

Abstract
We compared the effects of three missense mutations in the GABAA receptor γ2 subunit on GABAA receptor assembly, trafficking and function in HEK293T cells cotransfected with α1, β2, and wildtype or mutant γ2 subunits. The mutations R82Q and P83S were identified in families with genetic epilepsy with febrile seizures plus (GEFS+), and N79S was found in a single patient with generalized tonic-clonic seizures (GTCS). Although all three mutations were located in an N terminal loop that contributes to the γ+/β− subunit-subunit interface, we found that each mutation impaired GABAA receptor assembly to a different extent. The γ2(R82Q) and γ2(P83S) subunits had reduced α1β2γ2 receptor surface expression due to impaired assembly into pentamers, endoplasmic reticulum (ER) retention and degradation. In contrast, γ2(N79S) subunits were efficiently assembled into GABAA receptors with only minimally altered receptor trafficking, suggesting that N79S was a rare or susceptibility variant rather than an epilepsy mutation. Increased structural variability at assembly motifs was predicted by R82Q and P83S, but not N79S, substitution, suggesting that R82Q and P83S substitutions were less tolerated. Membrane proteins with missense mutations that impair folding and assembly often can be “rescued” by decreased temperatures. We coexpressed wildtype or mutant γ2 subunits with α1 and β2 subunits and found increased surface and total levels of both wildtype and mutant γ2 subunits after decreasing the incubation temperature to 30 °C for 24 hours, suggesting that lower temperatures increased GABAA receptor stability. Thus epilepsy-associated mutations N79S, R82Q and P83S disrupted GABAA receptor assembly to different extents, an effect that could be potentially rescued by facilitating protein folding and assembly.
Keywords: GABAA receptors, genetic generalized epilepsy, GABRG2(N79S) mutation, GABRG2(R82Q) mutation, GABRG2(P83S) mutation, loss of function, dominant negative effects, subunit interface, impaired receptor assembly
Introduction
Epilepsy is a common neurological disorder that affects about 1% of the world's population (Sander, 2003), and genetic epilepsy (GE) syndromes comprise ~30% of all cases (Steinlein, 2004; Reid et al., 2009). Many epilepsy-mutations in affected individuals in families with GEs have been found in ion channels, including γ-amino butyric acid (GABA) type A (GABAA) receptors, which are heteropentameric chloride ion channels that mediate the majority of inhibitory neurotransmission in the CNS. The receptor is composed of five subunits, and the predominant synaptic receptors are composed of two α subunits, two β subunits and one γ2 subunit. The most common epilepsy-associated GABAA receptor gene (GABR) is GABRG2, and epilepsy mutations in γ2 subunits have been shown to decrease receptor function by altering receptor biogenesis or channel function (Macdonald and Kang, 2009). Three GABRG2 mutations R82Q, P83S and N79S (numbered based on the immature γ2 subunit containing the signal peptide) were reported to be associated with generalized epilepsies and are all located in the same structural loop in the N terminus of γ2 subunits, suggesting that they might impair GABAA receptor function similarly.
R82Q is one of the best characterized epilepsy-associated GABRG2 mutations. It was originally found in a large family with genetic epilepsy with febrile seizures plus (GEFS+) (Wallace et al., 2001; Marini et al., 2003), contributing to childhood absence epilepsy and febrile seizures. A single nucleotide substitution caused a highly conserved arginine residue located within a loop between the α-helix and the β1-sheet (the α-β1 loop) in the extracellular N terminus to be replaced by a glutamine (Figure 1A), resulting in impaired surface expression of γ2 subunits and decreased GABAA receptor currents (Bianchi et al., 2002; Kang and Macdonald, 2004; Sancar and Czajkowski, 2004; Hales et al., 2005; Eugene et al., 2007; Frugier et al., 2007). Heterozygous knock-in mice carrying this mutation displayed spontaneous spike-wave discharges and thermal-induced seizures (Tan et al., 2007; Reid et al., 2013), consistent with R82Q being an epilepsy-causing mutation. However, whether this mutation has dominant negative effects on other GABAA receptor subunits and how it affects subunit-subunit interactions is still controversial (Hales et al., 2005; Frugier et al., 2007). A recent study showed that while loss of γ2 subunit function could account for the absence seizure phenotype, the R82Q mutation might be responsible for the febrile seizure phenotype (Reid et al., 2013), further suggesting that the R82Q mutation had effects in addition to haploinsufficiency.
Figure 1. Mutant residues were located in the α-β1 loop that contributes to the γ+/β− subunit-subunit interface.

A. Sequences of N-terminal α-helix, α-β1 loop and β1-sheet domains of human α(1-6), β(1-3), γ(1-3) and δ subunits from the GABAA receptor family were aligned with sequences of the nicotinic acetylcholine receptor α subunit (ACHA(7,9)), 5-hydroxytryptamine 3A receptor subunit (5HT3A) and glutamate-gated chloride channel GluCl α subunit (G5EBR3). Sites of missense mutations in the γ2 subunit were highlighted in red. In all sequences, identical residues were highlighted in dark gray and conserved residues were highlighted in light gray. The α-helix, α-β1 loop and β1-sheet domains were also represented across subunits above the alignments. B. On the left, a structural model of the α1β2γ2 GABAA receptor, as viewed from the synaptic cleft, was shown. Sites of missense mutations in γ2 subunit, located at the γ2(+)/β2(−) interface, were shown in space-filling representation, i.e., N79 in light blue, R82 in orange, and P83 in green, and the α-β1 loop where these three residues were located was shown in purple. Homologous motifs for α1β2γ2 receptor assembly at the respective complementary (−) interfaces (α1: red; β2: dark blue; γ2: yellow) and conserved tryptophan residues located in these motifs (α1W97, red; β2W91, dark blue; γ2W121, yellow) were also represented. On the right, an enlarged 45 ° side view of the γ+/β− subunit-subunit interface with a close-up of missense mutations in the α-β1 loop was also shown.
Recently, another epilepsy-associated GABRG2 mutation, P83S, which is also located within the α-β1 loop of the γ2 subunit, was identified in a three generation GEFS+ family (Lachance-Touchette et al., 2011). Although this mutation was found in all affected individuals in this family and was predicted to have damaging effects, it was reported that GABAA receptor channel function was not affected by the mutation, and the effects on receptor trafficking were not addressed. How this mutation contributes to epileptogenesis is therefore still uncertain.
Finally, it was reported that a GABRG2 mutation, N79S, also located in the α-β1 loop of the γ2 subunit, was found in a single patient with generalized tonic-clonic seizures (GTCS) (Shi et al., 2010). The mutation was reported to only modify the steepness of the GABA concentration-response curve (Migita et al., 2013).
All three mutations are located in the N terminal domain of γ2 subunits that forms part of the γ2+/β2− subunit interface (Figure 1B), suggesting that they may produce similar impairments of subunit oligomerization and receptor assembly (Hales et al., 2005). In the present study, we compared the effects of these three epilepsy-associated GABRG2 mutations on surface expression and function of α1β2γ2 receptors in transfected HEK293T cells and rat cortical neurons and found that they impaired assembly and trafficking of GABAA receptors by similar mechanisms but to different extents.
Materials and Methods
Expression vectors
The coding sequences of human α1, β2 and γ2 GABAA receptor subunits were cloned into pcDNA3.1 expression vectors (Invitrogen). All subunit residues were numbered based on the immature peptide. Mutant γ2 subunit constructs were generated using the QuikChange site-directed mutagenesis kit (Stratagene). An HA or FLAG epitope was inserted at a functionally silent site (between the 4th and 5th residue of the mature peptide) to facilitate our experiments (Connolly et al., 1996). Both γ2S and γ2L subunits (Connolly et al., 1999), two different splice isoforms, were used. For neuronal transfections, wildtype and mutant γ2L subunits were cloned into pLVX-IRES-ZsGreen vectors (Clontech).
