA interaction as well, could account for the isolation of FMRP targets that do not contain a WGGA cluster. The recent study by Ray et al. used a truncated form of FMRP that contained both KH domains but not the RGG box, which yielded a consensus sequence that we found to be prevalent in many of the datasets and may represent the motif targeted specifically by the KH domains. To determine if the non-QFM genes contained high levels of this putative KH domain binding motif, we analyzed the highest confidence FMRP targets (i.e. the three-way overlap datasets) by dividing each into two groups: genes that contain a WGGA-QFM and those that do not. We searched for enrichment of GACR or GACARG in the non-QFM genes as compared with the QFM-containing FMRP targets and found no significant enrichment of either motif (Supplementary Material, Table S4). Additionally, the pattern frequency per kilobase does not increase as the level of target enrichment increases (data not shown). These results demonstrate that although the GACR and GACARG motifs are common in FMRP targets as a whole, they are distributed evenly throughout the target genes. Based on this analysis, these KH-domain recognition motifs may indeed be responsible for FMRPs interaction with non-QFM targets, but are not found exclusively in these genes. This also suggests that, in some cases, the KH domains and RGG box may work in concert to specifically interact with target genes.
To address the possibility that the two domains recognize and bind targets jointly, we leveraged the similarities in KH domain structure between FMRP and two paralogs, FXR1 and FXR2. The KH domains of FMRP are highly conserved in FXR1 and FXR2 and exhibit RNA-binding activity indistinguishable from that of FMRP's KH domains. In contrast, the C-terminal region, where the RGG box is located in FMRP, is divergent in the FXR proteins and lacks definitive RGG boxes or G-quadruplex binding activity (30). If the KH and RGG box domains of FMRP function independently, then the shared targets of FMRP, FXR1 and FXR2 would represent targets bound by the KH domains, whereas FMRP targets not bound by FXR proteins would represent targets bound by FMRP's RGG box. Therefore, we looked for enrichment of mRNAs containing WGGA QFMs in FMRP-only targets as compared with shared FMR1-family targets, as identified in PAR-CLIP data from Ascano et al. The percentage of genes that contain a QFM was equal in shared targets and FMRP-only targets (data not shown), suggesting that there are not distinct pools of KH-bound and RGG-bound FMRP targets and further supporting a cooperative binding model.
FMRP target validation by external database overlap and gene ontology (GO)
Each study design has its own merits and drawbacks, which makes determining the most accurate FMRP target dataset difficult. Since our consensus target lists are derived strictly in a relative manner by comparing one study to another, we sought to support the validity of our approach and findings using commonly utilized, independent databases. Given FMRP's role in intellectual disability and ASD, we investigated the correlation between the different groups of data compiled here and several external sources including the OMIM database, the SFARI database, and GO analysis. First, we extracted genes associated with the terms ‘mental retardation’ (MR) and ‘intellectual disability’ (ID) from the OMIM database, which yielded 962 genes. We also obtained a gene list from the SFARI database containing 528 genes related to autism susceptibility. We performed the pattern enrichment analysis on these two gene sets and observed a minor enrichment of ACUK and WGGA in both, a minor enrichment of GACR in the OMIM genes, and a significant enrichment of GACR in the SFARI genes (Tables 1 and 2). We also performed the QFM analysis on these sets and found that both the OMIM and SFARI genes had WGGA patterns that were clustered together compared with other genes (permutation P-value = 0.0047 and 0.0081, respectively), although to a lesser extent than any of the FMRP target lists, likely due to the presence of non-FMRP targets that have been implicated in MR/ID or ASD and do not contain a G-quadruplex motif. Next, we examined the amount of overlap between the OMIM and SFARI gene lists compared with all the gene lists in our analysis. We found that all datasets except for Ascano-PAR were significantly overlapping with both the autism susceptibility genes and MR/ID genes, with the Darnell set being exceptionally significant in both cases (Table 4). To measure the relationship between FMRP targets and neuronal processes, we used GO and Bioconductor to annotate the genes from each list. We found at least one neuronal-specific process in the top 10 most significant GO terms in each consensus target list, with the Brown and Darnell consensus list containing the most (4 out of 10; Supplementary Material, Table S5), suggesting this dataset may contain the most relevant set of FMRP target genes in a neuronal context.
