

Abstract
Antibodies have become the fastest growing class of biological therapeutics, in part due to their exquisite specificity and ability to modulate protein-protein interactions with a high biological potency. The relatively large size and bivalency of antibodies, however, limits their use as therapeutics in certain circumstances. Antibody fragments, such as single-chain variable fragments and antigen binding-fragments, have emerged as viable alternatives, but without further modifications these monovalent formats have reduced terminal serum half-lives because of their small size and lack of an Fc domain, which is required for FcRn-mediated recycling. Using rational engineering of the IgG4 Fc domain to disrupt key interactions at the CH3-CH3 interface, we identified a number of point mutations that abolish Fc dimerization and created half-antibodies, a novel monovalent antibody format that retains a monomeric Fc domain. Introduction of these mutations into an IgG1 framework also led to the creation of half-antibodies. These half-antibodies were shown to be soluble, thermodynamically stable and monomeric, characteristics that are favorable for use as therapeutic proteins. Despite significantly reduced FcRn binding in vitro, which suggests that avidity gains in a dimeric Fc are critical to optimal FcRn binding, this format demonstrated an increased terminal serum half-life compared with that expected for most alternative antibody fragments.
Keywords: Antibody engineering, monomeric Fc, half-antibody, half-life extension, FcRn
Introduction
The ability of antibodies to recognize an almost unlimited number of antigens with high specificity has resulted in their becoming the fastest growing class of biological therapeutics.1,2 The most commonly used antibody class for therapy, immunoglobulin G (IgG), is based upon a protein structure consisting of two heavy and two light chains forming two Fab arms, containing variable (V) binding domains, attached by a flexible hinge region to the stem of the antibody, the Fc domain, resulting in a monospecific, bivalent molecule with a ‘Y’ shape.
The majority of approved therapeutic antibodies are of the IgG1 subclass,3 due in part to its ability to exert effector functions, such as antibody-dependant cell-mediated cytotoxicity and complement dependent cytotoxicity, through binding of Fc receptors.4,5 In certain therapeutic circumstances, however, such as targeting of the proto oncogenes hepatocyte growth factor receptor6 (HGFR or MET) or macrophage stimulating protein receptor7 (RON), receptor dimerization caused by bivalent antibodies is not desired. In these cases, a monomeric antibody format unable to dimerize and thus agonise cell surface receptors would be desirable. Antibody fragments lacking the Fc domain, such as single-chain variable fragments (scFv) and antigen binding-fragments (Fab), are alternatives,8 but these suffer from short serum half-lives9 due to both the lack of an Fc domain, which is required for FcRn mediated recycling,10-13 and their small size, which results in glomerular filtration. Protein engineering to generate a monovalent “half-antibody” would provide an attractive format large enough to exceed the theoretical renal filtration limit of 70 kDa,14,15 and also potentially maintain FcRn binding capabilities through a monomeric Fc domain. This would conceivably generate a smaller antibody fragment with increased diffusivity and capillary permeability,16 but with an improved serum half-life compared with most currently available monovalent options.
Recent studies have demonstrated that IgG4 molecules are able to undergo Fab-arm exchange, where heavy chains can be swapped between antibodies in vivo.17,18 IgG4 molecules have also been shown to have a small population of half-antibody in solution,19 suggesting that the bivalent form of IgG4 is less stable than IgG1. Although most efforts have concentrated on stabilizing the IgG4 hinge or CH3-CH3 interface to prevent arm exchange,18,20-24 a number of modifications have been identified that apparently increase the population of IgG4 half-antibody in vitro.19,25-27 In particular, a single point mutation, F405Q at the CH3-CH3 interface of a modified IgG4 antibody was reported to significantly increase the half-antibody population,19 while a combination of seven mutations at the interface of an IgG1 Fc domain has been shown to generate a stable monomeric Fc domain.28
In the work reported here, we build upon these findings and knowledge of energetically key interactions at the CH3-CH3 interface29 to investigate how a range of mutants at the CH3-CH3 interface of both IgG4 and IgG1 affect Fc dimerization, with the aim of generating a stable monomeric format. Our results demonstrate that a rational structure-based mutagenesis approach resulted in the identification of a number of point mutations that abolish Fc dimerization for both IgG4 and IgG1. This leads to a stable monovalent half-antibody with favorable in vitro characteristics, such as high levels of soluble expression, and a significant increase in terminal serum half-life compared with that expected for a scFv or Fab.
Results
Analysis of the CH3-CH3 interface
The CH3 domain consists of approximately 106 residues and the CH3-CH3 interface consists of 16 residues located on four anti-parallel β-sheets that make intermolecular contacts.30,31 Residues from the two internal β-sheets contribute significantly more to the stability of the dimer than those on the two external β-sheets.29 Our analysis grouped interface residues based on location, the strength and number of non-covalent intermolecular interactions in which each residue was involved and the potential steric hindrance that would be caused to the packing of the interface by replacement of the side chain. Using this method, we identified residues at the core of the interface (T366, L368, F405 and Y407) as being involved in significant interactions in this region, as well as four salt bridges (E356, D399, K392, R409 and K439) dispersed around the edge of the interface. This suggests that rational mutations at these locations may disrupt dimerization of the CH3 domain. In addition to these interactions, a third set of five residues (L351, S364, L368, K370 and T394) were identified as interacting in a homotypic manner (i.e., involved in interactions with their counterpart in the other CH3 domain) or being in a position that was deemed more likely to enable the insertion of a disruptive mutation (e.g., by insertion of like charges opposite each other). Finally, a fourth set of residues (Y349, S354 and E357) on the periphery of the CH3-CH3 interface were also determined to be likely to influence stabilization of the dimer. A selection of the aforementioned amino acids and their location at the interface are highlighted in Figure 1.

Figure 1. Cartoon representation of the CH3-CH3 interface of an antibody. (A) Representative image of the crystal structure of an antibody, with one heavy chain colored in green and the other in cyan. (B) Space filled representation of the residues T394 and L351, which are directly opposite their counterpart at the interface. (C) K370 sits in a compact region close to L368 and S364 which would potentially enable substitution of these residues with a positively charged amino acid to create a repulsive affect. (D) A selection of the amino acids involved in polar contacts at the interface. (E) A view looking through a space filled CH3 domain in cyan to visualize amino acids contributing to the core of the interface from the opposing CH3 domain colored in green.
To analyze the influence of single or multiple site-directed mutations at these positions, a set of five amino acids were chosen to be representative of each type of side chain: positive (arginine); negative (aspartate); large aromatic (tryptophan); small neutral (alanine); and hydrophilic (glutamine). Aliphatic side chains were avoided because it has been demonstrated previously that mutations increasing the hydrophobicity of antibody interfaces is more likely to thermodynamically stabilize an interface.32 On this basis, a total of 65 CH3 variants containing single, double and triple mutations were rationally designed, expressed and analyzed within an IgG4 hingeless Fc domain.