Cell culture and transfection
Human embryonic kidney cells (HEK293T) (ATCC, CRL-11268) were incubated at 37°C in humidified 5% CO2 incubator and maintained in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with fetal bovine serum (10%, Life technologies), and penicillin/streptomycin (100 IU/ml, Life technologies). Cells were transfected using the FuGENE 6 transfection reagent (Roche Applied Science) or polyethylenimine (PEI) reagent (40 kD, Polysciences) and harvested 36 hours after transfection. To express wildtype and mutant α1β2γ2 receptors, a total of 3 μg of subunit cDNAs were transfected at a ratio of 1:1:1 into 6 cm dishes for most experiments except for whole cell recording. In experiments studying the effects of low temperature, cells were incubated at 30°C for 24 hours beginning about 16 hours after transfection.
Rat cortical neurons were obtained from E18 embryos as previously described (Tian and Macdonald, 2012), incubated at 37°C in 5% CO2 incubator, and maintained in serum-free Neurobasal medium (Gibco) supplemented with B27 supplement (Gibco), glutamine (Gibco) and penicillin/streptomycin (Gibco, 20 U/ml). Cultured neurons were transfected at DIV5 using Lipofectamine 2000 (Invitrogen). One hour after transfection, culture medium containing DNA/Lipofectamine complex was replaced by fresh medium.
Western Blot, endoglycosidase H (Endo H) digestion, surface biotinylation and immunoprecipitation
After sonication, whole cell lysates of transfected HEK293T cells were collected in modified RIPA buffer (50 mM Tris (pH = 7.4), 150 mM NaCl, 1% NP-40, 0.2% sodium deoxycholate, 1 mM EDTA) and 1% protease inhibitor mixture (Sigma). Collected samples were subjected to gel electrophoresis using NuPAGE® (Invitrogen) precast gel and then transferred to PVDF-FL membranes (Millipore). Monoclonal anti-HA antibody (Covance or Cell signaling) and monoclonal anti-FLAG antibody (Sigma) were used to detect the epitope tag. Polyclonal anti-γ2 antibodies (Sysy or Millipore) were used to detect GABAA receptor γ2 subunits. Anti-sodium potassium ATPase antibody (Abcam) was used as a loading control. After incubation with primary antibodies, IRDye® (LI-COR Biosciences) conjugated secondary antibody was used at a 1:10,000 dilution, and the signals were detected using the Odyssey Infrared Imaging System (LI-COR Biosciences). The integrated intensity value of each specific band was calculated using the Odyssey 3.0 software (LI-COR Biosciences).
To remove immature N-linked glycans, cell lysates were incubated with the enzyme Endo H (NEBiolab) at 37°C for 3 hours. Treated samples were then subjected to SDS-PAGE and Western blot.
Surface proteins were collected using surface biotinylation as described before (Lo et al., 2010). Transfected cells were biotinylated using the membrane-impermeable reagent sulf-HNS-SS-biotin (1 mg/ml, Thermo Scientific) at 4°C for 1 h. Cells were lysed after being quenched with 0.1 M glycine. The biotin-labeled plasma membrane proteins were pulled down by High Binding Capacity NeutrAvidin beads (Thermo Scientific Pierce) after centrifugation and cleaved by sampling buffer (Invitrogen) containing 10% beta-mercaptoethanol.
Protein complexes containing FLAG-tagged GABAA receptor subunits were extracted in modified RIPA buffer with reduced amounts of detergents (50 mM Tris (pH=7.4), 150 mM NaCl, 1% Triton) and immunoprecipitated using EZview Red Anti-FLAG M2 affinity gel (Sigma) at 4°C overnight, then eluted with 3X FLAG peptide (Sigma).
Immunocytochemistry and confocal microscopy
Cultured cortical neurons were fixed by 4% paraformaldehyde/4% glucose in PBS for 15 min followed by 1h block with 10% BSA in PBS and were supplemented with 0.2% Triton for total staining. Coverslips were then incubated in mouse monoclonal anti-HA antibody (Covance) for 2 h, followed by incubation with Alexa 647-conjugated donkey anti-mouse IgG antibodies.
Confocal images were obtained using a Zeiss LSM 510 META inverted confocal microscope. Images were taken with 8 bit, 512×512 pixel resolution, and an average of four scans was taken to decrease the background noise. Pinholes were adjusted so that the sample thickness was smaller than 2 μm. Confocal experiments were performed in part through the use of the VUMC Cell Imaging Shared Resource.
Flow cytometry
High throughput flow cytometry was performed to investigate the surface expression of GABAA receptor subunits. Transfected cells were collected in phosphate-buffered saline containing 2% fetal bovine serum and 0.05% sodium azide as described before (Lo et al., 2008). Cell samples were incubated with an Alexa fluorophore (Invitrogen)-conjugated monoclonal anti-α1 antibody (Millipore), monoclonal anti-β2/β3 antibody (Millipore) or monoclonal anti-HA antibody (Covance), and then fixed by 2% paraformaldehyde. The fluorescence signals were read on a BD Biosciences FACSCalibur system. Nonviable cells were excluded from study based on the previously determined forward and side scatter profiles. The mean fluorescence value of each experimental condition was subtracted by that of mock-transfected condition and was then normalized to that of the control condition. Flow Cytometry experiments were performed in the VMC Flow Cytometry Shared Resource.
Whole cell voltage-clamp recordings
Whole cell voltage-clamp recordings were performed at room temperature on lifted HEK293T cells 36-48 hrs after transfection with GABAA receptor subunits as described previously (Hernandez et al., 2011). Cells were bathed in an external solution containing 142 mM NaCl, 1 mM CaCl2, 8 mM KCl, 6 mM MgCl2, 10 mM glucose, and 10 mM HEPES (pH 7.4, ~325 mOsM). Recording electrodes were pulled from thin-walled borosilicate capillary glass (World Precision Instruments) using a P2000 laser electrode puller (Sutter Instruments), fire-polished with a microforge (Narishige), and filled with an internal solution containing 153 mM KCl, 1 mM MgCl2, 10 mM HEPES, 5 mM EGTA, 2 mM Mg2+-ATP (pH 7.3, ~300 mOsM). All patch electrodes had a resistance of 1 – 1.6 MΩ. The combination of internal and external solutions yielded a chloride reversal potential of ~ 0 mV, and cells were voltage-clamped at −20 mV using an Axopatch 200B amplifier (Axon Instruments). A rapid exchange system (open tip exchange times ~ 400 μs), composed of a four-barrel square pipette attached to a Warner SF-77B Perfusion Fast-Step (Warner Instruments Corporation) and controlled by Clampex 9.0 software (Axon Instruments) was used to apply GABA to lifted whole cells. The channels were activated by 1 mM GABA for 4 s, followed by an extensive wash for 40 s, and then blocked by 10 μM Zn2+ for 10 s. GABA (1 mM) was then applied for 4 s in the presence of 10 μM Zn2+. Peak current amplitudes after the Zn2+ application were normalized to those before the Zn2+ application to calculate the sensitivity to Zn2+ blockade. All currents were low-pass filtered at 2 kHz, digitized at 5-10 kHz, and analyzed using the pCLAMP 9 software suite.