Table 4.
Overlap of genes from FMRP target datasets and independent databases
| FMRP target gene set (# genes in dataset) | SFARI database (528 genes) |
OMIM database (962 genes) |
||
|---|---|---|---|---|
| Gene overlap | P-value | Gene overlap | P-value | |
| Ascano-PAR (6658) | 216 | 0.23065 | 373 | 0.64051 |
| Ascano-RIP (921) | 38 | 0.03880 | 66 | 0.02199 |
| Brown (354) | 31 | 1.18 × 10−6 | 32 | 0.01746 |
| Darnell (807) | 94 | 3.45 × 10−29 | 101 | 4.83 × 10−14 |
| B_D_A-PAR (135) | 12 | 0.00077 | 19 | 0.00014 |
| D_A-PAR (516) | 16 | 0.00633 | 63 | 2.41 × 10−9 |
| B_D (180) | 16 | 9.63 × 10−5 | 22 | 0.00030 |
| B_D_A-RIP (53) | 20 | 6.62 × 10−7 | 22 | 0.00044 |
The level of gene overlap between each of the FMRP datasets and the SFARI and OMIM databases is shown. The common gene sets were generated from the following combinations: B_D_A-PAR, Brown/Darnell/Ascano PAR-CLIP; D_A-PAR, Darnell/Ascano PAR-CLIP; B _D, Brown/Darnell; B_D_A-RIP, Brown/Darnell/Ascano RIP-Chip. P-values were generated by Fisher's exact test.
Top ranked phenotype-associated FMRP targets
A drawback to the three-way consensus lists generated in this report is the exclusion of genes that are not present in all studies, even if good evidence of association with FMRP exists in two of the datasets. Furthermore, if genes targeted by FMRP already have known associations to neurodevelopmental disease, it would be helpful to highlight these interactions as perhaps the most relevant associations to investigate in FXS and related neurodevelopmental disorders. To address these issues, we first used a rank aggregation method (31) to generate a single top 100 ranked list derived from target enrichment data from the Brown, Darnell and Ascano-RIP datasets. Additionally, to link FMRP targets to neurodevelopmental phenotypes, we used the genes from this top 100 list to search the PubMed database for association with four terms: Fragile X, autism, mental retardation and intellectual disability. We then sorted the genes by the total number of publications that include both the gene and the search terms, thus creating a list of the 40 most highly enriched FMRP targets with published associations to a given phenotype (Table 5). It is important to note that although ∼60 of these 100 genes have no published association to neurodevelopmental disorders, they remain important candidates for future studies based on their high level of enrichment.
Table 5.
The top 40 target genes of FMRP-associated with phenotypes
| Rank | Gene | Fragile X | Autism | MR/ID | Total |
|---|---|---|---|---|---|
| 1 | TSC2 | 5 | 72 | 110 | 187 |
| 2 | MTOR | 37 | 75 | 65 | 177 |
| 3 | NAV1 | 0 | 15 | 35 | 50 |
| 4 | CREBBP | 0 | 3 | 38 | 41 |
| 5 | EHMT1 | 0 | 5 | 27 | 32 |
| 6 | TRIO | 0 | 18 | 9 | 27 |
| 7 | DST | 0 | 2 | 16 | 18 |
| 8 | ANKRD11 | 1 | 6 | 10 | 17 |
| 9 | CYFIP2 | 8 | 1 | 8 | 17 |
| 10 | ITPR1 | 1 | 2 | 11 | 14 |
| 11 | SMARCA4 | 1 | 1 | 12 | 14 |
| 12 | SKI | 1 | 1 | 9 | 11 |
| 13 | ANK3 | 1 | 6 | 3 | 10 |
| 14 | CHD8 | 0 | 6 | 3 | 9 |
| 15 | HERC2 | 0 | 4 | 4 | 8 |
| 16 | BCR | 1 | 1 | 5 | 7 |
| 17 | BSN | 1 | 2 | 4 | 7 |
| 18 | SPTAN1 | 0 | 1 | 5 | 6 |
| 19 | SCAP | 0 | 3 | 2 | 5 |
| 20 | HCFC1 | 0 | 0 | 5 | 5 |
| 21 | ATP2B2 | 0 | 4 | 1 | 5 |
| 22 | ARHGEF7 | 0 | 0 | 4 | 4 |
| 23 | COBL | 1 | 2 | 1 | 4 |
