

Abstract
Any given chromosomal activity (e.g., transcription) is governed predominantly by the local epiproteome. However, defining local epiproteomes has been limited by a lack of effective technologies to isolate discrete sections of chromatin and to identify with precision specific proteins and histone posttranslational modifications (PTMs). We report the use of the Cas9 and guide RNA (gRNA) components of the CRISPR system for gRNA-directed purification of a discrete section of chromatin. Quantitative mass spectrometry provides for unambiguous identification of proteins and histone PTMs specifically associated with the enriched chromatin. This CRISPR-based Chromatin Affinity Purification with Mass Spectrometry (CRISPR-ChAP-MS) approach revealed changes in the local epiproteome of a promoter during activation of transcription. CRISPR-ChAP-MS thus has broad applications for discovering molecular components and dynamic regulation of any in vivo activity at a given chromosomal location.
Keywords: epigenetics, proteomics, histone, epiproteome, posttranslational modification, affinity purification
Introduction
For the work presented, a “local epiproteome” refers to not only the histone PTMs at a specific chromosomal location that are involved in a particular activity,1 but also to the other proteins associated with the region in addition to the histones. Identifying the components of a specific epiproteome can provide unprecedented insight into the molecular and epigenetic mechanisms regulating an activity. For example, gene transcription could have various epiproteomes that regulate initiation, elongation and termination. A recently accomplished milestone for measuring local epiproteomes has been the development of affinity enrichment procedures to isolate small regions of chromatin.2-9 Purification of a small region of chromatin from the cellular milieu is one of the most challenging aspects of these approaches as the proteins and histone PTMs specifically isolated with the targeted chromatin typically constitute a small fraction of the identified proteins – most of which are non-specific associations.2,3,10 We developed two approaches using quantitative high resolution mass spectrometry that distinguish whether proteins and histone PTMs identified during epiproteome measurements are “specific” to the target chromatin or are “non-specific” contaminants.2,3 These quantitative approaches are critical components of our ChAP-MS (Chromatin Affinity Purification with Mass Spectrometry) platform of technologies that enable local epiproteome analysis. Included in this platform are the first generation ChAP-MS and second generation TAL-ChAP-MS approaches.2,3 The ChAP-MS approach, which used a targeted LexA protein as an affinity reagent, demonstrated the first unambiguous epiproteome measurement. The TAL-ChAP-MS approach achieved similar high resolution and specificity by using the genomic targeting ability of the TALEN (Transcription Activator-Like Effector Nuclease) system for local epiproteome isolation and analysis.3,11
Described here is the third generation technology termed CRISPR-ChAP-MS (Fig. 1). The prokaryotic viral defense system CRISPR (Clustered Regularly Interspaced Palindromic Repeats) has recently been developed as a genome-editing tool for eukaryotes.12 The core components of this system include the Cas9 nuclease, which is able to create double-strand breaks in DNA, and guide RNA (gRNA), which is bound by Cas9 and serves to direct this complex to a target sequence complementary to the gRNA. Using the Type II CRISPR system from Streptococcus pyogenes, we have harnessed the specific gene-targeting capability of the Cas9/gRNA complex to isolate and identify a specific local epiproteome. We utilized a catalytically inactive, protein A (PrA) tagged version of Cas9 along with a gRNA to target the promoter region of the GAL1 gene in Saccharomyces cerevisiae in order to validate the CRISPR-ChAP-MS technology (Fig. 1).