Structural modeling and simulation
Three-dimensional models of human GABAA receptor subunits were generated using the crystal structure of the C. elegans glutamate-gated chloride channel (GluCl) (Hibbs and Gouaux, 2011) as a template (PDB: 3rhw) using DeepView/Swiss-PdbViewer 4.02 (Schwede et al., 2003). The initial sequence alignments between human GABAA receptor subunits and C. elegans GluCl subunits were generated with full-length multiple alignments using ClustalW. Then full-length multiple alignments were submitted for automated comparative protein modeling implemented in the program suite incorporated in SWISS-MODEL (http://swissmodel.expasy.org/SWISS-MODEL.html) using human GABAA receptors sequences as target proteins and the C. elegans GluCl sequence as a template structure. To generate pentameric GABAA receptor homology models, α, β, and γ subunit structural models were assembled in a counter-clockwise β-α-β-α-γ order by superposition onto the C. elegans GluCl channel as a template. The resulting models were subsequently energy-optimized using GROMOS96 in default settings within the Swiss-PdbViewer. Side-chain prediction and conformational backbone variability of γ2 subunit mutation were implemented using Rosetta backrub flexible backbone design (Lauck et al., 2010) in the program suite incorporated in RosettaBackrub (https://kortemmelab.ucsf.edu/backrub/cgi-bin/rosettaweb.py). Structural models of the best-scoring low-energy backrub structures of wildtype and mutant γ2 subunits were represented.
Data analysis
Numerical data were reported as mean ± S.E. Statistical analysis was performed using GraphPad Prism. Statistically significant differences were taken as p < 0.05 using one way ANOVA followed by Dunnet's multiple comparison or by Student's t test.
Results
The three γ2 subunit mutations were located in the α-β1 loop that contributes to the γ2+/β2− subunit interface and all impaired surface levels of α1β2γ2 receptors but to different extents
The mutations N79S, R82Q, and P83S were located close to each other in the N terminus of γ2 subunits (Figure 1A). By comparing sequences of this region we found that the R82 and P83 residues were identical among different GABAA receptor subunits and other cys-loop receptor subunits (Figure 1A, dark grey bars), while the N79 residue was not conserved among the cys-loop receptor subunit families (i.e., acetylcholine receptor α subunit (ACHA), serotonin 3A receptor (5HT3A) subunit and Avermectin-sensitive glutamate-gated chloride channel α subunit (G5EBR3CA)).
We also built three-dimensional pentameric GABAA receptor homology models based on the crystal structure of the C. elegans GluCl channel (Figure 1B). We found that this cluster of γ2 subunit mutations was located in the loop between the α-helix and the β1-sheet (the α-β1 loop, purple) at the top of the N-terminal extracellular domain that contributes to the γ+/β− subunit interface in assembled receptors. The R82 residue (orange) was closer than the P83 residue (green) to the complementary (−) face of β2 subunits, while the N79 residue (light blue) was located behind the interface. Of note it has been shown that the γ2 subunit mutation R82Q disrupts salt bridges at the γ+/β− subunit interface (Frugier et al., 2007). Furthermore, in silico analysis using Polyphen-2 (Adzhubei et al., 2010) and SIFT (Ng and Henikoff, 2001), software programs that predict whether or not protein structure would tolerate mutations based on sequence conservation and local structural features, predicted that that both R82Q and P83S substitutions would not be tolerated and might damage protein structure, while the N79S substitution would be tolerated.
To examine the effects of these three mutations on surface expression of receptors, we cotransfected HEK293T cells with α1, β2, and wildtype or mutant γ2LHA or γ2SHA subunits at a 1:1:1 α1:β2:γ2 subunit ratio and evaluated surface and total levels of wildtype and mutant γ2HA subunits by flow cytometry. For γ2LHA subunits containing the R82Q or P83S mutation, we found substantial reductions of surface γ2LHA subunit levels and small reductions of total γ2LHA subunit levels (Figure 2A top). For coexpressed α1β2γ2L(R82Q)HA or α1β2γ2L(P83S)HA subunits, surface HA levels were decreased to 0.14 ± 0.01 (p < 0.001, n = 12) and 0.26 ± 0.02 (p < 0.001, n = 13), respectively, relative to that of coexpressed α1β2γ2LHA subunits (1.00, n = 17), while the surface HA level of coexpressed α1β2γ2L(N79S)HA subunits was not decreased significantly (0.92 ± 0.05 , p > 0.05, n = 13). With coexpression of α1β2γ2L(R82Q)HA and α1β2γ2L(P83S)HA subunits, total HA levels were slightly but significantly decreased to 0.79 ± 0.04 (p < 0.001, n = 11) and 0.84 ± 0.06 (p < 0.01, n = 12), respectively, relative to that of coexpressed α1β2γ2LHA subunits (1.00, n = 16), while the total HA level of coexpressed α1β2γ2L(N79S)HA was not affected significantly (0.96 ± 0.05, p > 0.05, n = 12).
Figure 2. Surface expression of mutant γ2 subunits was reduced to different extents.

A. α1β2 and α1β2γ2HA (wildtype or mutant γ2S or γ2L) subunits were coexpressed in HEK293T cells. Surface and total γ2HA subunit levels were evaluated through flow cytometry. The mock-subtracted mean fluorescence value of γ2HA subunits under different experimental conditions were normalized to those obtained with cotransfection of wildtype α1β2γ2HA subunits (n ≥ 5, mean ± SEM). Differences compared to cotransfection of wildtype α1β2γ2HA subunits were analyzed by the one way ANOVA test followed by Dunnett's multiple comparison test. (*** p < 0.001; ** p < 0.01; * p < 0.05). B. Wildtype or mutant γ2L subunits were coexpressed with α1β2 subunits in HEK293T cells. Surface protein samples were collected through surface biotinylation and blotted by anti-γ2 and anti-ATPase antibodies (not shown). Cell lysates from transfected cells were loaded as the total fraction. Band intensity of the γ2LHA subunit was normalized to the ATPase signal (n ≥ 4, mean ± SEM). Differences compared to cotransfection of wildtype α1β2γ2L subunits were analyzed by the one way ANOVA test followed by Dunnett's multiple comparison test. (*** p < 0.001; ** p < 0.01; * p < 0.05). C. Wildtype or mutant γ2LHA subunits were expressed in rat cortical neurons in pLVX-IRES-ZsGreen vectors and stained by anti-HA antibody. Surface (without permeabilization) and total (with permeabilization) staining patterns were revealed by confocal imaging. Scale bar = 20 μm. Inset scale bar = 2 μm.
We found similar results with mutant γ2S subunits (Figure 2A bottom). For coexpressed α1β2γ2S(N79S)HA, α1β2γ2S(R82Q)HA and α1β2γ2S(P83S)HA subunits, surface HA levels were decreased to 0.88 ± 0.01 (p < 0.001, n = 5), 0.10 ±0.01 (p < 0.001, n = 9) and 0.11 ± 0.01 (p < 0.001, n = 10), respectively, compared to that for coexpressed α1β2γ2SHA subunits (1.00, n = 11). Total HA levels of coexpressed α1β2γ2S(R82Q)HA and α1β2γ2S(P83S)HA subunits were decreased to 0.76 ± 0.07 (p < 0.05, n = 8) and 0.68 ± 0.06 (p < 0.005, n = 11), respectively, relative to that for coexpressed α1β2γ2SHA subunits (1.00, n = 11). The total HA level of coexpression of α1β2γ2S(N79S)HA subunits was not decreased significantly (0.92 ± 0.15, p > 0.05, n = 5).
To control for any artifact produced by the HA epitope-tag, we also coexpressed untagged α1β2γ2L subunits and examined surface γ2L levels by surface biotinylation (Figure 2B). Similar to the flow cytometry results, compared to wildtype γ2L subunits (1.00, n = 6) we found that surface levels of γ2L(R82Q) and γ2L(P83S) subunits were reduced to 0.17 ± 0.06 (p < 0.001, n = 5) and 0.24 ± 0.05 (p < 0.001, n = 4), respectively, and that the surface level of γ2L(N79S) subunits was reduced slightly but significantly to 0.78 ± 0.08 (p < 0.05, n = 5). Total expression of γ2L(R82Q) and γ2L(P83S) subunits was also decreased to 0.60 ± 0.08 (n = 8, p < 0.01) and 0.56 ± 0.10 (n = 6, p < 0.01), respectively, but total expression of γ2L(N79S) subunits was not reduced significantly (0.86 ± 0.10, p > 0.05, n = 8).