| 24 | ALS2 | 1 | 1 | 2 | 4 |
| 25 | MAPK8IP1 | 0 | 0 | 4 | 4 |
| 26 | PRPF8 | 1 | 1 | 1 | 3 |
| 27 | TRAPPC10 | 0 | 0 | 2 | 2 |
| 28 | CUX1 | 0 | 1 | 1 | 2 |
| 29 | TSHZ1 | 0 | 0 | 2 | 2 |
| 30 | CIC | 0 | 0 | 2 | 2 |
| 31 | FOXK2 | 0 | 0 | 2 | 2 |
| 32 | HERC1 | 0 | 1 | 1 | 2 |
| 33 | MYT1L | 0 | 1 | 1 | 2 |
| 34 | PI4KA | 0 | 1 | 0 | 1 |
| 35 | BAI2 | 0 | 1 | 0 | 1 |
| 36 | GPRIN1 | 0 | 1 | 0 | 1 |
| 37 | MAST4 | 0 | 1 | 0 | 1 |
| 38 | LPHN1 | 0 | 0 | 1 | 1 |
| 39 | JAK1 | 0 | 0 | 1 | 1 |
| 40 | FASN | 0 | 1 | 0 | 1 |
A list of top targets of FMRP was generated based on enrichment rank within all three studies (Brown, Darnell and Ascano-RIP) and their association with the published literature using the search terms: Fragile X, autism and mental retardation (MR)/intellectual disability (ID). The numbers below each of these search terms represents the number of abstracts in PubMed that include both the target gene and the specific search term, while the far right column is the total number of publications for each gene and all search terms. Note: One gene, LARGE, was excluded from this list since the term ‘large’ appears in numerous publications unrelated to the gene, artificially inflating the number of publications returned.
DISCUSSION
We critically analyzed three of the largest studies of empirically derived FMRP targets to determine mRNAs that are found to consistently interact with the protein and identify any specific recognition elements within those targets. The recognition motifs ACUK and WGGA were found to be enriched in the full target list generated by the Ascano-PAR assay, confirming their findings by our independent analysis methods. However, when the Ascano-RIP validated candidates were examined, arguably the more stringent and stably bound target set, the ACUK sequence was no longer in excess. In fact, our analysis of all datasets, including the consensus lists, showed there is not only a lack of ACUK enrichment, but a deficiency of the pattern in each dataset. A possible reason for the enrichment of ACUK exclusively in the PAR-CLIP data is based on the fact that the assay utilizes an in vivo incorporation of the uridine analog 4-thiouridine (4SU) into nascent RNA transcripts to identify the precise location of RNA:protein interaction. The ACUK sequence contains at least one, and possibly two uridine nucleotides, which imparts a greater probability of 4SU incorporation and, therefore, a greater chance of creating a crosslink between an RNA and protein without regard for the strength of the initial interaction. This could result in the co-immunoprecipitation of RNAs that may not have a significant association with FMRP. Additionally, this bond, induced by UV at 365 nm, was shown to be a stronger link than that of the 254 nm UV crosslinking used by Darnell et al. in the HITS-CLIP assay (9,32), which may also cause the co-precipitation of genes that are not interacting at physiologically relevant levels. Along with differences in bioinformatic filtering, this may partially account for the large difference in FMRP-associated RNAs discovered by these two similar assays. These findings indicate that ACUK is, at best, a weak recognition sequence of FMRP and is not more common in the top candidates of any of these studies, even another CLIP assay.