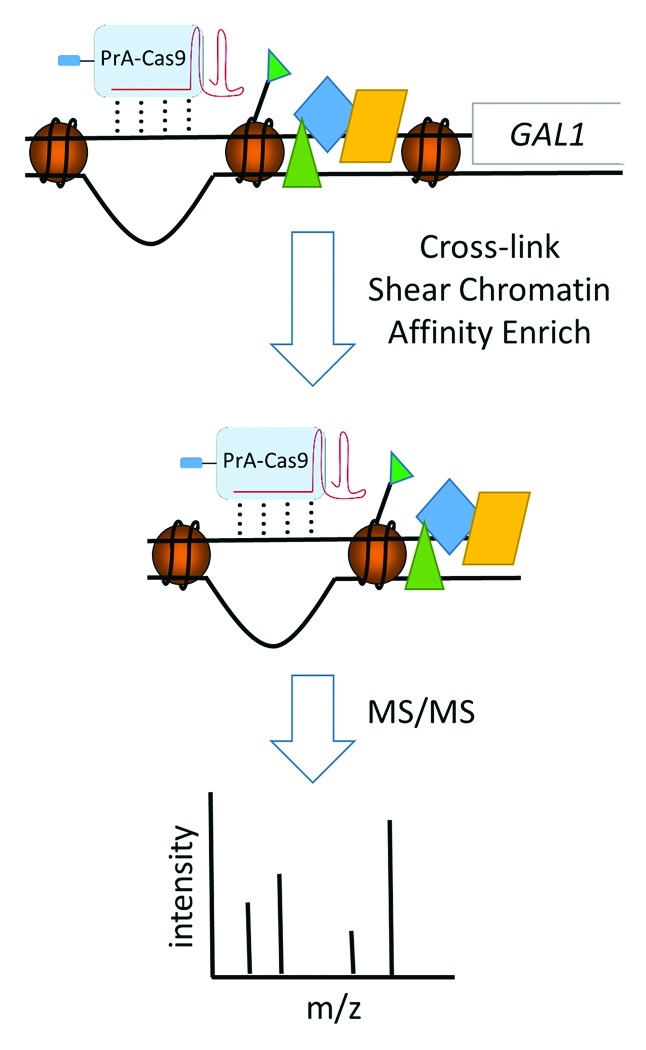
Figure 1. CRISPR-ChAP-MS approach. PrA-tagged Cas9 bound to gRNA is targeted to a specific region of chromatin. Following chemical cross-linking, the chromatin is sheared to approximately 1 kb in size and subjected to affinity isolation with IgG-coated beads. Isolated chromatin containing PrA-tagged Cas9/gRNA is then analyzed with high resolution mass spectrometry to identify specifically associated proteins and histone posttranslational modifications.
To isolate the targeted chromatin, cells were treated with formaldehyde to stabilize interactions,3 chromatin was sheared to fragments approximately 1 kb in length, and the target chromatin was affinity purified using the PrA tag. Affinity tagged versions of Cas9 have been shown to target chromatin for partial enrichment13; however, a quantitative analysis of the specifically bound proteins and histone PTMs has not been reported. Here using our CRISPR-ChAP-MS approach that does provide for quantitative identification of specifically bound proteins and histone PTMs, the GAL1 promoter chromatin from yeast was isolated under transcriptionally active conditions and subjected to a label-free mass spectrometric workflow to identify the specific components of the local epiproteome. Relative to the first and second generations of the ChAP-MS technological platform, CRISPR-ChAP-MS shows an enhanced ability to isolate targeted chromatin, which is critical for epiproteome analysis. The TAL-based approach also requires design of a specific TAL protein for each sequence targeted whereas CRISPR-ChAP-MS only requires site-directed mutagenesis to alter the gRNA for genomic targeting, which provides a faster and more cost effective approach that can be multiplexed to target additional sites.