These results suggested that R82Q, P83S and N79S substitutions were all in the same loop structure contributing to the γ2+/β2− subunit interface and all affected surface levels of receptors, but to different extents. The R82Q and P83S substitutions likely disrupted subunit oligomerization or receptor assembly more severely than the N79S substitution, thus producing a much more substantial decrease of surface γ2 subunits.
In neurons, the R82Q and P83S mutations impaired surface trafficking of γ2 subunits, but the N79S mutation had minimal if any effect
To study how the mutant subunits were expressed in neurons, we transfected wildtype or mutant γ2LHA subunits into rat cortical neurons and labeled the transfected γ2LHA subunits using anti-HA antibody (Figure 2C). Wildtype and mutant γ2LHA subunits were cloned into pLVX-IRES-ZsGreen vectors, and the ZsGreen signal was used to identify transfected neurons. Without cell permeabilization, the surface expression and localization of γ2L subunits could be visualized. For γ2LHA and γ2L(N79S)HA subunits, the HA signal could be seen outlining the soma and dendrites of ZsGreen positive cells (Figure 2C, left top). In contrast, the surface HA signals for γ2L(R82Q)HA and γ2L(P83S)HA subunits were almost absent (Figure 2C, left bottom). With cell permeabilization, however, wildtype and all three mutant γ2LHA subunits were well detected in both soma and dendrites (Figure 2C, right). Thus, similar to HEK293T cells, γ2L(R82Q)HA and γ2L(P83S)HA subunit levels were reduced on the cell surface of neurons, but γ2L(N79S) HA subunits had surface levels similar to those obtained for wildtype γ2LHA subunits. While there was no apparent reduction of surface γ2L(N79S) HA subunits in neurons, this is only a qualitative method that is not suitable for detecting small changes, and thus, we cannot exclude small effects of the N79S mutation on γ2L subunit surface level in neurons.
Mutant subunits disrupted GABAA receptor function and/or changed GABAA receptor composition
Since all three mutations decreased surface levels of γ2 subunits, although to different extents, we determined how receptor function was affected using whole cell patch clamp recordings. Wildtype or mutant γ2L subunits were coexpressed with α1 and β2 subunits in HEK293T cells at a 1:1:0.1 α1:β2:γ2L subunit ratio, and macroscopic peak currents were evoked by applying a saturating GABA concentration (1 mM) for 4 s using a rapid exchange system (Figure 3A, left traces). Current density of receptors containing γ2(N79S) subunits was only slightly, but significantly, reduced (1003 ± 16.53 pA/pF, n = 10, p < 0.05) compared with wildtype receptors (1163 ± 50.95 pA/pF, n = 21). Current densities for receptors containing γ2(R82Q) subunits (394.7 ± 35.95 pA/pF, n = 11, p < 0.001) or γ2(P83S) subunits (140.4 ± 16.36 pA/pF, n = 10, p < 0.001) were substantially decreased compared with wildtype receptors (Figure 3B), consistent with the results described above showing that R82Q and P83S mutations reduced the surface levels of γ2L subunits much more extensively than the N79S mutation.
Figure 3. Mutant receptors showed decreased whole cell current amplitudes and increased Zn2+sensitivity.

A. Wildtype or mutant γ2L subunits were coexpressed with α1β2 subunits in HEK293T cells. GABAA receptor currents in response to 4 s applications of 1 mM GABA alone (left traces) or coapplied with 10 μM Zn2+ (right traces) to lifted cells containing wildtype and mutant γ2 subunits were shown. Subunit identity and length of GABA application (black line) were indicated above the current traces. Scale bars = 1 nA and 0.4 nA. B. Mean current densities (pA/pF, top panel) and Zn2+ inhibition (%, bottom panel) from cells coexpressing wildtype or mutant γ2L subunits were calculated. *** indicated p < 0.001, * indicated p < 0.05, compared with wildtype.
The reduction in current density produced by the mutations suggested that the mutant subunits may not be effectively assembled into receptor pentamers. GABAA receptors composed of α1 and β2 subunits can form in the absence of γ2 subunits, and it is possible that the currents recorded in the presence of the mutant γ2 subunits were due, at least in part, to surface α1β2 receptors. While α1β2 receptors can form, they have different physiological and pharmacological properties including increased sensitivity to Zn2+ inhibition. To evaluate the possibility of α1β2 receptor formation in the presence of mutant γ2 subunits, we determined the Zn2+ sensitivity of currents from receptors formed with coexpression of α1, β2, and wildtype or mutant γ2L subunits. Whole-cell currents evoked by co-application of 1 mM GABA with or without 10 μM Zn2+ were recorded (Figure 3A, right traces). The fractional Zn2+ inhibition of currents evoked from cells coexpressing α1β2γ2(R82Q) or α1β2γ2(P83S) subunits was significantly higher than inhibition of currents from cells coexpressing α1β2γ2 or α1β2γ2(N79S) subunits (WT: 9 ± 1%, n = 16; N79S: 6 ± 2%, n = 10, p > 0.05; R82Q: 49 ± 4%, n = 11, p < 0.001; P83S: 84 ± 2%, n = 10, p < 0.001) (Figure 3B). Because the sensitivity of GABAA receptor currents to Zn2+ inhibition depends on subunit composition, these results suggested that mutant γ2(R82Q) and γ2(P83S) subunits were incompletely incorporated into ternary α1β2γ2L receptors, leading to increased expression of Zn2+-sensitive binary α1β2 receptors and decreased expression of relatively Zn2+ insensitive α1β2γ2L receptors on the cell surface, thus resulting in decreased GABAA receptor currents.
In contrast, although the peak current amplitude was slightly and significantly reduced, cells coexpressing α1β2γ2(N79S) subunits displayed currents that were Zn2+ insensitive. Peak currents evoked from α1β2γ2L receptors are much larger than those from α1β2 receptors. Since the N79S mutation only slightly reduced peak current amplitude, it is likely that there was only a small reduction of incorporation of γ2(N79S) subunits into ternary α1β2γ2L(N79S) receptors, and thus the dominant Zn2+-insensitive α1β2γ2L(N79S) receptor currents would have masked any small increase of Zn2+-sensitive α1β2 receptor currents.
Mutant γ2L(R82Q) and γ2L(P83S) subunits impaired formation of stable trafficking-competent oligomers with partnering subunits and were retained in the ER and degraded
During biogenesis of ternary GABAA receptor pentamers, subunit dimers form but are not trafficked to the cell surface (Klausberger et al., 2001a). Further, it has been demonstrated that γ2L subunits alone and βγ2 subunit complexes did not form trafficking-competent receptors and were trapped in the ER (Connolly et al., 1996; Tretter et al., 1997a). As all three mutations are located in the α-β1 loop that contributes to the γ2+/β2− subunit-subunit interface, and surface and total expression of γ2 subunits carrying these mutations were decreased to different levels (Figure 2), we studied how receptor biogenesis was affected. To explore how these mutations affected expression/stability of γ2 subunits in HEK293T cells, we expressed wildtype and mutant γ2LHA subunits alone, with only β2 subunits or with both α1 and β2 subunits to study their expression as single subunits, dimeric oligomers and ternary receptors (Figure 4A). Whole cell lysates were obtained and Western blots were performed to study the expression pattern of γ2L subunits. We blotted for wildtype and mutant γ2LHA subunits using anti-HA antibody, quantified the band intensity, normalized it to that of ATPase, and normalized the HA/ATPase ratio to that obtained with expression of wildtype α1β2γ2LHA subunits. The expression differences between single subunits, dimeric oligomers and ternary receptors were compared for each wildtype or mutant group.
Figure 4. Mutant γ2(R82Q) and γ2(P83S) subunits showed immature glycosylation patterns and decreased stability.