The other proposed FMRP recognition sequence, WGGA, was found to be modestly enriched in the FMRP targets of several datasets and consensus lists, though several other lists did not show enrichment, leaving the validity of the sequence as a standalone recognition element uncertain. We hypothesized that if the WGGA sequences were closely clustered such that they could potentially form a two tetrad G-quadruplex structure, a known target of FMRP, this could explain the apparent lack of enrichment of the motif in several datasets, while others are enriched for it. Our analysis shows this to be an extremely plausible theory, as WGGA motifs were very highly clustered in FMRP targets compared with the rest of the genes in the genome. To demonstrate specificity of this phenomenon to WGGA, analysis of a different four-nucleotide sequence with a single ambiguous position (ACUK) showed almost no indication of pattern clustering. Based on these findings, we postulate that the distribution of WGGA motifs, rather than the aggregate number, is important for recognition. For example, the consensus list from the overlap of Brown and Darnell, which is one of the strongest target lists as determined by SFARI and OMIM overlap as well as GO term annotation, was not enriched for the WGGA pattern compared with other genes. However, the WGGA patterns that were present in this set were found to be highly clustered based on our analysis. This conclusion is supported by the lack of correlation between FMRP target enrichment levels and the number of WGGA patterns present when gene length is considered. These computational results require empirical confirmation, but they are highly suggestive of a scenario where G-quadruplex formation is possible, especially in light of FMRP's demonstrated proclivity for the structure.
The patterns GAC, GACR and GACARG were identified as FMRP binding motifs via in vitro experiments by Ray et al. and we found that each of these motifs is prevalent in the high-confidence FMRP targets from in vivo studies, particularly the GACR motif, which was significantly enriched in almost all FMRP datasets compared with other genes. Thus, our data supports these binding motifs as recognition elements of FMRP. Understanding the limited specificity that can be imparted by short recognition sequences, we assessed the possibility that the GAC sequence is a requisite motif within a secondary structure element. Ray et al. deliberately minimized RNA secondary structure while preparing their RNA sequence library. Accordingly, we found no evidence for consistent RNA secondary structure when we assessed the top sequences bound by FMRP in the Ray et al. data set (data not shown). We also found no correlation between the presence of GAC and kissing complexes, nor any consistent position of the GAC motif within predicted kissing complexes. These data support a model in which FMRP binds unstructured GAC but also likely requires binding to additional recognition elements to specify target mRNAs.
Because the GAC motif was established using a truncated FMRP with only KH domains, this provided us the opportunity to analyze the high-confidence FMRP targets that did not contain a clustered WGGA for the presence of this KH-specific motif to potentially explain their association with FMRP as KH domain mediated. However, we found very little evidence for enrichment of the pattern in non-QFM target genes compared with QFM-containing targets, indicating that although the KH-specific patterns are common in FMRP targets, they are not more prevalent in non-G-quadruplex containing genes. Similarly, QFMs were not enriched in targets bound only by FMRP over targets that were also bound by FXR1 and FXR2, which share KH domains homologous to FMRP's but lack functional RGG boxes. Taken together, these data suggest that FMRP targets are established through a more complex interaction than independent binding by the KH or RGG box domains alone.
Our G-quadruplex analysis identified the presence of increased QFMs in the Darnell dataset, whereas Darnell et al. (10) found no enrichment of such motifs in the same data. Our analyses differed in several ways; namely, the pattern used to explore the data and our permutation analysis. The aim of our investigation was to determine the distribution of WGGA sequences in FMRP targets compared with non-targets using the general requirements for G-quadruplex formation proposed by the scientific community, and we performed this using the pattern shown in Fig. 5; this differs slightly from the parameters used by Darnell et al. In addition to this difference, the FMRP target set was compared with one set of random non-targets by Darnell et al., whereas we generated 10 000 000 random non-target gene sets for comparison, and found none with more QFMs than the FMRP targets. This permutation analysis provides a great deal of confidence in our results and suggests the FMRP targets are indeed highly enriched for WGGA QFMs in all datasets compared with the rest of the genes in the genome.