Results and Discussion
To validate the CRISPR-ChAP-MS approach, the promoter chromatin of the GAL1 gene was targeted for enrichment in S. cerevisiae. This region of chromatin is an attractive target for validation studies as one can supply yeast with galactose in place of glucose to rapidly and synchronously stimulate transcriptional activation of GAL1 – thereby setting a transcriptionally active chromatin state for epiproteome analysis. Cells were transformed with plasmids expressing a nuclease inactive and PrA-tagged version of Cas9 (pPrA-Cas9) and/or expressing a gRNA specific to the promoter region of the GAL1 gene (pgRNA-GAL1). Similar expression of PrA-Cas9 in glucose and galactose was demonstrated by Western-blotting (Fig. 2A). To evaluate whether expression of this PrA-Cas9/gRNA complex affected transcription of GAL1, cDNA was prepared from cells in glucose (transcriptionally repressed GAL1) and galactose (transcriptionally active GAL1) and was analyzed by quantitative real-time PCR (qPCR). GAL1 transcription was similar in cells expressing PrA-Cas9/gRNA compared with cells expressing only PrA-Cas9, indicating that expression of PrA-Cas9/gRNA does not drastically alter transcriptional activation (Fig. 2B). To determine whether this expressed PrA-Cas9/gRNA complex was bound to and could enrich chromatin at the GAL1 promoter region, ChIP was performed to PrA-Cas9 in cells from glucose and galactose cultures and analyzed relative to an unguided PrA-Cas9 by qPCR. GAL1 promoter chromatin was enriched in a gRNA dependent manner with PrA-Cas9 from both glucose (4.9-fold) and galactose (70-fold) growth conditions (Fig. 2C). There was no detectable enrichment of chromatin 2 kb upstream or downstream of the GAL1 target site by qPCR (Fig. 2C), indicating that chromatin purification was localized to the gRNA target region. In previous studies using TAL proteins targeted to the same chromatin region,2 a ∼6-fold enrichment was observed under galactose growth conditions and no enrichment under glucose growth. Therefore, the enrichment observed with PrA-Cas9/gRNA in galactose-containing media is greater than an order of magnitude higher relative to a TAL targeted to this region of chromatin.

Figure 2. A PrA-tagged Cas9/gRNA complex can specifically enrich a small chromatin section. (A) Using Western-blotting to the PrA-tag, similar expression of PrA-Cas9 was shown in both glucose and galactose-containing media. Western-blotting to histone H4 was used as a loading control. S. cerevisiae were transformed with either a plasmid expressing PrA-tagged Cas9 (pPrA-Cas9) and/or a plasmid expressing gRNA specific to a sequence in the promoter of GAL1 (pgRNA-GAL1). (B) Real-time reverse transcription PCR showed similar galactose-induced transcription of the GAL1 gene relative to ACT1 in cells expressing PrA-Cas9 ± gRNA-GAL1. Transcript levels of GAL1 are reported as a ratio of detection in galactose relative to glucose-containing media. (C) PrA-Cas9/gRNA complex specifically enriched GAL1 promoter chromatin under transcriptionally active conditions. Using ChIP to the PrA-tag on Cas9, enrichment at each indicated target relative to actin was measured in cells containing the PrA-Cas9/gRNA complex in comparison to those with only the PrA-Cas9. The genomic targets were: GAL1 for the genomic target of the gRNA-GAL1, 2000 base-pairs up- and downstream of the gRNA-GAL1 target, and four off-target (OT) sites for the PrA-Cas9/gRNA-GAL1 complex containing varying levels of sequence similarity to the gRNA-GAL1 target (± protospacer-adjacent motif (PAM motif)). Error bars are standard error from triplicate analyses. (*) Indicates significant (P < 0.05) enrichment in cells containing the PrA-Cas9/gRNA complex relative to those with only the PrA-Cas9 (not targeted).