A. Wildtype or mutant γ2LHA subunits were expressed alone, coexpressed with β2 subunits only, or coexpressed with both α1 and β2 subunits in HEK293T cells. The total lysates of transfected cells were collected and blotted by anti-HA and anti-ATPase antibodies. When γ2LHA subunits were coexpressed with both α1 and β2 subunits, a top band around 47 kD (black arrow) appeared in addition to the bottom band (grey arrow) around 42 kD. Band intensity of the γ2LHA subunit was normalized to the ATPase signal and then normalized to that with cotransfection of wildtype α1β2γ2LHA subunits (n = 4, mean ± SEM). Results obtained from transfection of wildtype or mutant γ2LHA subunits alone or cotransfection of wildtype or mutant γ2LHA subunits with β2 subunits were compared to those from cotransfection of corresponding wildtype or mutant γ2LHA subunits and α1β2 subunits and were analyzed by one way ANOVA test followed by Dunnett's multiple comparison test. (*** p < 0.001; ** p < 0.01; * p < 0.05). B. Wildtype or mutant γ2LHA subunits were coexpressed with α1β2 subunits in HEK293T cells. The total lysates of transfected cells were collected, digested by Endo H, and blotted by anti-HA antibody. The two bands of γ2LHA subunits before Endo H digestion were labeled by black and grey arrows. The intensity of the Endo H sensitive band (bottom band after digestion) was normalized to the total band intensity (top band and bottom band together after digestion) and analyzed by one way ANOVA test followed by Dunnett's multiple comparison test. (*** p < 0.001)
For wildtype γ2L subunits, total level was greatly increased with coexpression with α1 and β2 subunits compared to expression of γ2L subunits alone or with only β2 subunits (Figure 4A, lanes 1, 5, 9), suggesting that coexpression of wildtype α1β2γ2L subunits formed stable oligomers while expression of single subunits and coexpression of γ2L and β2 subunits did not form stable oligomers and were degraded (n = 4, p < 0.01). Similarly, the total level of γ2L(N79S) subunits was also increased when coexpressed with α1 and β2 subunits, compared to expressed alone or with only β2 subunits (Figure 4A, lanes 2, 6, 10) (n = 4, p < 0.01). In contrast, total levels of γ2L(R82Q) (Figure 4A, lanes 3, 7, 11) and γ2L(P83S) (Figure 4A, lanes 4, 8, 12) subunits were not significantly increased when coexpressed with α1 and β2 subunits (n = 4, p > 0.05). These results suggested that most γ2L(R82Q) and γ2L(P83S) subunits did not form stable oligomers with α1 and β2 subunits and were likely degraded as single subunits or intermediate oligomers.
Interestingly, we found that when coexpressed with α1 and β2 subunits, γ2L and γ2L(N79S) subunits displayed an additional band with increased molecular mass (Figure 4A, lanes 9, 10), which was present but relatively weak for coexpressed γ2L(R82Q) and γ2L(P83S) subunits (Figure 4A, lane 11, 12: top band, ~47 kD; bottom band, ~42 kD). During protein biogenesis and trafficking, glycans are attached to nascent peptides in the ER and then subjected to several rounds of processing. To examine whether this shift of molecular mass reflected different glycosylation patterns, we coexpressed α1 and β2 subunits with wildtype or mutant γ2LHA subunits and digested with Endo H, which cleaves only immature glycans added in the ER but not mature glycans added in the Golgi apparatus (Figure 4B). After Endo H digestion, we found that the γ2L and γ2L(N79S) subunits were relatively resistant to Endo H digestion, while in contrast, mutant γ2L(R82Q) and γ2L(P83S) subunits showed high sensitivity to Endo H digestion. The lower band (grey arrow), which was the dominant expression pattern of γ2L(R82Q) and γ2L(P83S) subunits, was almost gone after Endo H digestion. We compared the proportion of Endo H sensitive bands (bottom bands after digestion) and found that the Endo H sensitive proportions for γ2L(R82Q) (0.80 ± 0.02, p < 0.001) and γ2L(P83S) (0.84 ± 0.05, p < 0.001), but not γ2L(N79S) (0.30 ± 0.10, p > 0.05), subunits were significantly larger than for wildtype γ2L subunits (0.28 ± 0.06, n = 4). Consistent with findings mentioned above (Figure 4A), the amount and size of undigested mutant γ2L(R82Q) and γ2L(P83S) subunits were different from the undigested wildtype γ2L and γ2L(N79S) subunits. The lower molecular mass and smaller amount of undigested mutant γ2L(R82Q) and γ2L(P83S) subunits indicated they were immature and nonstable. As none of these three residues are located near the predicted glycosylation sites, it is unlikely that the mutation itself affect the glycosylation patterns. Taken together, the mature glycosylation pattern and increased expression levels of γ2L and γ2L(N79S) subunits demonstrated that the majority of them formed stable trafficking-competent receptors with α1 and β2 subunits, which were successfully delivered to the Golgi apparatus and cell surface. In contrast, the immature glycosylation pattern and decreased total levels of γ2L(R82Q) and γ2L(P83S) subunits suggested that although some of them could still form stable trafficking-competent receptors when coexpressed with partnering subunits, most of them did not and were trapped in the ER and degraded like single subunits or dimeric oligomers.
γ2L(R82Q) and γ2L(P83S) subunits were incorporated into pentamers inefficiently
It has been shown that the extracellular N termini of different GABAA receptor subunits interact during receptor assembly (Klausberger et al., 2001a). We found that the mutations were located near to or contributing to the γ2+/β2− subunit interface (Figure 1B, left panel), and thus we wondered whether the assembly of γ2(N79S), γ2(R82Q), and γ2(P83S) subunits into trafficking-competent receptors was interrupted. Previous studies reported that the α-β1 loop structure on the plus interface of γ2 subunits (γ2+) directly interacts with the minus interface of β2 subunits (β2−), and that the R82Q mutation impaired the interaction of γ2 and β2 subunits mediated by this α-β1 loop (Hales et al., 2005). To better understand how these mutations affected the subunit-subunit oligomerization during receptor assembly, we coexpressed α1 and β2FLAG subunits with wildtype or mutant γ2LHA subunits, pulled down β2 subunits and their subunit binding partners using anti-FLAG beads and blotted associated γ2L subunits using anti-HA antibody (Figure 5A). The amount of γ2L(N79S)HA subunit associated with β2FLAG subunits was reduced slightly, but not significantly, compared to associated wildtype γ2LHA subunits (0.94 ± 0.05, n = 6, p > 0.05) (Figure 5B). In contrast, the amounts of γ2L(R82Q) and γ2L(P83S) subunits associated with β2 subunits were substantially reduced, indicating that fewer γ2L subunits were assembled into αβγ pentamers (R82Q: 0.47 ± 0.05, n = 6, p < 0.001; P83S: 0.71 ± 0.05, n = 6, p < 0.001, respectively) (Figure 5B).
Figure 5. Mutant γ2(R82Q) and γ2(P83S) subunits were incorporated inefficiently into receptor pentamers.