Using the independent databases OMIM and SFARI, we showed that genes associated with MR and ASD overlap significantly with all of the FMRP target lists except the Ascano-PAR data, which is likely due to the large number of genes identified by this method, some of which may be interacting incidentally or not associated with MR and ASD. Additionally, GO analysis revealed a high level of neurological processes in several datasets, especially in the common genes derived from the Brown and Darnell lists, where four of the top five most significant terms were related to neurological processes, as well as having the highest total of any dataset. This suggests that this consensus list may represent the most consistent targets of FMRP in the brain, since two vastly different experimental approaches identified these targets and both used neuronal tissue. The Ascano datasets had no GO terms specific to neurological processes in the 10 most significant annotations, which is likely the result of using a tissue type other than brain rather than a reflection of the quality of the data. Additionally, It is important to note that while each of these studies have identified some number of true FMRP ligands, it is perhaps only a subset of all the genes targeted by the protein since transcript abundance, cellular conditions and experimental limitations likely dictate the mRNAs bound by FMRP. This could result in real interactions that were undetectable by any one of these study designs and thus would be excluded from the target lists.
Comparison of the target lists from each study to one another revealed an extremely high overlap between the Brown and Darnell lists, while either Ascano dataset, although overlapping substantially, was ∼100 orders of magnitude less significant in comparison. Despite using relatively dissimilar assays, the Brown and Darnell studies used mouse brain tissue to identify targets of endogenous FMRP. In contrast, the Ascano data were generated by evaluating interactions of heterologously expressed FMRP in HEK293 cell lines, which likely accounts for the weaker correlation with the other two studies. RNA-seq data suggest that human brain tissue and HEK293 lines express 8579 genes in common, which accounts for ∼90% of all gene expression in HEK293 cells (9). Importantly, however, there are over 4400 genes expressed in human brain tissue that are not detected in HEK293 lines, meaning HEK293 cells express only 66% of the genes present in the brain. Each of these 4400 genes represents a potential target of FMRP that was not interrogated in any of the Ascano data and is a very reasonable explanation for the diminished overlap with the Brown and Darnell target lists. Additionally, these 4400 genes almost certainly represent many that are neuron-specific and perhaps the most important to evaluate for FMRP interaction, while the overlapping genes are more likely to be common to many or all cell types. Furthermore, while the expression level of FMR1 was found to be similar between brain tissue and the experimental system used by Ascano et al., the endogenous expression level of all other genes may differ considerably in each tissue, thereby confounding the evaluation of the level of enrichment and true relationships between FMRP and its targets. Since many genes are expressed in a tissue-specific manner, and Fragile X syndrome is a neuropsychiatric disorder, neuronal tissue is the most relevant experimental system in which to assess binding targets of FMRP. We recommend that any future attempts to evaluate targets of FMRP or other neuronal RNA-binding proteins should be performed using brain tissue.
The FMRP target lists generated here may be particularly useful for investigating the role of FMRP in the etiology of related neurodevelopmental diseases. Studies have revealed that de novo point mutations occur in FMRP target genes more frequently in autism patients than unaffected siblings, and FMRP targets may account for up to half of all autism susceptibility genes (7,33–35). Additionally, FMRP targets have recently been implicated in schizophrenia, where an elevated level of single nucleotide mutations or small indels was found in FMRP targets of affected individuals (6,8). Our high-confidence consensus lists provide a well-defined set of FMRP-associated genes that will help link diseases influenced by FMRP function to specific genes and molecular pathways, such as the newly discovered association between FMRP targets and schizophrenia. Given these emerging relationships, these lists also serve as a method for prioritizing the multitude of variants of unknown significance revealed by whole exome/genome sequencing of autism and schizophrenia patients, and will help guide follow-up functional studies.