To determine if enrichment using CRISPR-ChAP was specific, a series of potential off-target sites were analyzed (Fig. 2C). The four most similar sites in the genome to the first 12 base-pairs of the 20 base-pairs targeted at GAL1 by gRNA-GAL1 were analyzed by qPCR-ChIP for PrA-Cas9/gRNA binding. The first 12 base-pairs of the 20 base-pair target sequence strongly influence gRNA-directed binding specificity.14 The off-target (OT) sites contained 14/20 (OT1), 15/20 (OT2), 15/20 (OT3) and 13/20 (OT4) of sequence identity relative to the GAL1 target DNA. Two of the four off-target sites showed 3.2- and 4.4-fold (OT1 and OT2) enrichment with PrA-Cas9/gRNA (Fig. 2C). For most effective targeting of Cas9 to a genomic region, the 20 base-pair target region needs to contain a protospacer adjacent motif (PAM motif) immediately 5′ to the target DNA. In the type II S. pyogenes system used in this work, the PAM motif is NGG.12,15 Accordingly, OT1 and OT2 that showed Cas9/gRNA enrichment contained a PAM motif, while OT 3 and OT4 did not. This demonstrated that off-target binding of the PrA-Cas9/gRNA complex targeting GAL1 is enhanced with a PAM motif and can provide ∼4-fold off-target enrichment. This ∼4-fold off-target enrichment is much lower than the 70-fold enrichment observed for transcriptionally active GAL1 promoter chromatin and will not prohibit large scale proteomic approaches as the specifically bound proteins/PTMs will dominate the mass spectrometric data collection. The enrichment of GAL1 promoter chromatin under glucose growth conditions was 4.9-fold whereas off-target binding can contribute up to ∼4-fold enrichments. Therefore, purification of GAL1 promoter chromatin under glucose growth conditions was not pursued for large scale proteomic analysis, but rather the 70-fold enriched chromatin from galactose growth conditions was used for subsequent proteomic studies. The Cas9/gRNA and previously reported TAL results illustrate that DNA-binding affinity reagents may differentially access chromatin in different states; thus, care has to be taken to test for specific enrichment prior to proteomic studies.2 Accordingly, not all genomic regions may be amendable to targeting strategies such as CRISPR-ChAP-MS. One may have to identify adjacent accessible genomic regions to identify sites amendable for targeting and enrichment. As the CRISPR-based approaches become more engineered for DNA-binding specificity, off-target issues will have less impact on the CRISPR-ChAP-MS approach.16
To demonstrate the utility of the CRISPR-ChAP-MS approach, the GAL1 promoter chromatin was enriched from 1x1011 cells that were grown in media containing galactose. As a control for quantitative mass spectrometric identification of proteins as “specific” or “non-specific” to the purification, CRISPR-ChAP-MS was performed with PrA-Cas9 expressing cells either with or without gRNA-GAL1. Of particular importance for purification of small regions of chromatin, an experimentally-determined amount of formaldehyde cross-linking and sonication must be used to ensure that a native chromatin region can be isolated and analyzed.2,3,10 Cells were cross-linked with 1.25% formaldehyde, lysed under cryogenic conditions with a ball mill, and after thawing were sonicated in purification buffer to yield chromatin fragments ~1 kb in length. Dynabeads coated with IgG were used to affinity purify the PrA-Cas9/gRNA complex or control PrA-Cas9 with any associated proteins and posttranslationally modified histones. For large scale enrichment procedures, the GAL1 chromatin was found to be enriched by 15.5 ± 1.2-fold relative to ACT1 by qPCR. Isolated proteins were resolved by SDS-PAGE (Fig. 3A), excised from the entire gel lane in 2 mm bands and subjected to in-gel trypsin digestion. Tryptic peptides were analyzed by high resolution mass spectrometry with a Thermo Velos Orbitrap mass spectrometer as reported.2 Proteins and histone PTMs (acetylation and mono-, di- and trimethylation of lysine) were identified with Mascot (Tables S1 and S2).
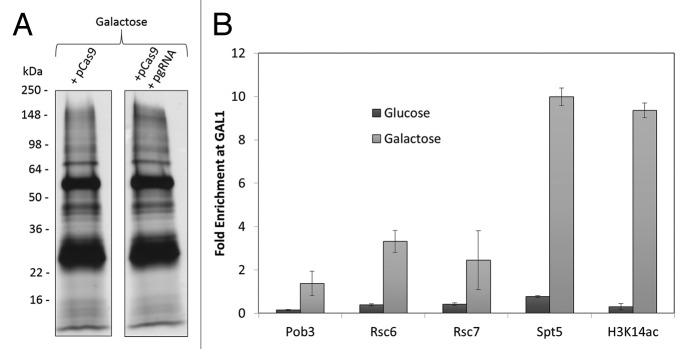
Figure 3. CRISPR-ChAP-MS analysis of transcriptionally active promoter chromatin. (A) Chromatin was affinity purified on IgG-beads from cells grown in galactose-containing media that expressed PrA-Cas9 as a control and cells that expressed PrA-Cas9/gRNA targeted to the promoter region of GAL1. Co-enriched proteins were resolved by SDS-PAGE and identified with high resolution mass spectrometry. Label-free proteomics was used to determine whether a protein or histone PTM was specifically enriched with the promoter chromatin. (B) Enrichment of GAL1 relative to ACT1 by ChIP to PrA-tagged versions of the proteins or to the histone PTM (normalized for nucleosome occupancy) was measured in cells grown in galactose in comparison to cells grown in glucose. Error bars are standard error from technical triplicate analyses.