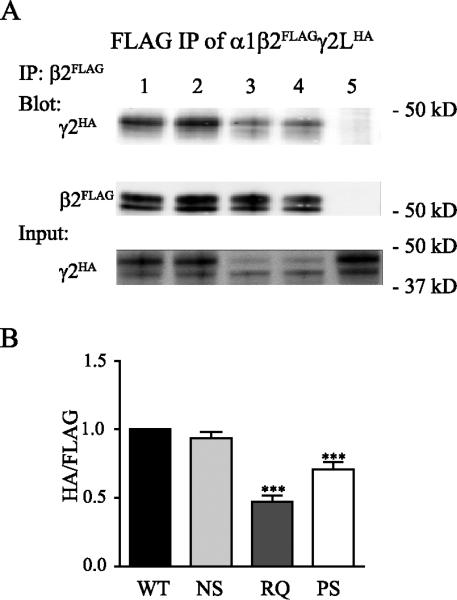
A. Wildtype or mutant γ2LHA subunits were coexpressed with α1 and β2FLAG subunits in HEK293T cells. In whole cell lysates, β2FLAG subunits and associated subunits were pulled down by anti-FLAG beads and blotted on Western blots by anti-FLAG and anti-HA antibodies. The total lysates were loaded as input and blotted by anti-HA antibody. B. The amount of wildtype or mutant γ2LHA subunits associated with β2FLAG subunits when coexpressed with α1 and β2FLAG subunits were compared (n = 6, mean ± SEM). Differences compared to cotransfection of wildtype α1β2γ2LHA subunits were analyzed by the one way ANOVA test followed by Dunnett's multiple comparison test. (*** p < 0.001; ** p < 0.01; * p < 0.05)
It is possible that the reduced association was caused by the reduced amount of mutant γ2L(R82Q) and γ2L(P83S) subunits. However, we found that while the main bands of γ2L(R82Q) and γ2L(P83S) subunits in the whole cell lysate had lower molecular mass than wildtype γ2L and γ2L(N79S) subunits, consistent with immature glycosylation (Figure 4 and 5A bottom, Input), most of the γ2L(R82Q) and γ2L(P83S) subunits associated with β2 subunits were of the same higher molecular mass as the wildtype γ2L and γ2L(N79S) subunits (Figure 5A top, IP). Taking these results with previous findings (Figure 4), we suggest that when forming α1β2γ2 receptors, mutant γ2(R82Q) and γ2(P83S) subunits inefficiently assembled into pentamers to form mature receptors and unassembled subunits were trapped in the ER and degraded. Nonetheless, a small portion of the mutant γ2L(R82Q) and γ2L(P83S) subunits were still incorporated into stable pentamers that were trafficked beyond the ER and reached the cell surface. Thus, the reduced amount of mutant γ2L(R82Q) and γ2L(P83S) subunits is the result rather than the cause of reduced subunit/subunit interaction. In contrast, the γ2L(N79S) subunits were much more efficiently assembled into receptors and trafficked to the cell surface, consistent with the finding that this mutation only produced a small reduction of surface level (Figures 2A, B; 3A, B).
Mutant subunits impaired trafficking of partnering subunits and/or changed receptor composition
Our results above demonstrated that processing and assembly of αβγ receptors were reduced, but not abolished totally, by the presence of either γ2(R82Q) and γ2(P83S) subunits and to a much lesser extent by the γ2(N79S) subunit. Increased Zn2+ sensitivity of receptors containing either γ2(R82Q) and γ2(P83S) subunits also suggested a changed receptor stoichiometry. However, as mutant subunits were still expressed, they could form unstable trafficking-incompetent intermediate oligomers with partnering subunits in the ER, which could impede their assembly and trafficking. To further investigate which type of GABAA receptors were trafficked to the surface and if mutant γ2L subunits had a dominant negative effect by decreasing the trafficking of partnering α and β subunits, we coexpressed α1 and β2 subunits with wildtype or mutant γ2LHA subunits and evaluated surface and total expression of α1 and β2 subunits by flow cytometry (Figure 6).
Figure 6. Over-expression of mutant γ2 subunits decreased surface levels of partnering subunits.

A. B. Wildtype or mutant γ2LHA (A) or γ2SHA (B) subunits were coexpressed with α1β2 subunits in HEK293T cells. Surface and total levels of α1 or β2 subunits were evaluated using flow cytometry. The mock-subtracted mean fluorescence value of each subunit under different experimental conditions were normalized to those obtained with cotransfection of wildtype α1β2γ2LHA (A) (n ≥ 9, mean ± SEM) or α1β2γ2SHA (B) (n ≥ 5, mean ± SEM) subunits. Differences compared to wildtype α1β2γ2 receptor condition were analyzed by the one way ANOVA test followed by Dunnett's multiple comparison test. (*** p < 0.001; ** p < 0.01; * p < 0.05).
In the absence of wildtype γ2L subunits, the α1 subunit surface level was slightly increased (1.21 ± 0.05, n = 17, p < 0.001), and the β2 subunit surface level was greatly increased (2.82 ± 0.21, n = 14, p < 0.001) relative to their surface levels in the presence of γ2L subunits (Figure 6A), compatible with a change of receptor stoichiometry from 2α2β1γ to 2α3β (Tretter et al., 1997b; Baumann et al., 2002). Surface α1 subunit levels were slightly, but not significantly, reduced with coexpression of α1 and β2 subunits with either γ2L(N79S) (0.92 ± 0.05, n =13, p > 0.05) or γ2L(R82Q) (0.92 ± 0.04, n = 11, p > 0.05) subunits and only slightly, but significantly, reduced with γ2L(P83S) subunits (0.85 ± 0.03, n = 12, p < 0.05) (Figure 6A). Surface β2 levels were not increased with coexpression of γ2L(N79S) (0.86 ± 0.07, n = 11, p > 0.05), were slightly, although insignificantly, increased with coexpression of γ2L(P83S) subunits (1.38 ± 0.07, n = 11, p > 0.05) and were significantly increased with coexpression of γ2L(R82Q) subunits (1.52 ± 0.13, n = 9, p < 0.05) (Figure 6A). Total levels of α1 and β2 subunits also exhibited similar trends (Figure 6A). Again, although we did not find altered surface receptor stoichiometry for coexpressed α1β2γ2L(N79S) subunits, there could have been a slight increase of surface α1β2 receptors that was not detected. The changed surface receptor stoichiometry in the presence of R82Q or P83S mutations suggested increased expression of surface α1β2 receptors, consistent with the increased Zn2+ sensitivity of GABA evoked currents (Figure 3), but the increase was lower than that obtained with total removal of the γ2L subunit (α1β2 subunit coexpression condition). The slight reduction rather than an increase of surface α1 subunits as well as the small increase of surface β2 subunits when coexpressed with mutant γ2L(R82Q) or γ2L(P83S) subunits indicated these mutant subunits might have dominant negative effects to suppress assembly of α1 and β2 subunits.
In contrast to γ2L subunits, γ2S subunits can be trafficked to the surface by themselves (Boileau et al., 2010). Thus, an excess of γ2S subunits might impede trafficking of partnering subunits less than γ2L subunits did. To identify any dominant negative effects of mutant γ2S subunits, we determined surface and total levels of α1 and β2 subunits coexpressed with wildtype or mutant γ2SHA subunits. In the absence of γ2S subunits, the α1 subunit surface level was not changed (0.92 ± 0.05, n = 11, p > 0.05), but the β2 subunit surface level was increased (2.09 ± 0.17, n = 11, p < 0.001), compared to surface levels in the presence of wildtype γ2S subunits, also suggesting assembly of αβ receptors (Figure 6B). Interestingly, in the presence of the mutations, α1 subunit surface levels were all significantly decreased (N79S, 0.76 ± 0.06, n = 5, p < 0.001; R82Q, 0.54 ± 0.02, n = 8, p < 0.001; P83S, 0.57 ± 0.03, n =11, p < 0.001) compared to wildtype receptors, while β2 subunit surface levels were not increased (N79S, 0.77 ± 0.03, n = 5, p > 0.05; R82Q, 0.94 ± 0.04, n = 8, p > 0.05; P83S, 0.88 ± 0.08, n = 11, p > 0.05). Total levels of α1 and β2 subunits also exhibited similar trends (Figure 6B). The β2 subunits could not be trafficked to the surface without α1 subunits. Since we found a significant decrease of surface α1 subunits without an increase of surface β2 subunits when coexpressing mutant γ2S subunits, it is likely that the mutations caused a decrease of αβγ receptors that was offset by an increase of αβ receptors. Taken together, these results demonstrated that both γ2 subunit R82Q and P83S mutations caused disrupted pentameric receptor processing or trafficking, not only due to inefficient receptor assembly but also due to trapping partnering subunits in the ER hindering their assembly and trafficking; whereas the N79S mutation had similar, but much smaller, effects on receptor assembly and trafficking.