Size exclusion chromatography
To analyze the affect of these mutations on dimerization of hingeless IgG4 Fc domains, initial screening of the approximate molecular weight of all mutant Fc domains was performed by size exclusion chromatography (SEC). Representative data showing that Fc domain mutants could be resolved based on SEC retention time is shown in Figure 2. Determination of average partition coefficient, kav, for each from column calibration enabled determination of predicted molecular weight based on retention time (Table 1). The spread of SEC retention times is very broad, with the majority of elution profiles appearing monodisperse, although some have a predicted molecular weight between that of monomer and dimer. This suggests that a significant number of the samples analyzed are in rapid-exchange between monomer and dimer, such that on the time-scale of SEC they appear monodisperse with the exact retention time depending on the proportion of time the molecule spends as monomer or dimer. Because of the exposure of a hydrophobic surface in the monomeric Fc domains, it is plausible that the spread of retention times could also be due to column binding artifacts. To rule this out, more accurate multi-angle laser light scattering (MALLS) solution molecular weights were determined for ten of the mutants, confirming the findings from SEC (Table 1).

Figure 2. Representative HPLC chromatograms showing how mutations can influence the monomer-dimer equilibrium. (A) IgG4 Fc wild type (solid line), IgG4 Fc T366Q (dashed line) and IgG4 Fc T366WL368W (dotted line) are dimeric, in monomer-dimer equilibrium and monomeric respectively. (B) Variants showing predominantly monomeric behavior, IgG4 D399R (solid line), IgG4 T364RL368R (dashed line), and IgG4 T349D (dotted line). (C) Variants showing equilibrium or mixed behavior, IgG4 Y407D (solid line), IgG4 T394R (dashed line), and IgG4 E356RK392D (dotted line). (D) Variants showing dimeric behavior, IgG4 K439D (solid line), IgG4 T366DY407D (dashed line), and IgG4 E356R (dotted line). In each of (B), (C), and (D) chromatograms are shown relative to IgG4 Fc wild type (solid line), and IgG4 Fc T366WL368W (dotted line) in a gray trace.
Table 1. A summary of the IgG4 Fc mutants analyzed by SEC.
| IgG4Fc Mutant | Retention time (min) | Calibrated weight (kDa) | MALLS weight (kDa) | |
|---|---|---|---|---|
| Dimer | E356RK392DR409D | 19.6 | 59.0 | |
| T366W | 19.7 | 59.5 | 53 | |
| T366D | 20.3 | 54.0 | ||
| K439D | 20.5 | 52.5 | ||
| K370W | 20.5 | 52.5 | ||
| K392AK439A | 20.5 | 52.5 | ||
| K439A | 20.6 | 51.5 | ||
| WT | 20.6 | 51.5 | 52 | |
| R409A | 20.6 | 51.5 | ||
| D399W | 20.7 | 51.0 | ||
| S364W | 20.7 | 51.0 | ||
| S354D | 20.7 | 51.0 | ||
| K370A | 20.7 | 51.0 | ||
| E356AK392A | 20.7 | 51.0 | ||
| K392D | 20.8 | 50.0 | ||
| E356A | 20.8 | 50.0 | ||
| E356R | 20.8 | 50.0 | ||
| R409D | 20.9 | 49.0 | ||
| D399A | 21.0 | 48.0 | ||
| S354W | 21.0 | 48.0 | ||
| Monomer-dimer equilibrium | T366DY407D | 20.7 | 51.0 | |
| D399WR409W | 21.0 | 48.0 | ||
| D399AK439A | 21.1 | 47.5 | ||
| T366QY407Q | 21.1 | 47.5 | ||
| F405A | 21.1 | 47.5 | 50 | |
| E356RR409D | 21.1 | 47.5 | ||
| L351W | 21.1 | 47.5 | ||
| K392DK439D | 21.2 | 46.5 | ||
| Y349D | 21.4 | 44.5 | ||
| L368W | 21.5 | 44.0 | ||
| Y407Q | 21.6 | 43.5 | ||
| T366Q | 21.7 | 42.0 | ||
| E356RK392D | 21.8 | 41.5 | ||
| E356AD399A | 22.0 | 40.0 | ||
| T394W | 22.0 | 40.0 | ||
| Y407A | 22.1 | 39.5 | ||
| L351WT394W | 22.2 | 38.5 | ||
| Monomer | Y407D | 22.0 | 40.0 | |
| T394R | 22.1 | 39.5 | ||
| T366R | 22.3 | 37.5 | 35 | |
| R409W | 22.3 | 37.5 | ||
| E357W | 22.4 | 37.0 | ||
| Y407R | 22.4 | 37.0 | 32 | |
| D399R | 22.5 | 36.5 | ||
| T366RY407R | 22.5 | 36.5 | 32 | |
| F405AY407A | 22.6 | 36.0 | ||
| T366QF405QY407Q | 22.7 | 35.0 | ||
| T394D | 22.8 | 34.0 | 28 | |
| F405Q | 22.9 | 33.5 | ||
| S364R | 22.9 | 33.5 | ||
| L351DT394D | 23.0 | 33.0 | ||
| L368R | 23.0 | 33.0 | ||
| L351D | 23.0 | 33.0 | 29 | |
| S364RL368R | 23.1 | 32.0 | ||
| L351R | 23.1 | 32.0 | 30 | |
| F405R | 23.1 | 32.0 | 29 | |
| L351RT394R | 23.2 | 31.5 | ||
| S364WL368W | 23.3 | 31.0 | ||
| E357R | 23.4 | 30.0 | ||
| D399RK439D | 23.4 | 30.0 | ||
| E356RD399R | 23.4 | 30.0 | ||
| T366WL368W | 23.7 | 28.0 | ||
| L351RS364RT394R | 25.1 | 26.0 |
The samples are split into three groups and ordered by retention time with calibration of the column used to estimate molecular weight. The calculated molecular weight from SEC-MALLS is also shown for those samples for which data was collected.
Separating the mutants into three nominal groups based on SEC retention time, MALLS predicted molecular weight, and the appearance of the chromatogram (such as an apparently monodisperse sample, or an obvious mixture of species due to peak broadening or double peaks) results in 19 dimers, 17 in monomer-dimer equilibrium and 26 mutants that have a molecular weight indicative of a predominantly monomeric species. Introduction of a wild-type IgG4 hinge to the monomeric IgG4 Fc domains resulted in some dimerization (1–25%). This was abolished upon substitution of the hinge cysteines for glutamine (data not shown), suggesting that the hinge region does not play a significant role in Fc dimerization other than stabilizing it through disulphide bonds once the dimer has formed.
The Fc domain of IgG1 is known to be a more stable dimer than the IgG4 Fc domain due to a single amino acid difference at position 409.22 To determine whether the monomeric Fc mutations identified for IgG4 would also result in the formation of monomeric IgG1 Fc domains, 21 mutants were analyzed as IgG1 hingeless Fc domains by SEC, with 11 of these exhibiting monomeric behavior (Table S1). Site-directed mutagenesis of the IgG1 Fc mutants to produce an IgG4-like interface (K409R) resulted in 5 of 6 of the dimeric IgG1 mutant Fc domains reverting to monomer upon introduction of this mutation (Table S1). This agrees with previous findings that a K409R mutation can invoke IgG4-like behavior into an IgG1 Fc domain22 and offers a potential means of generating a larger number of monomeric IgG1 Fc domains.