To determine which proteins were specifically enriched with the GAL1 promoter chromatin, a quantitative mass spectrometric approach was used to compare proteins identified with PrA-Cas9/gRNA and PrA-Cas9 alone. This reported approach uses normalized spectral abundance factors to represent the relative level of each protein in each sample, which can then be cross-compared to identify those proteins/PTMs enriched with PrA-Cas9/gRNA.2,17,18 Using this approach, 86 out of 1832 identified proteins were found to enrich with PrA-Cas9/gRNA (Table S1). The label-free approach used provided a short list of 86 proteins to validate as specific interactors, which is vastly less than the 1832 proteins identified. One approach to further narrow the list of putative interactors is to use established protein annotation of protein function. A total of 11 of the 86 proteins were related to transcription (Reb1, Spt5, Toa2, Baf1, Sin3, H2B2, Ume1, Pob3, Rsc6, Rpa14, Rsc7), while the other 75 were common contaminants found in affinity enrichments.2,3 In addition to proteins, acetylation of lysine 14 on histone H3 (H3K14) and H3K23 were found enriched with the GAL1 promoter chromatin (Table S2). Both H3K14ac and H3K23ac are correlated with active transcriptional states of chromatin. ChIP for a subset of these proteins/PTMs (Spt5, Pob3, Rsc6, Rsc7 and H3K14ac) was used to verify that these proteins/PTMs are components of the epiproteome at the targeted region of GAL1 promoter chromatin (Fig. 3B). H3K14ac was identified at the GAL1 promoter region with our first and second generation ChAP technologies,2,3 while Pob3, Spt5, Rsc6 and Rsc7 are unique to this study. Pob3 forms a complex with Spt16 to make yFACT, which promotes chromatin rearrangement to allow progression of RNA polymerase. Spt16 was identified with our first and second generation ChAP technologies at GAL12,3; thus, providing compelling evidence that yFACT is localized to this chromatin region during transcriptional activation. Spt5 is an elongation factor that aids RNA polymerase II, while the RSC complex of proteins serves as a chromatin remodeler that is involved with transcription. Taken together our results present a snapshot of the dynamics of transcriptional activation within a 1 kb viewing window at the GAL1 promoter chromatin. This analysis demonstrates that the CRISPR-ChAP-MS approach can be used to identify a local epiproteome.
The CRISPR-ChAP-MS approach provides a new tool to study epigenetic regulation. Researchers can now identify proteins and histone PTMs at 1 kb resolution using proteomic approaches that do not depend on a priori knowledge of the protein/PTM target, which distinguishes this method from traditional ChIP. Key to success with chromatin enrichment procedures is the quantitative mass spectrometry used to determine which identified proteins/PTMs are “specific” to the isolated chromatin. These mass spectrometric approaches can be label-free, as used here and in our TAL-based second generation ChAP methodology,2 or utilize more expensive isotope labeling methods, as used in our LexA-based first generation methodology.3 Relative to the TAL-based and LexA-based ChAP methodology, our PrA-Cas9/gRNA approach showed greatly enhanced enrichment of targeted chromatin, which is instrumental for analyzing low copy cellular entities like specific chromatin sections. However, similar to what was observed with the previous approaches, there was an inability to sufficiently purify GAL1 chromatin under repressive growth conditions. This is presumably due to occlusion of the Cas9 binding site. This highlights an important need for testing target sites for enrichment by ChIP before pursuing a proteomic analysis of a particular genomic site of interest. At the same time, this highlights a possible caveat for using the CRISPR system in any of its useful forms, including genome editing. Furthermore, the Cas9/gRNA system is easily manipulated by simply altering the gRNA sequence, which provides for adaptability and multiplexing approaches. Recent and future efforts to further engineer the specificity of the Cas9/gRNA system will only expand the capabilities of the CRISPR-ChAP-MS approach.16 This technology should be amendable to cell culture and in vivo systems that provide for expression of the Cas9/gRNA machinery. The CRISPR-ChAP-MS approach suggests far-reaching applicability for identifying molecular components driving chromosomal activities.