Decreased temperature increased surface and total levels of wildtype and mutant γ2L subunits
Membrane proteins with missense mutations that impair trafficking have been shown to have their function “rescued” at lower temperatures, presumably due to slowed protein processing that facilitates subunit folding and receptor assembly and/or slowed subunit degradation or receptor internalization (Denning et al., 1992; Thomas et al., 2003; Varga et al., 2008; Guo et al., 2012). We thus explored the effects of decreased temperature on the total and surface expression of wildtype and mutant receptors. We coexpressed wildtype or mutant γ2LHA subunits with α1 and β2 subunits in HEK293T cells and determined total expression of wildtype and mutant γ2LHA subunits after incubation at 37°C or 30°C for 24 h by Western blot using anti-HA antibody (Figure 7A). We quantified the band intensity, normalized it to that of ATPase, and normalized the HA/ATPase ratio for mutant γ2LHA subunits to that obtained with expression of α1, β2 and wildtype γ2LHA subunits. The expression difference between 37°C and 30°C was compared for each wildtype or mutant condition. We found substantially increased total wildtype and mutant γ2LHA subunit levels when incubated at 30°C for 24 h compared to those at 37°C (WT: (37°C: 1.00, 30°C: 2.08 ± 0.28, p < 0.05); N79S: (37°C: 0.91 ± 0.12, 30°C: 1.55 ± 0.17, p < 0.05); R82Q: (37°C: 0.37 ± 0.06, 30°C: 0.54 ± 0.08, p < 0.001); P83S: (37°C: 0.36 ± 0.06, 30°C: 0.57 ± 0.03, p < 0.05); n = 5), indicating that the stability of both wildtype and mutant γ2LHA subunits was increased at a lower temperature.
Figure 7. Decreased temperature stabilized both wildtype and mutant γ2 subunits.

A. Wildtype or mutant γ2LHA subunits were coexpressed with α1 and β2 subunits in HEK293T cells and incubated at 37°C or 30°C for 24 hours. The total lysates of transfected cells were collected and blotted by anti-HA and anti-ATPase antibodies. Band intensity of the γ2LHA subunit was normalized to the ATPase signal, then normalized to that of wildtype α1β2γ2LHA subunits (n = 5, mean ± SEM). Differences between 37°C and 30°C incubation were analyzed by t test. (*** p < 0.001; ** p < 0.01; * p < 0.05). B. Wildtype or mutant γ2LHA subunits were coexpressed with α1 and β2 subunits in HEK293T cells and incubated at 37°C or 30°C for 24 hours. Surface protein samples were collected through surface biotinylation and blotted by anti-α1, anti-γ2 and anti-ATPase antibody. Band intensities of the α1 and γ2L subunits were normalized to the ATPase signal. The values for the 30°C incubation were further normalized to those obtained at 37°C (n = 4, mean ± SEM) to calculate the fold increase. The significance of fold increase was analyzed by t test. (*** p < 0.001; ** p < 0.01; * p < 0.05).
We then determined whether surface levels of receptor subunits were also increased by reduced temperature (Figure 7B). We coexpressed α1β2γ2LHA subunits in HEK293T cells and examined the surface levels of α1 subunits and wildtype and mutant γ2LHA subunits after a 24 h incubation at 37°C or 30°C by surface biotinylation using anti-α1 and anti-γ2 antibodies. We normalized the surface expression levels at 30°C to those at 37°C. Interestingly, we found a moderate, but not significant, increase of surface α1 subunit levels with all conditions (WT: 1.33 ± 0.25; N79S: 1.63 ± 0.32; R82Q: 1.40 ± 0.24; P83S: 1.36 ± 0.29; n = 4, p > 0.05), but a significant large increase of surface γ2L subunit levels (WT: 2.16 ± 0.34; N79S: 2.26 ± 0.20; R82Q: 1.86 ± 0.16; P83S: 1.93 ± 0.21; n = 4, p < 0.05). These results suggest that the biogenesis of wildtype and mutant γ2L subunits was facilitated by lower temperatures. Although we found a moderate but not significant increase of surface α1 subunit levels, it is possible that at a lower temperature the rate of α1β2γ2L receptor assembly is relatively slow compared to that of γ2L homopentamer assembly, leading to a large increase of surface γ2L homopentamers and a small increase of surface α1β2γ2L receptors.
Discussion
The R82Q and P83S mutations were located in the α-β1 loop at the γ2+/β2− subunit-subunit interface and disrupted receptor assembly and trafficking
Biogenesis of cys-loop receptors is complex and inefficient (Gorrie et al., 1997). After the synthesis of single subunits, intermediate dimers form, but only pentamers with correct subunit folding and assembly will pass the ER quality control, be further trafficked to and processed by the Golgi apparatus and then be trafficked to the cell surface (Tretter et al., 1997a; Klausberger et al., 2001a). Inappropriately folded and unassembled subunits are quickly degraded (Gorrie et al., 1997). Our data suggested that the γ2 subunit mutations R82Q and P83S decreased to similar extents the efficiency of pentamer formation. Mutant γ2 subunits that were not incorporated into pentamers were trapped in the ER and likely degraded. Due to the inefficient assembly of mutant γ2 subunit-containing pentameric receptors, receptors with a different stoichiometry (α1β2 dimeric receptors) were able to be assembled. We previously reported two other mutations within structural loops contributing to interface interactions with similar fates: the β3 subunit mutation G32R located at the γ+/β− subunit interface (Gurba et al., 2012), and the γ2 subunit mutation R177G located at the α+/γ− interface (Audenaert et al., 2006). Both GABRB3(G32R) and GABRG2(R177G) mutations were shown to decrease surface levels of mutant γ2L subunits and increase surface levels of αβ heteropentamers and/or β3 homopentamers, indicating a common molecular mechanism shared by this group of mutations.
Our findings in this study were contrary to those in a previous study, which reported that receptor function as well as Zn2+ sensitivity of GABAA receptors containing mutant γ2(P83S) subunits were normal (Lachance-Touchette et al., 2011). This conflict may have been due to the different ratios of subunit cDNAs used for transfection. In contrast to a 1:1:2 α1:β2:γ2 cDNA ratio used in their study, we used a 1:1:0.1 cDNA ratio. Unpublished data from our laboratory has shown that GABA-evoked peak currents in HEK cells transfected with 1:1:0.1 (α1:β2:γ2) ratio were equal to those obtained with a 1:1:1 ratio, although the surface level of γ2 subunits was much lower. Thus, our data indicated that transfection ratios greater than 1:1: 1 are oversaturating for whole cell recordings, which could mask the deficits caused by mutations.