Analytical ultracentrifugation
To verify the classification of mutants as dimeric, monomeric or in monomer-dimer equilibrium, sedimentation velocity analytical ultracentrifugation (SV-AUC) was performed as an accurate means of determining intact solution molecular weights. The apparent major species for wild type IgG4 Fc domain gave a sedimentation coefficient of 3.7 S with a smaller component of 2.4 S representing 1.2% of the population as measured by UV absorbance. Conversion to molar mass distribution, c(M), gave the 3.7 and 2.4 S components apparent solution molecular weights of 51.2 and 27.4 kDa, respectively, which is in agreement with the expected molecular mass of homodimer and monomer. A Y349D mutant gave a sedimentation coefficient of 3.5 S, equating to an apparent molecular weight of 43 kDa. In agreement with SEC data, this suggests that this mutant is in rapid exchange between monomer and dimer. A T394D mutant gave a predominant sedimentation coefficient of 2.4 S. Conversion to c(M) resulted in an apparent solution molecular weight of 26.8 kDa, consistent with a monomeric species. No species with a sedimentation coefficient consistent with dimer was observed for this particular mutant. Graphical summaries of the size distribution analysis, c(s), are shown in Figure 3.
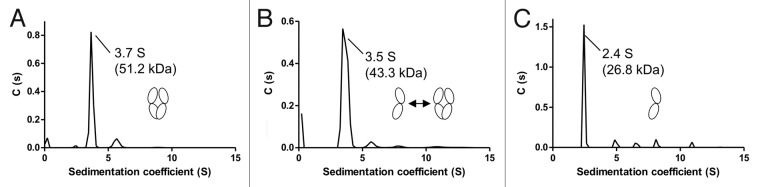
Figure 3. SV-AUC profiles showing continuous size distribution against sedimentation coefficient and apparent solution molecular weight. Apparent molecular weights have been determined from the sedimentation coefficients with cartoon images representing the multimeric state of the main population. (A) IgG4 Fc WT has a mass indicative of a dimeric species with only a small fraction of monomer detectable at 2.4 S. (B) IgG4 Y349D has broadened profile consistent with a rapid equilibrium between monomeric and dimeric species and an average sedimentation coefficient of 3.5S (C) IgG4 Fc T394D has a calculated mass representing monomer with no detectable dimer at 3.7 S.
To further investigate the potential concentration dependence of the monomeric nature of the Fc domain mutants and the extent of reversible self-association, further SV-AUC analysis was performed for eight hingeless IgG4 Fc domains: wild-type; Y349D; L368R; F405Q; S364R; T394D; Y407R; and F405R. Initial screening at 0.5 mg/ml confirmed that wild-type was predominantly dimeric with Y349D exhibiting behavior consistent with a monomer-dimer equilibrium. Self-association constants, keq, of 2.1 nM and 2.4 μM, respectively, were calculated for these Fc domains, which is in the range of previously determined self-association constants for these proteins from orthologous techniques.19 No significant reversible self-association was detected for the remaining mutants at 0.5 mg/ml. A concentration series for L368R gave expected size distribution plots for a monomer at 0.25 and 0.5 mg/ml, but reversible self-association was observed at 1.56 mg/ml (Fig S1). SEDANAL33 curve fit of L368R to a model of a monomer-dimer equilibrium fit all data points well, resulting in a keq of 670 μM.
Structural models of the mutants L368R, F405Q, S364R, T394D, Y407R, and F405R were prepared by homology modeling and energy minimisation. Hydrodynamic bead modeling software, SOMO,34 was used to determine theoretical sedimentation coefficient values, S20w, for each variant. Experimentally determined sedimentation coefficients fit to a single non-interacting species for each mutant at 0.5 mg/ml were converted to S20w using SEDNTERP.35 Comparison of theoretical and experimentally determined sedimentation values showed that L368R, F405Q, Y407R and S364R had values with less than 1% difference. This suggests that these mutants behave in a comparable manner to L368R and will show weak self-association at concentrations above ~1.5 mg/ml. When fit to a monomer-dimer model the sedimentation value for L368R was ~4% lower than the SOMO determined value, suggesting that the sedimentation value for a true monomer is 4% lower than the SOMO determined values. For F405R and T394D, the experimental sedimentation values were 3.2 and 4.0% lower than the SOMO calculated values, indicating that these mutants are likely to show no signs of reversible self-association. To confirm this, a concentration series for T394D gave expected size distribution plots for a monomer at 0.1, 0.5, 2.0 and 5.5 mg/ml, indicating that this particular mutant does not undergo reversible self-association at all concentrations analyzed (Fig S1).
Modeling
To understand the affect of the monomeric mutations on the packing of the CH3-CH3 interface, energy minimised models of dimeric Fc versions of the mutants were compared. Superposition of all models shows no significant perturbation of the backbone structure of the Fc domain; however, significant rearrangement of the packing of amino acid side chains at the CH3-CH3 interface compared with wild-type IgG4 Fc domain can be identified for all monomeric mutants analyzed. As a representation of the disruption to the interface, (Fig. S3) highlights the disruption of key interactions at the interface in the T394D mutant compared with wild-type IgG4 Fc. In addition to the positioning of an aspartate residue directly opposite itself at the interface, which was the initial rationale behind the design of this mutation, the T394D mutation also is predicted to cause substantial rearrangement of the side chains of K370, K392, D399, R409 and K439, as well as more minor modifications in the positioning of other side chains at the interface. Such perturbations would be predicted to be sufficient to abolish dimerization of this mutant Fc domain, explaining the observed behavior of the mutant.
Conformational stability
In a dimeric Fc, intermolecular interactions between the CH3 domains and the carbohydrates of the CH2 domain stabilize the Fc domain.36 CH2 and CH3 domains have typical thermal unfolding temperatures of 60–70 and 80–85°C, respectively. To ensure that the monomeric Fc domains retain a conformational stability that is indicative of a stable folded protein, thermal unfolding was performed by differential scanning calorimetry (DSC). The monomeric hingeless IgG4 Fc domains selected for AUC analysis all showed a 10–15°C decrease in the melting temperature of the CH2 domain compared with a wild-type IgG4 Fc domain (data not shown) with all mutants having minimum melting temperature above 55°C.
To confirm these findings in the context of a monovalent half-antibody rather than monomeric Fc domain, glycosylated and aglycosylated versions of an anti-Fas monovalent half-antibody incorporating the T394D mutation were analyzed (Fig. 4). The wild-type antibody has a typical thermal stability profile for an antibody37 with a minimal melting temperature of 69°C, while the CH2 domain of the half-antibody shows an approximate 9°C decrease in stability with a further 5°C decrease in the aglycosylated version.
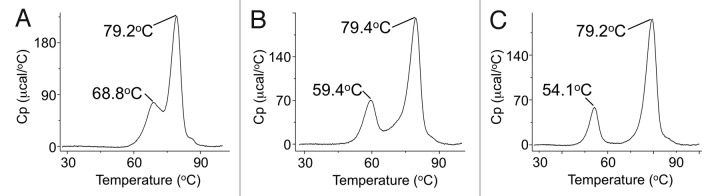
Figure 4. DSC thermal stability profiles. (A) Wild-type IgG4 antibody, (B) monovalent half-antibody and (C) monovalent aglycosylated half-antibody. The initial peak is unfolding of the CH2 domain followed by a second unfolding event for all other domains. The CH2 domain of the monovalent half-antibody has an approximate 9°C decrease in stability with a further 5°C decrease for the aglycosylated format.