Overall, we compiled FMRP target lists based on the overlap of several different datasets, and each has value in various experimental settings. All consensus lists generated here are genes found to consistently associate with FMRP by several different methods and research groups, which provides a higher level of confidence in the interaction. Most of these datasets were enriched for GACR, depleted of ACUK, and several were enriched for the WGGA pattern. The distribution of WGGA within FMRP targets appears to be important and we provide evidence that this is consistent with the formation of G-quadruplexes by this sequence. Though WGGA may not be the only sequence element recognized by FMRP, the presence of clusters of this motif in mRNAs will improve the identification of putative FMRP targets and provides a mechanistic and testable basis for the interaction. Tissue type and experimental method used doubtlessly play a role in the FMRP:mRNA interactions that are captured, and the consensus lists generated here should be applied to future endeavors with the knowledge of which datasets were used to derive them. Lastly, these consensus lists should prove to be a valuable resource for investigating the genetic causes of autism and schizophrenia because of the connection between FMRP-associated genes and these diseases.
MATERIALS AND METHODS
Dataset construction and acquisition
Ensembl was used to construct a whole genome dataset and was limited to genes that code for proteins and have both a 5′ UTR and a 3′UTR. There were 18 409 such genes and we use this dataset for all pattern frequency analyses. The FMRP target datasets generated by Ascano et al., Darnell et al. and Brown et al. were obtained via the Supplementary Material provided by the journal in which each was published. Each of these datasets contained genes that were not present in our whole genome dataset, and these genes were excluded from analysis. Information about each dataset used is summarized in Supplementary Material, Table S1.
Significance tests for target list overlap
For significance of overlap between two gene sets, both the Fisher's exact test and a permutation test were conducted. For significance of overlap between three datasets, only a permutation test was conducted. In each case, the full dataset from each study was used without regard for whether a gene was present or absent from the Ensembl dataset. The number of common genes in a random permutation was calculated as the overlap between two random gene lists that contained an equal number of genes as the two actual gene lists. The permutation P-value was calculated as the number of times the number of common genes obtained from the permutations exceeded the number of common genes between the two actual gene lists, divided by the number of permutations (N = 1 000 000). A similar methodology was followed to calculate the permutation P-value for the overlap between three datasets.
Significance tests for pattern enrichment
The significance of the difference in mean number of patterns (e.g. WGGA) occurring in a gene set (e.g. Darnell) and the rest of the genome was assessed using the t-test, Wilcox rank sum test and permutation tests. All t-tests were two tailed. The permuted P-value for enrichment was calculated as the number of times the permuted mean difference exceeded the actual mean difference divided by the number of permutations (N = 1 000 000). A similar methodology was followed to obtain the P-values for QFM analysis. All analyses were conducted in R (36).
Significance tests for QFM analysis
The QFM permutation value was calculated as the number of times the occurrences of QFM patterns in a random gene list, containing the same number of genes as the actual gene list, exceeded the occurrences of QFM patterns in the actual gene list divided by the number of permutations (N = 10 000 000).
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the National Institutes of Health (HD24064 to S.T.W., 1RO1MH085617 to G.J.B., 1T32MH087977 to J.A.S. and B.R.A., and 1F32NS083205 to B.R.A.).