Materials and Methods
Cloning, Western-blotting, real-time reverse transcription PCR and chromatin immunoprecipitation (ChIP)
Cas9 was subcloned from Addgene plasmid 44246 (http://www.addgene.org/CRISPR/; Cross Lab) into pPrA-LexA (TRP1 selection)3 - fusing Cas9 with a PrA (Protein A) tag to make pPrA-Cas9. Addgene plasmid 43803 (http://www.addgene.org/CRISPR/; Cross Lab) was used to express the gRNA (URA3 selection). The gRNA sequence in the plasmid was mutated in two steps using a Stratagene site-directed mutagenesis kit to produce the following sequence matching 20 base-pairs in the GAL1 promoter region: 5′ATTTGAAGGT TTGTGGGGCC. Three S. cerevisiae strains (W303 matA) were created by transforming the resulting plasmids: pgRNA-GAL1, pPrA-Cas9, and pPrA-Cas9 + pgRNA-GAL1. Western-blotting, real-time reverse transcription PCR and chromatin immunoprecipitation (ChIP) were as described.2,3 Off-target sites tested in Figure 2C were: OT1 5′ATGAAAAAAT TAGTGGGGCC, OT2 5′ATACGTAGTC TTGTGGGGCC, OT3 5′TACGGAAGGT TGGTGGGGCC, OT4 5′TATGTCGCGT TTGTGGGGCC.
CRISPR-ChAP-MS
S. cerevisiae with pPrA-Cas9 or pPrA-Cas9 + pgRNA-GAL1 were grown to mid-log phase in synthetic yeast media (minus tryptophan and minus tryptophan/uracil respectively) with 3% galactose and subjected to 1.25% formaldehyde cross-linking for 6 min. Cross-linking was quenched with 125 mM glycine for 5 min. Cells were collected by centrifugation and lysed under cryogenic conditions.3 Lysate from 1010 cells was re-suspended in purification buffer (25 mM HEPES-KOH, 0.5 mM EGTA, 1 mM EDTA, 10% glycerol, 0.02% NP-40, 150 mM KCl, 1X Sigma fungal protease inhibitor cocktail, 4 µg/mL Pepstatin A, 2 mM PMSF) at 5 mL / gram cell lysate. Re-suspended cell lysate was subjected to sonication with a Bioruptor to shear chromatin to ∼1 kb in size as described.2,3 PrA-tagged Cas9/gRNA complex and associated proteins were affinity purified on 144 mg of IgG-coated Dynabeads.2,3 IgG-coated beads were incubated with lysate for 7 h at 4 °C with constant agitation. Beads were collected with magnets and washed twice in purification buffer, once with purification buffer with 1 M NaCl / 1 M urea, and once in purification buffer. Proteins were eluted from the washed beads with 0.5 N ammonium hydroxide / 0.5 mM EDTA for 5 min at room temperature. Eluted proteins were lyophilized, re-suspended in Laemmli loading buffer, resolved by 4–20% gradient SDS-PAGE, and visualized by colloidal Coomassie-staining. Gel lanes were sliced into 2 mm sections and subjected to in-gel trypsin digestion.3 Tryptic peptides were analyzed by high resolution tandem mass spectrometry with a Thermo Velos Orbitrap mass spectrometer coupled to a Waters nanoACQUITY LC system.2,3 Proteins and histone PTMs (lysine acetylation and methylation) were identified with Mascot (Tables S1 and S2). To determine if a protein was “specific” or “non-specific” to the purification, a previously reported quantitative mass spectrometry approach was utilized.2 In brief, a normalized spectral abundance factor (NSAF) value was calculated for each protein in the PrA-Cas9 and PrA-Cas9/gRNA purifications. The NSAF value is the number of spectral counts assigned to a given protein (normalized by the molecular weight of that protein) divided by the sum of all normalized spectral counts of all proteins identified in the specific purification.17 A fold-change of normalized NSAF values was then used to identify proteins specific to the PrA-Cas9/gRNA purification (Table S1). The data represents one biological replicate.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to acknowledge mass spectrometric support from the UAMS Proteomics Facility. This work was supported by National Institutes of Health grants R01GM106024, R01GM098922, R01GM081766, R33CA173264, UL1RR029884, P30GM103450, and P20GM103429
References