On the basis of the in vitro characterization described above, the T394D mutant was selected as the monomeric mutant to be used for all subsequent analyses because it fulfilled key criteria, i.e., it was monomeric at concentrations up to 5.5 mg/ml, had thermal stability greater than 54°C, had similar expression yields to wild-type, showed no major signs of aggregation during purification and was monomeric as both IgG4 and IgG1 Fc domains.
Arm exchange analysis
Understanding the potential for IgG4-based therapeutics to exchange arms with endogenous IgG4 is important because the exchange may result in formation of undesirable heterodimeric molecules.17 To determine whether a monomeric format incorporating the T394D mutation could undergo arm exchange with wild-type IgG4, an in vitro SEC assay was performed in a similar manner as previously reported.38,39 A wild-type IgG4 antibody was mixed with either a wild-type IgG4 hinged Fc domain or a monomeric IgG4 hinged Fc domain in the presence or absence of 0.5 mM reduced glutathione (GSH), which is known to induce arm exchange in vitro.18,38,39 SEC was used to resolve the resulting species, clearly showing that in the presence of GSH the antibody and wild-type IgG4 hinged Fc domain undergo arm exchange to form a heterodimer of intermediate size, while the monomeric IgG4 Fc does not undergo arm exchange after incubation at 37°C for 24 or 96 h (Fig. 5). This confirms that the T394D mutation is sufficiently disruptive to abolish dimerization both with itself and with a wild-type Fc domain.

Figure 5. HPLC traces analyzing Fab arm exchange. (A) Overlay of separate runs for a wild-type IgG4, represented by the black cartoon image, and an IgG4 wild-type hinged Fc domain, represented by a white cartoon image. (B) A mixture of IgG4 antibody and IgG4 wild-type hinged Fc after incubation at 37°C for 96 h in the presence of 0.5 mM GSH. The wild-type Fc undergoes arm exchange with the IgG4 antibody to produce an intermediate sized heterodimer, as indicated by the cartoon representation. (C) The same respective incubation as (B) but in the absence of GSH showing no arm exchange. (D) Overlay of separate runs for the IgG4 antibody and a monomeric hinged Fc domain with the T394D mutation. (E) A mixture of IgG4 antibody and monomeric Fc after incubation at 37°C for 96 h in the presence of 0.5 mM GSH. No arm exchange is detectable. (F) The same respective incubation as (E) but in the absence of GSH.
FcRn binding kinetics
The ability of antibodies to bind FcRn is critical to their long serum half-life, and it is postulated that two molecules of FcRn can bind to the Fc domain of an antibody, one to each chain.40,41 For a monovalent half-antibody, the ratio will be reduced to 1:1, which may affect half-life in vivo. To assess this, the mouse FcRn binding kinetics of the wild-type IgG4 and monovalent half-antibody were determined by surface plasmon resonance (SPR). Fitting of the data to a heterogeneous ligand model,40,42 which incorporates a high and low affinity binding site on FcRn, clearly shows that a marked increase in off rate for the half-antibody can be attributed primarily to a 5000-fold increase in kd2 (Table 2; Fig. S1). Determination of steady-state binding constants shows an approximate four-fold decrease in affinity of the monovalent half-antibody to mouse FcRn with a KD of 301 nM compared with 69 nM for wild-type IgG4. A similar pattern was observed for half-antibody binding to human FcRn (data not shown).
Table 2. Rate constants for binding to FcRn.
| ka1 (1/Ms) | kd1 (1/s) | KD1 (M) | ka2 (1/Ms) | kd2 (1/s) | KD2 (M) | |
|---|---|---|---|---|---|---|
| Wild-type IgG4 | 3.11E+04 | 1.14E-02 | 3.67E-07 | 1.54E+05 | 2.56E-06 | 1.66E-11 |
| Monovalent IgG4 | 7.84E+04 | 3.87E-02 | 4.94E-07 | 2.28E+04 | 1.37E-02 | 6.01E-07 |
Biacore data for wild-type IgG4 and monovalent IgG4 binding to mouse FcRn was fit to a heterogeneous ligand model. This assumes there is both a low and high affinity binding site on FcRn to which an antibody can bind. The half-antibody had a greater dissociation rate constant (kd2) compared with the wild-type IgG4 by ~1800 fold and a greater association rate constant (ka2) by 7-fold, resulting in a decreased affinity (KD2) of 601 nM for the half antibody vs. 16.6 pM for the wild-type IgG4.
Although we cannot rule out the possibility of impaired FcRn binding due to structural rearrangement or increased flexibility at the binding site covering the CH2 and CH3 domain, the T394D mutation is spatially far removed from this site and, based on energy minimised modeling, has no affect on the global structure of the Fc domain. It would, therefore, seem likely that the reduced FcRn binding property of the monovalent half-antibody is primarily due to reduced avidity for FcRn.
Pharmacokinetic study
To assess the in vivo properties of the monovalent half-antibody format a pharmacokinetic (PK) study was performed with wild-type and half-antibody versions of an anti-Fas IgG4 antibody. Mouse serum drug titers (Fig. 6) and associated numerical analysis (Table 3) show a significantly reduced serum half-life for both glycosylated and aglycosylated versions of the half-antibody (21 h) compared with wild-type IgG4 (13 d). Because we used an anti-human Fas antibody that is not mouse cross-reactive, we believe that this decrease does not represent any target mediated factors and is, therefore, an inherent property of the half-antibody. The reduction in in vivo half-life is in agreement with the measured in vitro decrease in affinity to mouse FcRn for the monovalent half-antibody. The monovalent half-antibody, however, shows an approximate 10- to 20-fold improvement in half-life compared with Fabs, which have a typical half-life of 0.5 to 3 h in rodents.43-46 A similar comparative PK study reported in the patent literature showed an improved half-life for a monovalent half-antibody engineered through multiple CH3 interface mutations compared with a Fab, but with a more modest half-life of 6 h for the monovalent half-antibody.27
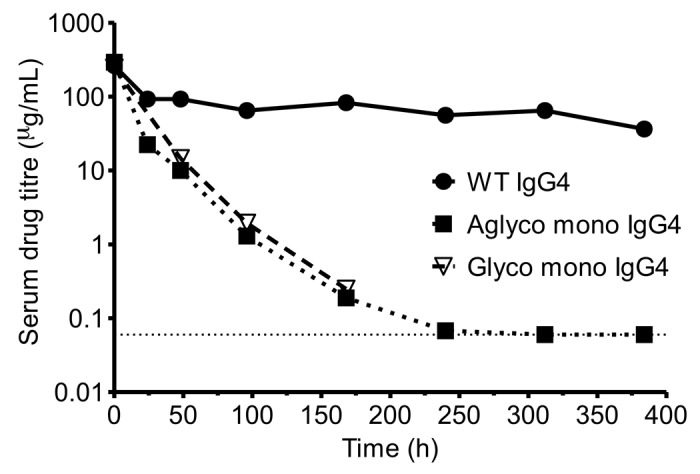
Figure 6. Mouse PK profiles of the monovalent half-antibody format. Serum concentrations of a wild type anti-Fas IgG4 antibody, monovalent aglycosylated anti-Fas IgG4 antibody and monovalent glycosylated anti-Fas IgG4 antibody over a period of 16 d following a 10 mg/kg body weight IV bolus dose. Drug titer determined by immunoassay with the dotted horizontal line representing the lower limit of quantification.