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Emory University's research IT service center for access to its high performance computer cluster; Michael Santoro and Karen Connelly for helpful discussions; David Cutler for review of the manuscript and useful suggestions; and Warren lab members for thoughtful comments.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Verkerk A.J., Pieretti M., Sutcliffe J.S., Fu Y.H., Kuhl D.P., Pizzuti A., Reiner O., Richards S., Victoria M.F., Zhang F.P., et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 2.Pieretti M., Zhang F.P., Fu Y.H., Warren S.T., Oostra B.A., Caskey C.T., Nelson D.L. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. 1991;66:817–822. doi: 10.1016/0092-8674(91)90125-i. [DOI] [PubMed] [Google Scholar]
- 3.Coffee B., Keith K., Albizua I., Malone T., Mowrey J., Sherman S.L., Warren S.T. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am. J. Hum. Genet. 2009;85:503–514. doi: 10.1016/j.ajhg.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ashley C.T., Jr, Wilkinson K.D., Reines D., Warren S.T. FMR1 protein: conserved RNP family domains and selective RNA binding. Science. 1993;262:563–566. doi: 10.1126/science.7692601. [DOI] [PubMed] [Google Scholar]
- 5.Brown V., Jin P., Ceman S., Darnell J.C., O'Donnell W.T., Tenenbaum S.A., Jin X., Feng Y., Wilkinson K.D., Keene J.D., et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107:477–487. doi: 10.1016/s0092-8674(01)00568-2. [DOI] [PubMed] [Google Scholar]
- 6.Fromer M., Pocklington A.J., Kavanagh D.H., Williams H.J., Dwyer S., Gormley P., Georgieva L., Rees E., Palta P., Ruderfer D.M., et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iossifov I., Ronemus M., Levy D., Wang Z., Hakker I., Rosenbaum J., Yamrom B., Lee Y.H., Narzisi G., Leotta A., et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Purcell S.M., Moran J.L., Fromer M., Ruderfer D., Solovieff N., Roussos P., O'Dushlaine C., Chambert K., Bergen S.E., Kahler A., et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506:185–190. doi: 10.1038/nature12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ascano M., Jr, Mukherjee N., Bandaru P., Miller J.B., Nusbaum J.D., Corcoran D.L., Langlois C., Munschauer M., Dewell S., Hafner M., et al. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature. 2012;492:382–386. doi: 10.1038/nature11737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Darnell J.C., Van Driesche S.J., Zhang C., Hung K.Y., Mele A., Fraser C.E., Stone E.F., Chen C., Fak J.J., Chi S.W., et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Darnell J.C., Jensen K.B., Jin P., Brown V., Warren S.T., Darnell R.B. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell. 2001;107:489–499. doi: 10.1016/s0092-8674(01)00566-9. [DOI] [PubMed] [Google Scholar]
- 12.Miyashiro K.Y., Beckel-Mitchener A., Purk T.P., Becker K.G., Barret T., Liu L., Carbonetto S., Weiler I.J., Greenough W.T., Eberwine J. RNA cargoes associating with FMRP reveal deficits in cellular functioning in Fmr1 null mice. Neuron. 2003;37:417–431. doi: 10.1016/s0896-6273(03)00034-5. [DOI] [PubMed] [Google Scholar]
- 13.Bassell G.J., Warren S.T. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008;60:201–214. doi: 10.1016/j.neuron.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Santoro M.R., Bray S.M., Warren S.T. Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Ann. Rev. Pathol. 2012;7:219–245. doi: 10.1146/annurev-pathol-011811-132457. [DOI] [PubMed] [Google Scholar]
- 15.Menon L., Mader S.A., Mihailescu M.R. Fragile X mental retardation protein interactions with the microtubule associated protein 1B RNA. RNA. 2008;14:1644–1655. doi: 10.1261/rna.1100708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Menon L., Mihailescu M.R. Interactions of the G quartet forming semaphorin 3F RNA with the RGG box domain of the fragile X protein family. Nucleic Acids Res. 2007;35:5379–5392. doi: 10.1093/nar/gkm581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schaeffer C., Bardoni B., Mandel J.L., Ehresmann B., Ehresmann C., Moine H. The fragile X mental retardation protein binds specifically to its mRNA via a purine quartet motif. EMBO J. 2001;20:4803–4813. doi: 10.1093/emboj/20.17.4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Phan A.T., Kuryavyi V., Darnell J.C., Serganov A., Majumdar A., Ilin S., Raslin T., Polonskaia A., Chen C., Clain D., et al. Structure-function studies of FMRP RGG peptide recognition of an RNA duplex-quadruplex junction. Nat. Struct. Mol. Biol. 2011;18:796–804. doi: 10.1038/nsmb.2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joachimi A., Benz A., Hartig J.S. A comparison of DNA and RNA quadruplex structures and stabilities. Bioorganic Med. Chem. 2009;17:6811–6815. doi: 10.1016/j.bmc.2009.08.043. [DOI] [PubMed] [Google Scholar]