- 1.Dai B, Rasmussen TP. Global epiproteomic signatures distinguish embryonic stem cells from differentiated cells. Stem Cells. 2007;25:2567–74. doi: 10.1634/stemcells.2007-0131. [DOI] [PubMed] [Google Scholar]
- 2.Byrum SD, Taverna SD, Tackett AJ. Purification of a specific native genomic locus for proteomic analysis. Nucleic Acids Res. 2013;41:e195. doi: 10.1093/nar/gkt822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Byrum SD, Raman A, Taverna SD, Tackett AJ. ChAP-MS: a method for identification of proteins and histone posttranslational modifications at a single genomic locus. Cell Rep. 2012;2:198–205. doi: 10.1016/j.celrep.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Déjardin J, Kingston RE. Purification of proteins associated with specific genomic Loci. Cell. 2009;136:175–86. doi: 10.1016/j.cell.2008.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akiyoshi B, Nelson CR, Ranish JA, Biggins S. Quantitative proteomic analysis of purified yeast kinetochores identifies a PP1 regulatory subunit. Genes Dev. 2009;23:2887–99. doi: 10.1101/gad.1865909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoshino A, Fujii H. Insertional chromatin immunoprecipitation: a method for isolating specific genomic regions. J Biosci Bioeng. 2009;108:446–9. doi: 10.1016/j.jbiosc.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 7.Griesenbeck J, Boeger H, Strattan JS, Kornberg RD. Affinity purification of specific chromatin segments from chromosomal loci in yeast. Mol Cell Biol. 2003;23:9275–82. doi: 10.1128/MCB.23.24.9275-9282.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Unnikrishnan A, Gafken PR, Tsukiyama T. Dynamic changes in histone acetylation regulate origins of DNA replication. Nat Struct Mol Biol. 2010;17:430–7. doi: 10.1038/nsmb.1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamperl S, Brown CR, Garea AV, Perez-Fernandez J, Bruckmann A, Huber K, Wittner M, Babl V, Stoeckl U, Deutzmann R, et al. Compositional and structural analysis of selected chromosomal domains from Saccharomyces cerevisiae. Nucleic Acids Res. 2014;42:e2. doi: 10.1093/nar/gkt891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Byrum SD, Taverna SD, Tackett AJ. Quantitative analysis of histone exchange for transcriptionally active chromatin. J Clin Bioinforma. 2011;1:17. doi: 10.1186/2043-9113-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scholze H, Boch J. TAL effectors are remote controls for gene activation. Curr Opin Microbiol. 2011;14:47–53. doi: 10.1016/j.mib.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 12.Mali P, Esvelt KM, Church GM. Cas9 as a versatile tool for engineering biology. Nat Methods. 2013;10:957–63. doi: 10.1038/nmeth.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fujita T, Fujii H. Efficient isolation of specific genomic regions and identification of associated proteins by engineered DNA-binding molecule-mediated chromatin immunoprecipitation (enChIP) using CRISPR. Biochem Biophys Res Commun. 2013;439:132–6. doi: 10.1016/j.bbrc.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 14.Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31:822–6. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DiCarlo JE, Norville JE, Mali P, Rios X, Aach J, Church GM. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 2013;41:4336–43. doi: 10.1093/nar/gkt135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol. 2013;31:233–9. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zybailov B, Mosley AL, Sardiu ME, Coleman MK, Florens L, Washburn MP. Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J Proteome Res. 2006;5:2339–47. doi: 10.1021/pr060161n. [DOI] [PubMed] [Google Scholar]
- 18.Byrum SD, Larson SK, Avaritt NL, Moreland LE, Mackintosh SG, Cheung WL, Tackett AJ. Quantitative Proteomics Identifies Activation of Hallmark Pathways of Cancer in Patient Melanoma. J Proteomics Bioinform. 2013;6:43–50. doi: 10.4172/jpb.1000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.