Table 3. Non-compartmental and two-compartmental analysis of pharmacokinetic parameters for a wild-type IgG4, glycosylated monovalent IgG4 and aglycosylated monovalent IgG4.
| Non-compartmental analysis | ||||
|---|---|---|---|---|
| Parameter | Unit | Mono agly IgG4 | Mono gly IgG4 | WT IgG4 |
| Half-life | days | 0.86 | 0.86 | 13.89 |
| Cmax | ug/ml | 293.26 | 244.71 | 262.31 |
| AUCINF | day*ug/ml | 131.83 | 178.93 | 1896.33 |
| Clearance | ml/day/kg | 75.85 | 55.89 | 5.27 |
| Two-compartmental modeling | ||||
| Half-life | days (S.D.) | 0.85 (0.08) | 0.87 (0.08) | 13.36 (4.12) |
| Clearance | ml/day/kg (S.D.) | 119.6 (12.1) | 103.9 (11.3) | 5.32 (1.1) |
Discussion
The work described here demonstrates that mutations at the CH3-CH3 interface of an antibody can affect the extent of Fc dimerization, either leading to rapid monomer-dimer exchange or abolishing dimerization altogether. Through a number of orthologous biophysical techniques, we identified 26 IgG4 Fc domain mutants, many of which contain single point mutations, that are almost exclusively monomeric under the conditions tested. In particular, a T394D mutation showed no measurable self-association up to a concentration of at least 5.5 mg/ml. Despite the increased stability of an IgG1 Fc domain resulting from a lysine instead of arginine at position 409,22 11 of these mutations also resulted in the formation of stable monomeric IgG1 Fc domains. Monomeric IgG1 Fc domains have recently been reported elsewhere, but with the need for a combination of seven mutations at the interface rather than single mutants.28
Our data suggests that monomeric Fc domains inherently have a lower conformational stability than the dimer. In particular, it is the thermal stability of the CH2 domain that is reduced, presumably due to the loss of carbohydrate interactions that are known to stabilize this domain.36 The use of single mutations to generate monomeric Fc domains has a less detrimental effect on conformational stability, however, and may also reduce the potential immunogenicity of these molecules compared with those with multiple mutations.
PK analysis clearly demonstrates a reduced terminal serum half-life of approximately 21 h for the half-antibody format compared with wild-type antibody; however, this still represents at least a 10-fold increase in half-life over a typical Fab domain.43-46 The reduced half-life compared with a wild-type antibody is due primarily to an approximate 5000-fold increase in dissociation rate for the high affinity binding site on FcRn (kd2), and is not due to increased degradation of the monomeric Fc domain by serum proteases (data not shown). This marked increase in dissociation rate is likely due to reduced avidity through a 1:1 rather than 2:1 binding ratio. This supports the suggestion that two FcRn binding sites on the Fc domain are required for efficient rescue from catabolism.47 A study of monomeric binding of Fc to FcRn using a heterodimeric Fc domain with only one FcRn binding site has previously shown that, although equal amounts of wild-type and heterodimeric Fc initially enter the cell, intracellular trafficking is markedly different.48 Upon internalization and delivery to early endosomes, a greater proportion of wild-type Fc remains bound to FcRn and is ultimately recycled back to the cell surface. Conversely, more of the heterodimeric Fc molecule is transferred to the lysosomal degradation pathway along with other fluid-phase components. Although the use of Fc mutations to increase pH-dependant FcRn binding affinity49,50 may offer an opportunity to increase the serum persistence of monovalent half-antibodies further, it is apparent that there is not a simple linear relationship between in vitro FcRn binding and in vivo half-life for monomeric Fc domains.
Compared with alternative antibody fragments with potential as therapeutics, the stability, solubility, high expression levels and prolonged half-life of the monomeric half-antibody make it a promising format for further development. In addition, the use of monomeric Fc domains as fusion partners or cleavable tags offers the potential to enhance the ease of expression, purification and detection of a range of proteins without the added complexity that dimerization may engender.
Materials and Methods
Molecular analysis
Analysis of the CH3-CH3 interface was performed using the high resolution crystal structure of a human IgG1 Fc (PDB accession number 1H3U51) or IgG4 Fc domain (PDB accession number 1ADQ52) using PyMol software.53 Residues involved in intermolecular contacts were defined as those residues with any pair of atomic groups closer than the sum of their van der Waal’s radii plus 0.5 Å.54 The potential disruptiveness of site-directed mutants at the interface were grouped based on typical non-covalent interaction strengths for any salt bridges (1–3 kcal/mol), hydrogen bonds (1–3 kcal/mol), hydrophobic interactions (0.7 kcal/mol), aromatic interactions (1–3 kcal/mol) and van der Waals forces (0.5–1 kcal/mol)55,56 that the particular amino acid was involved in at the interface, as well as theoretical steric clashes predicted by the PyMol mutagenesis wizard upon substitution to a different amino acid side chain.
Cloning, expression and protein purification
To generate hinged or hingeless human IgG1 or IgG4 Fc domains, the relevant domains were amplified by PCR from pre-existing antibody constructs and cloned into pEU expression vectors57 containing an OriP element. Mutant CH3 domains were engineered by oligonucleotide-directed mutagenesis performed following the QuikChange II Site-Directed Mutagenesis protocol (Stratagene). For the anti-Fas antibody, EP12r_E01,58 a λ light chain and three versions of an IgG4 heavy chain were constructed in pEU vectors. The heavy chains were: wild-type IgG4; monovalent IgG4 with hinge cysteines substituted (C226Q/C229Q/T394D); and monovalent aglycosylated IgG4 with hinge cysteines substituted (C226Q/C229Q/N297Q/T394D).
Transient expression of recombinant Fc domains and antibodies was performed in CHO cells with the protein secreted into the medium. Proteins were purified by protein A affinity chromatography. Mutated Fc domains for monomer-dimer analysis were concentrated and buffer exchanged into phosphate buffered saline (PBS). Fc domains and antibody samples for other in vitro and in vivo analyses were further purified using either a Superdex 75 or 200 16/60 PG column (GE Healthcare) equilibrated with PBS and eluted with an isocratic flow. Protein purity was analyzed by SDS-PAGE with typical yields of approximately 50–200 mg of > 95% pure protein per original liter of culture.
Size exclusion chromatography and light scattering
Purified Fc domains were analyzed by SEC using a Superdex 75 10/300 GL column (GE Healthcare) run in PBS on an Agilent 1100 series HPLC. Data was analyzed using ChemStation software (Agilent). Column calibration was performed using a set of low molecular weight standards (GE Healthcare). The void volume, Vo, for the column was determined by measuring the elution volume, Ve, of the 2 MDa oligosaccharide Blue Dextran. Kav for each protein was determined using the following formula59 with a column volume, Vc, of 24 ml: Kav = (Ve-Vo)/(Vc-Vo)
Multi-angle laser light scattering was performed in-line with fractionation (SEC-MALLS), which was performed as above. Light scattering and differential refractive index were detected using the DAWN-HELEOS and Optilab rEX instruments respectively and the data analyzed using Astra V software (Wyatt Technology).