- 20.Dolzhanskaya N., Sung Y.J., Conti J., Currie J.R., Denman R.B. The fragile X mental retardation protein interacts with U-rich RNAs in a yeast three-hybrid system. Biochem. Biophys. Res. Comm. 2003;305:434–441. doi: 10.1016/s0006-291x(03)00766-6. [DOI] [PubMed] [Google Scholar]
- 21.Chen L., Yun S.W., Seto J., Liu W., Toth M. The fragile X mental retardation protein binds and regulates a novel class of mRNAs containing U rich target sequences. Neuroscience. 2003;120:1005–1017. doi: 10.1016/s0306-4522(03)00406-8. [DOI] [PubMed] [Google Scholar]
- 22.Ray D., Kazan H., Cook K.B., Weirauch M.T., Najafabadi H.S., Li X., Gueroussov S., Albu M., Zheng H., Yang A., et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013;499:172–177. doi: 10.1038/nature12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kikin O., D'Antonio L., Bagga P.S. QGRS Mapper: a web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res. 2006;34:W676–W682. doi: 10.1093/nar/gkl253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stegle O., Payet L., Mergny J.L., MacKay D.J., Leon J.H. Predicting and understanding the stability of G-quadruplexes. Bioinformatics. 2009;25:i374–i382. doi: 10.1093/bioinformatics/btp210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burge S., Parkinson G.N., Hazel P., Todd A.K., Neidle S. Quadruplex DNA: sequence, topology and structure. Nucleic Acids Res. 2006;34:5402–5415. doi: 10.1093/nar/gkl655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bourdoncle A., Estevez Torres A., Gosse C., Lacroix L., Vekhoff P., Le Saux T., Jullien L., Mergny J.L. Quadruplex-based molecular beacons as tunable DNA probes. J. Am. Chem. Soc. 2006;128:11094–11105. doi: 10.1021/ja0608040. [DOI] [PubMed] [Google Scholar]
- 27.Darnell J.C., Fraser C.E., Mostovetsky O., Stefani G., Jones T.A., Eddy S.R., Darnell R.B. Kissing complex RNAs mediate interaction between the Fragile-X mental retardation protein KH2 domain and brain polyribosomes. Genes Dev. 2005;19:903–918. doi: 10.1101/gad.1276805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bon M., Micheletti C., Orland H. McGenus: a Monte Carlo algorithm to predict RNA secondary structures with pseudoknots. Nucleic Acids Res. 2013;41:1895–1900. doi: 10.1093/nar/gks1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Darnell J.C., Warren S.T., Darnell R.B. The fragile X mental retardation protein, FMRP, recognizes G-quartets. Mental Retardation Dev. Disabilities Res. Rev. 2004;10:49–52. doi: 10.1002/mrdd.20008. [DOI] [PubMed] [Google Scholar]
- 30.Darnell J.C., Fraser C.E., Mostovetsky O., Darnell R.B. Discrimination of common and unique RNA-binding activities among Fragile X mental retardation protein paralogs. Hum. Mol. Genet. 2009;18:3164–3177. doi: 10.1093/hmg/ddp255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kolde R., Laur S., Adler P., Vilo J. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics. 2012;28:573–580. doi: 10.1093/bioinformatics/btr709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hafner M., Landthaler M., Burger L., Khorshid M., Hausser J., Berninger P., Rothballer A., Ascano M., Jr, Jungkamp A.C., Munschauer M., et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neale B.M., Kou Y., Liu L., Ma'ayan A., Samocha K.E., Sabo A., Lin C.F., Stevens C., Wang L.S., Makarov V., et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O'Roak B.J., Vives L., Girirajan S., Karakoc E., Krumm N., Coe B.P., Levy R., Ko A., Lee C., Smith J.D., et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanders S.J., Murtha M.T., Gupta A.R., Murdoch J.D., Raubeson M.J., Willsey A.J., Ercan-Sencicek A.G., DiLullo N.M., Parikshak N.N., Stein J.L., et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Team R.C. R: A Language and Environment for Statistical Computing. Vienna, Austria: 2012. http://www.R-project.org/ [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.