For analysis of arm exchange, anti-Fas wild-type IgG4 antibody was mixed in a 1:1 molar ratio with either wild-type IgG4 hinged Fc or monomeric IgG4 hinged Fc (incorporating mutations C226Q/C229Q/T394D) in either the presence or absence of 0.5 mM GSH. Samples were incubated for 24 or 96 h at 37°C followed by SEC analysis using a BioSep-SEC-S 2000 column on an Agilent 1100 series HPLC with the run performed in PBS running buffer. Data was analyzed using ChemStation software (Agilent).
Analytical ultracentrifugation
SV-AUC was undertaken on a Beckman Coulter XL-A AUC instrument at 20°C with sample concentrations between 28 and 42 μM and PBS buffer as a reference. A final rotor speed of 40,000 rpm was selected with 300 nm wavelength scans performed at 6 min intervals for a total of 200 scans. The data obtained was assessed using the SEDFIT program60 to obtain the continuous size distribution profile of the sedimentation coefficient values, reported in Svedberg units (S). An average partial specific volume of 0.73 ml/g at 20°C was used in the analysis. The computer program SEDNTERP35 was used to calculate the buffer density and viscosity of PBS.
To measure reversible self-association, a concentration series was performed between 0.5 and 1.56 mg/ml for the following IgG4 Fc domains: wild-type, Y349D and L368R. Self-association constants were determined by curve fitting using SEDANAL.33 Hydrodynamic bead modeling software, SOMO,34 was used to calculate S20w sedimentation coefficient values from homology models of mutant Fc domains L368R, F405Q, S364R, T394D, Y407R and F405R. Experimentally determined sedimentation coefficient values for each mutant fit to a single non-interacting species at 0.5 mg/ml were converted to S20w using SEDNTERP35 and compared with theoretical S20w values from bead modeling.
Modeling
Homology models of mutant Fc domains were generated using the Discovery Studio 2.5.5 software package (Accelrys Inc.). Templates for homology modeling were identified using the BLAST NCBI database. Sequence alignments were optimized and three-dimensional homology models built using MODELER.61 The homology models were submitted to side-chain optimization and minimization steps, followed by model validation.
Conformational stability
DSC to determine thermal stability was performed using a VP-Capillary DSC system (Microcal LLC) with samples heated from 25 to 100°C at a scan rate of 95°C per hour. Data was analyzed using Origin 7 software.
FcRn binding kinetics
The kinetics of soluble wild-type IgG4 and aglycosylated monovalent IgG4 binding with mouse FcRn was determined using a T200 SPR biosensor (GE Healthcare). Soluble mouse FcRn was amine coupled to a CM5 sensor chip at a surface density of approximately 175 RU. Reference flow cells were also prepared using the same amine coupling procedure but without the addition of FcRn. Assay running buffer and sample diluent was 50 mM sodium phosphate, 100 mM sodium chloride, 0.05% P20, pH 6.0. A 3-fold dilution series of both the wild-type and monovalent half-antibody was prepared from 1 µM to 1.37 nM. Samples were allowed to associate for 3 min and dissociate for 2 min, after which the FcRn surface was regenerated with a 30 sec injection of PBS, pH 7.4. Experiments were conducted at 25°C using a flow rate of 30 µl/min. Sensorgram data was both reference subtracted and buffer subtracted before being fit to a heterogeneous ligand model.40,42
Pharmacokinetic study
BALB/c mice were given a 10 mg/kg body weight intravenous bolus dose of one of three versions of an anti-human Fas antibody: wild-type IgG4; glycosylated monovalent IgG4; or an aglycosylated monovalent IgG4. Each group contained five mice with plasma samples collected at 5 min, 1, 2, 4, 7, 10, 13 and 16 d for the wild-type IgG4 and aglycosylated monovalent IgG4 and at 5 min, 2, 4 and 7 d for the glycosylated monovalent IgG4. Serum titers were assayed using an immunoassay with capture of the antibodies using an anti-human IgG4 Fc polyclonal antibody and detection using an anti-human lambda light chain monoclonal antibody. For each group WinNonlin software (Pharsight) was used to calculate pharmacokinetic parameters using non-compartmental analysis or two-compartmental modeling.
Supplementary Material
Acknowledgments
The authors would like to thank Lutz Jermutus for his critical review of the manuscript and MedImmune for their generous funding of all aspects of the work presented in this paper. In addition the authors would like to thank members of the MedImmune DNA Chemistry team for the provision of oligonucleotides and for sequence confirming all of the DNA constructs.
Glossary
Abbreviations:
- IgG
immunoglobulin G
- Fc
fragment crystallizable
- scFv
single chain variable fragment
- Fab
antigen binding fragment
- FcRn
neonatal fragment crystallizable receptor
- SEC
size exclusion chromatography
- MALLS
multi angle laser light scattering
- SV-AUC
sedimentation velocity analytical ultracentrifugation
- DSC
differential scanning calorimetry
- GSH
reduced glutathione
- PK
pharmacokinetic
- CHO
Chinese hamster ovary
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Pavlou AK, Belsey MJ. The therapeutic antibodies market to 2008. Eur J Pharm Biopharm. 2005;59:389–96. doi: 10.1016/j.ejpb.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 2.Parren PW, van de Winkel JG. An integrated science-based approach to drug development. Curr Opin Immunol. 2008;20:426–30. doi: 10.1016/j.coi.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 3.Reichert JM. Monoclonal antibodies as innovative therapeutics. Curr Pharm Biotechnol. 2008;9:423–30. doi: 10.2174/138920108786786358. [DOI] [PubMed] [Google Scholar]
- 4.Jefferis R. Antibody therapeutics: isotype and glycoform selection. Expert Opin Biol Ther. 2007;7:1401–13. doi: 10.1517/14712598.7.9.1401. [DOI] [PubMed] [Google Scholar]
- 5.Salfeld JG. Isotype selection in antibody engineering. Nat Biotechnol. 2007;25:1369–72. doi: 10.1038/nbt1207-1369. [DOI] [PubMed] [Google Scholar]
- 6.Pacchiana G, Chiriaco C, Stella MC, Petronzelli F, De Santis R, Galluzzo M, et al. Monovalency unleashes the full therapeutic potential of the DN-30 anti-Met antibody. J Biol Chem. 2010;285:36149–57. doi: 10.1074/jbc.M110.134031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Toole JM, Rabenau KE, Burns K, Lu D, Mangalampalli V, Balderes P, et al. Therapeutic implications of a human neutralizing antibody to the macrophage-stimulating protein receptor tyrosine kinase (RON), a c-MET family member. Cancer Res. 2006;66:9162–70. doi: 10.1158/0008-5472.CAN-06-0283. [DOI] [PubMed] [Google Scholar]
- 8.Brekke OH, Løset GA. New technologies in therapeutic antibody development. Curr Opin Pharmacol. 2003;3:544–50. doi: 10.1016/j.coph.2003.05.002. [DOI] [PubMed] [Google Scholar]
- 9.Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol. 2009;157:220–33. doi: 10.1111/j.1476-5381.2009.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghetie V, Hubbard JG, Kim JK, Tsen MF, Lee Y, Ward ES. Abnormally short serum half-lives of IgG in beta 2-microglobulin-deficient mice. Eur J Immunol. 1996;26:690–6. doi: 10.1002/eji.1830260327. [DOI] [PubMed] [Google Scholar]
- 11.Israel EJ, Wilsker DF, Hayes KC, Schoenfeld D, Simister NE. Increased clearance of IgG in mice that lack beta 2-microglobulin: possible protective role of FcRn. Immunology. 1996;89:573–8. doi: 10.1046/j.1365-2567.1996.d01-775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Junghans RP, Anderson CL. The protection receptor for IgG catabolism is the beta2-microglobulin-containing neonatal intestinal transport receptor. Proc Natl Acad Sci U S A. 1996;93:5512–6. doi: 10.1073/pnas.93.11.5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin WL, West AP, Jr., Gan L, Bjorkman PJ. Crystal structure at 2.8 A of an FcRn/heterodimeric Fc complex: mechanism of pH-dependent binding. Mol Cell. 2001;7:867–77. doi: 10.1016/S1097-2765(01)00230-1. [DOI] [PubMed] [Google Scholar]
- 14.Pavenstädt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003;83:253–307. doi: 10.1152/physrev.00020.2002. [DOI] [PubMed] [Google Scholar]
- 15.Wartiovaara J, Ofverstedt LG, Khoshnoodi J, Zhang J, Mäkelä E, Sandin S, et al. Nephrin strands contribute to a porous slit diaphragm scaffold as revealed by electron tomography. J Clin Invest. 2004;114:1475–83. doi: 10.1172/JCI22562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thurber GM, Schmidt MM, Wittrup KD. Antibody tumor penetration: transport opposed by systemic and antigen-mediated clearance. Adv Drug Deliv Rev. 2008;60:1421–34. doi: 10.1016/j.addr.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Labrijn AF, Buijsse AO, van den Bremer ET, Verwilligen AY, Bleeker WK, Thorpe SJ, et al. Therapeutic IgG4 antibodies engage in Fab-arm exchange with endogenous human IgG4 in vivo. Nat Biotechnol. 2009;27:767–71. doi: 10.1038/nbt.1553. [DOI] [PubMed] [Google Scholar]
- 18.van der Neut Kolfschoten M, Schuurman J, Losen M, Bleeker WK, Martínez-Martínez P, Vermeulen E, et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science. 2007;317:1554–7. doi: 10.1126/science.1144603. [DOI] [PubMed] [Google Scholar]
- 19.Rose RJ, Labrijn AF, van den Bremer ET, Loverix S, Lasters I, van Berkel PH, et al. Quantitative analysis of the interaction strength and dynamics of human IgG4 half molecules by native mass spectrometry. Structure. 2011;19:1274–82. doi: 10.1016/j.str.2011.06.016. [DOI] [PubMed] [Google Scholar]
- 20.Angal S, King DJ, Bodmer MW, Turner A, Lawson AD, Roberts G, et al. A single amino acid substitution abolishes the heterogeneity of chimeric mouse/human (IgG4) antibody. Mol Immunol. 1993;30:105–8. doi: 10.1016/0161-5890(93)90432-B. [DOI] [PubMed] [Google Scholar]
- 21.Bloom JW, Madanat MS, Marriott D, Wong T, Chan SY. Intrachain disulfide bond in the core hinge region of human IgG4. Protein Sci. 1997;6:407–15. doi: 10.1002/pro.5560060217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Labrijn AF, Rispens T, Meesters J, Rose RJ, den Bleker TH, Loverix S, et al. Species-specific determinants in the IgG CH3 domain enable Fab-arm exchange by affecting the noncovalent CH3-CH3 interaction strength. J Immunol. 2011;187:3238–46. doi: 10.4049/jimmunol.1003336. [DOI] [PubMed] [Google Scholar]
- 23.Schuurman J, Perdok GJ, Gorter AD, Aalberse RC. The inter-heavy chain disulfide bonds of IgG4 are in equilibrium with intra-chain disulfide bonds. Mol Immunol. 2001;38:1–8. doi: 10.1016/S0161-5890(01)00050-5. [DOI] [PubMed] [Google Scholar]
- 24.Peters SJ, Smales CM, Henry AJ, Stephens PE, West S, Humphreys DP. Engineering an improved IgG4 molecule with reduced disulfide bond heterogeneity and increased Fab domain thermal stability. J Biol Chem. 2012;287:24525–33. doi: 10.1074/jbc.M112.369744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Labrijn AF, Loverix S, Parren PW, Van de Winkel JG, Schuurman J, Lasters I. Antibody variants having modifications in the constant region. 2008; WO 2010/063785.
- 26.Parren PW, Schuurman J, Vink T, Bleeker WK, Van de Winkel JG, Van Berkel PH, et al. Recombinant monovalent antibodies and methods for production thereof. 2006; PCT/DK2006/000669:WO 2007/059782.
- 27.Zhou H, Kannan G, Sun N. Monomeric antibody fc. 2009; PCT/US2010/057662:WO 2011/063348 A1.
- 28.Ying TL, Chen WZ, Gong R, Feng Y, Dimitrov DS. Soluble monomeric IgG1 fc. J Biol Chem. 2012;287:19399–408. doi: 10.1074/jbc.M112.368647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dall’Acqua W, Simon AL, Mulkerrin MG, Carter P. Contribution of domain interface residues to the stability of antibody CH3 domain homodimers. Biochemistry. 1998;37:9266–73. doi: 10.1021/bi980270i. [DOI] [PubMed] [Google Scholar]
- 30.Deisenhofer J. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-A resolution. Biochemistry. 1981;20:2361–70. doi: 10.1021/bi00512a001. [DOI] [PubMed] [Google Scholar]
- 31.Miller S. Protein-protein recognition and the association of immunoglobulin constant domains. J Mol Biol. 1990;216:965–73. doi: 10.1016/S0022-2836(99)80014-X. [DOI] [PubMed] [Google Scholar]
- 32.Teerinen T, Valjakka J, Rouvinen J, Takkinen K. Structure-based stability engineering of the mouse IgG1 Fab fragment by modifying constant domains. J Mol Biol. 2006;361:687–97. doi: 10.1016/j.jmb.2006.06.073. [DOI] [PubMed] [Google Scholar]
- 33.Stafford WF, Sherwood PJ. Analysis of heterologous interacting systems by sedimentation velocity: curve fitting algorithms for estimation of sedimentation coefficients, equilibrium and kinetic constants. Biophys Chem. 2004;108:231–43. doi: 10.1016/j.bpc.2003.10.028. [DOI] [PubMed] [Google Scholar]
- 34.Rai N, Nöllmann M, Spotorno B, Tassara G, Byron O, Rocco M. SOMO (SOlution MOdeler) differences between X-Ray- and NMR-derived bead models suggest a role for side chain flexibility in protein hydrodynamics. Structure. 2005;13:723–34. doi: 10.1016/j.str.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 35.Laue TM, Shah BD, Ridegway TM, Pelletier SL. Computer-aided interpretation of analytical sedimentation data for proteins. In: Harding SE, Rowe AJ, Horton JC, eds. Ultracentrifugation in Biochemistry and Polymer Science: Royal Soceity of Chemistry, Cambridge, 1992:90-125. [Google Scholar]
- 36.Mimura Y, Church S, Ghirlando R, Ashton PR, Dong S, Goodall M, et al. The influence of glycosylation on the thermal stability and effector function expression of human IgG1-Fc: properties of a series of truncated glycoforms. Mol Immunol. 2000;37:697–706. doi: 10.1016/S0161-5890(00)00105-X. [DOI] [PubMed] [Google Scholar]
- 37.Vermeer AW, Norde W. The thermal stability of immunoglobulin: unfolding and aggregation of a multi-domain protein. Biophys J. 2000;78:394–404. doi: 10.1016/S0006-3495(00)76602-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rispens T, den Bleker TH, Aalberse RC. Hybrid IgG4/IgG4 Fc antibodies form upon ‘Fab-arm’ exchange as demonstrated by SDS-PAGE or size-exclusion chromatography. Mol Immunol. 2010;47:1592–4. doi: 10.1016/j.molimm.2010.02.021. [DOI] [PubMed] [Google Scholar]
- 39.Rispens T, Ooijevaar-de Heer P, Bende O, Aalberse RC. Mechanism of immunoglobulin G4 Fab-arm exchange. J Am Chem Soc. 2011;133:10302–11. doi: 10.1021/ja203638y. [DOI] [PubMed] [Google Scholar]
- 40.Martin WL, Bjorkman PJ. Characterization of the 2:1 complex between the class I MHC-related Fc receptor and its Fc ligand in solution. Biochemistry. 1999;38:12639–47. doi: 10.1021/bi9913505. [DOI] [PubMed] [Google Scholar]
- 41.Raghavan M, Wang Y, Bjorkman PJ. Effects of receptor dimerization on the interaction between the class I major histocompatibility complex-related Fc receptor and IgG. Proc Natl Acad Sci U S A. 1995;92:11200–4. doi: 10.1073/pnas.92.24.11200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vaughn DE, Bjorkman PJ. High-affinity binding of the neonatal Fc receptor to its IgG ligand requires receptor immobilization. Biochemistry. 1997;36:9374–80. doi: 10.1021/bi970841r. [DOI] [PubMed] [Google Scholar]
- 43.Chapman AP, Antoniw P, Spitali M, West S, Stephens S, King DJ. Therapeutic antibody fragments with prolonged in vivo half-lives. Nat Biotechnol. 1999;17:780–3. doi: 10.1038/11717. [DOI] [PubMed] [Google Scholar]
- 44.Nguyen A, Reyes AE, 2nd, Zhang M, McDonald P, Wong WL, Damico LA, et al. The pharmacokinetics of an albumin-binding Fab (AB.Fab) can be modulated as a function of affinity for albumin. Protein Eng Des Sel. 2006;19:291–7. doi: 10.1093/protein/gzl011. [DOI] [PubMed] [Google Scholar]
- 45.Pepinsky RB, Walus L, Shao Z, Ji B, Gu S, Sun Y, et al. Production of a PEGylated Fab’ of the anti-LINGO-1 Li33 antibody and assessment of its biochemical and functional properties in vitro and in a rat model of remyelination. Bioconjug Chem. 2011;22:200–10. doi: 10.1021/bc1002746. [DOI] [PubMed] [Google Scholar]
- 46.Valentine JL, Arnold LW, Owens SM. Anti-phencyclidine monoclonal Fab fragments markedly alter phencyclidine pharmacokinetics in rats. J Pharmacol Exp Ther. 1994;269:1079–85. [PubMed] [Google Scholar]
- 47.Kim JK, Tsen MF, Ghetie V, Ward ES. Localization of the site of the murine IgG1 molecule that is involved in binding to the murine intestinal Fc receptor. Eur J Immunol. 1994;24:2429–34. doi: 10.1002/eji.1830241025. [DOI] [PubMed] [Google Scholar]
- 48.Tesar DB, Tiangco NE, Bjorkman PJ. Ligand valency affects transcytosis, recycling and intracellular trafficking mediated by the neonatal Fc receptor. Traffic. 2006;7:1127–42. doi: 10.1111/j.1600-0854.2006.00457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dall’Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn) J Biol Chem. 2006;281:23514–24. doi: 10.1074/jbc.M604292200. [DOI] [PubMed] [Google Scholar]
- 50.Dall’Acqua WF, Woods RM, Ward ES, Palaszynski SR, Patel NK, Brewah YA, et al. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: biological consequences. J Immunol. 2002;169:5171–80. doi: 10.4049/jimmunol.169.9.5171. [DOI] [PubMed] [Google Scholar]
- 51.Krapp S, Mimura Y, Jefferis R, Huber R, Sondermann P. Structural analysis of human IgG-Fc glycoforms reveals a correlation between glycosylation and structural integrity. J Mol Biol. 2003;325:979–89. doi: 10.1016/S0022-2836(02)01250-0. [DOI] [PubMed] [Google Scholar]
- 52.Corper AL, Sohi MK, Bonagura VR, Steinitz M, Jefferis R, Feinstein A, et al. Structure of human IgM rheumatoid factor Fab bound to its autoantigen IgG Fc reveals a novel topology of antibody-antigen interaction. Nat Struct Biol. 1997;4:374–81. doi: 10.1038/nsb0597-374. [DOI] [PubMed] [Google Scholar]
- 53.DeLano WL. The PyMOL User's Manual. DeLano Scientific, Palo Alto, Ca, USA: 2002. [Google Scholar]
- 54.Tsai CJ, Lin SL, Wolfson HJ, Nussinov R. A dataset of protein-protein interfaces generated with a sequence-order-independent comparison technique. J Mol Biol. 1996;260:604–20. doi: 10.1006/jmbi.1996.0424. [DOI] [PubMed] [Google Scholar]
- 55.Andrews PR, Craik DJ, Martin JL. Functional group contributions to drug-receptor interactions. J Med Chem. 1984;27:1648–57. doi: 10.1021/jm00378a021. [DOI] [PubMed] [Google Scholar]
- 56.Carver F, Hunter C, Seward E. Structure-activity relationship for quantifying aromatic interactions. Chem Commun. 1998:775–6. doi: 10.1039/a800567b. [DOI] [Google Scholar]
- 57.Persic L, Roberts A, Wilton J, Cattaneo A, Bradbury A, Hoogenboom HR. An integrated vector system for the eukaryotic expression of antibodies or their fragments after selection from phage display libraries. Gene. 1997;187:9–18. doi: 10.1016/S0378-1119(96)00628-2. [DOI] [PubMed] [Google Scholar]
- 58.Chodorge M, Züger S, Stirnimann C, Briand C, Jermutus L, Grütter MG, et al. A series of Fas receptor agonist antibodies that demonstrate an inverse correlation between affinity and potency. Cell Death Differ. 2012;19:1187–95. doi: 10.1038/cdd.2011.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Laurent TC, Killander J. A theory of gel filtration and its exeperimental verification. J Chromatogr A. 1964;14:317–30. doi: 10.1016/S0021-9673(00)86637-6. [DOI] [Google Scholar]
- 60.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J. 2000;78:1606–19. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sali A, Potterton L, Yuan F, van Vlijmen H, Karplus M. Evaluation of comparative protein modeling by MODELLER. Proteins. 1995;23:318–26. doi: 10.1002/prot.340230306. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.