Our study explored how the R82Q and P83S mutations affected receptor biogenesis. We found that the surface level of mutant subunits was greatly decreased (Figure 2), while the total level was also slightly, but significantly, decreased (Figure 2, 4). This was similar to in the findings with homozygous knock-in mice carrying the R82Q mutation (Tan et al., 2007). While we did not compare the rates of subunit synthesis or degradation, we have previously demonstrated that two other epilepsy-associated GABAA receptor subunit missense mutations GABRA1(A322D) and GABRG2(Q390X) altered the degradation rates, but not the synthesis rates, of the subunits (Gallagher et al., 2007; Kang et al., 2010). Recently we found that another subunit-interface-located missense mutation GABRG2(R177G) increased the degradation rate of mutant subunits (Todd et al., submitted). Thus, stability rather than synthesis of GABAA receptor subunits was regulated by the ER quality control machinery. We also explored how the R82Q and P83S mutations affected assembly of α1, β2 and γ2 subunits into receptors. It has been reported that the γ2 subunit mutation R82Q disrupted the binding of β2 subunits and a GST-fused γ2 subunit peptide N-terminal fragment containing this mutation (Hales et al., 2005). However, another group reported that the mutation did not decrease β3γ2 subunit association (Frugier et al., 2007). In the current study, we found a substantial decrease in β2γ2 subunit association when coexpressing α1β2γ2(R82Q) or α1β2γ2(P83S) subunits. Compared to wildtype γ2 subunits, only a small proportion of mutant γ2(R82Q) and γ2(P83S) subunits were assembled into pentamers, trafficked to the Golgi apparatus and inserted into the surface membrane, while the majority of them were trapped in the ER and degraded. Meanwhile, the mutant γ2(R82Q) and γ2(P83S) subunits still formed unstable intermediate oligomers with partnering subunits (data not shown), thus also impeding their assembly and trafficking (Figure 6). This is consistent with our structural simulation (discussed below) showing that R82Q and P83S mutations caused substantial structural rearrangements in several distinct domains involved in receptor assembly. The finding that some mutant γ2(R82Q) and γ2(P83S) subunits were still successfully incorporated into pentamers is also in agreement with the finding that small currents with normal kinetic properties were formed with coexpression of α1β2γ2(R82Q) subunits (Bianchi et al., 2002) and with a recent report showing that some mutant γ2(R82Q) subunits reached the cell surface and triggered endocytosis of the receptor (Chaumont et al., 2013). The discrepancy with the previous study could have been caused by use of different subtypes of β subunits or by the immunoprecipitation process. In the previous study (Frugier et al., 2007), different amounts of wildtype and mutant γ2 subunits were pulled down, while many mutant γ2 subunits that were not incorporated into pentamers were already degraded. In our study, we tried to pull down the same amount of β2 subunits, including γ2 subunits that were both associated and not associated, representing αβγ and αβ pentamers respectively. It is possible that the decreased association was caused by the paucity of mutant γ2 subunits. Nevertheless, in that case, the dominant form of γ2 subunits associated with β2 subunits should be the immature pattern rather than the mature pattern observed (Figure 5A). Thus we believe that the primary defect in receptor biogenesis was during subunit assembly, but we could not exclude some contributions from other steps.
Although also located in the γ2 subunit α-β1 loop, the N79S mutation had only small effects on receptor assembly. Different from R82Q and P83S mutations associated with epilepsy families, the N79S mutation was identified only in one patient (Shi et al., 2010). Similar to a recent finding demonstrating that γ2(N79S) subunits had no effects on GABAA receptor function except to modify the steepness of the GABA concentration-response curve (Migita et al., 2013), we found that the N79S mutation produced significant but minimal reduction of receptor assembly. We identified a small reduction of surface γ2(N79S) subunits by flow cytometry and surface biotinylation and decreased peak current amplitudes through whole cell recordings in HEK cells. We also found a small reduction of surface α1 subunits, indicating that the N79S mutation also slightly affected the assembly and trafficking of partnering subunits. We did not quantify the fluorescence intensity of immunostaining in neurons because it was not sensitive to small changes. As the defects were minimal and the majority of γ2(N79S) subunits assembled into stable pentamers that were trafficked beyond the ER and expressed on the surface as functional α1β2γ2 receptors, the γ2(N79S) mutation had only small effects on receptor biogenesis. Our model of receptor structure predicted that the N79 residue does not face the γ2+/β2− subunit interface but rather is adjacent to the interface (Figure 1B), and thus the N79S mutation may disrupt to a lesser extent the subunit interaction required to form trafficking-competent pentamers. These findings suggest that while GABRG2(N79S) decreases receptor surface expression and peak whole cell current amplitude, the magnitudes of effects are small and unlikely to be the major disease-causing factor. Considering that GABRG2(N79S) was identified in only one patient without evidence of co-segregation with an epilepsy syndrome or sporadic occurrence and is absent in the NHLBI exome variant server (http://evs.gs.washington.edu/EVS/), we suggest that rather than being an epilepsy associated mutation, GABRG2(N79S) might be a relatively benign rare variant or might increase seizure susceptibility and that other unidentified mutations or variants may be responsible for the GTCSs experienced by the patient. However, due to the lack of genetic evidence, we will continue to refer to GABRG2(N79S) as a “mutation” in this paper.
Structural simulation predicts that mutation-induced changes in protein structure impaired subunit oligomerization
Not all missense mutations will significantly affect protein structure and function, and a considerable number of mutations are well tolerated for protein folding. Proper folding and trafficking of GABAA receptors requires specific sequences and structural motifs within subunits that contribute to selective oligomerization among their γ+/β−, β+/α-, and α+/γ− interfaces. For example, it has been reported that γ2 subunit residues 130-143 in between the β2-β3 and β3-β4 loops interacted directly with α1 subunit residues, γ2 subunit residues 122-131 at the beginning of β2-β3 loop interacted with the β3 subunit (Klausberger et al., 2000; Klausberger et al., 2001b) and γ2 subunit residues 106-121 in the β1-β2 loop and β2 sheet were important for formation of the α1+/γ2− subunit interface (Sarto et al., 2002) (Figure 8C). Many of these sequences lie in homologous regions of α, β, and γ subunits (Figure 1B, homologous assembly motifs in α, β, and γ subunits were shown in red, dark blue and yellow loops, respectively). Structural rearrangements of assembly motifs could strongly impair association of partnering subunits, formation of correct subunit/subunit interfaces, oligomerization of pentameric receptors, and receptor trafficking to the cell surface.
Figure 8. Structural simulation predicted mutation-induced changes in subunit structure.

A. Superpositions of structural models for up to 10 of the best-scoring low-energy generated backbones of wildtype and mutated N79S, R82Q and P83S γ2 subunits were made. In ribbon representation, the native secondary structure was shown in gray, and the mutated secondary structures were represented by other colors. Structural domains as shown in panel C were also represented. Sites of missense mutations in γ2 subunits were shown in space-filling representation in the inserts: N79 in blue, R82 in orange, and P83 in green. B. Local side-chain rearrangements observed for mutated N79S, R82Q and P83S γ2 subunit residues were displayed. Neighboring residues within an 11 Å radius were shown in stick representation and color by element (CPK representation). Sites of wildtype or mutated N79S, R82Q or P83S residues were labeled in blue, orange or green circles. C. A table of predicted amino acids contributing to side-chain rearrangements for mutated residues at positions 79, 82 and 83 was categorized by structural domains. The mutated residues were shown in red, and identical residues among the Cys-loop family were labeled with an asterisk.
We propose that even though the N79S, R82Q and P83S mutations in the γ2 subunit are located at or near the γ2+/β2− subunit/subunit interface in the same general subunit domain, they might have different impacts on receptor structure, and thus impair the assembly and trafficking of partnering subunits quite differently. Using flexible backbone simulations we characterized structural conformational changes (Figure 8A) produced by introducing an N79S, R82Q or P83S mutation in γ2 subunits (Figure 8, box inserts). We found that all mutations were predicted to cause structural rearrangements to neighboring residues that were within 7Å from their respective mutation site. Increased structural variability was observed by the presence of alternative secondary backbone conformations at specific structural loops (Figure 8A; wildtype in gray, mutation-associated alternative backbones in other colors, and Figure 8C), and disordered side-chains of residues surrounding the mutation sites at the α-β1 loop were also observed (Figure 8B).
We also compared the mutation-induced structural differences by analyzing the carbon alpha root mean squared deviation (Cα RMSD) from the wildtype structure caused by each substitution (Figure 8C). Cα RMSD provides Cα-Cα comparisons between two structurally aligned models; the larger the Cα RMSD, the more the mutant structure deviates from the wildtype structure. The R82Q mutation had a much larger Cα RMSD than P83S and N79S mutations for any structural loop. With a Cα RMSD of 0.73 ± 0.05 Å for residues D65-G102 in the α-helix, α-β1 loop and β1 sheet, the R82Q substitution had a less preserved native conformation than P83S (Cα RMSD of 0.16 ± 0.02 Å) or N79S (Cα RMSD of 0.14 ± 0.02 Å) substitutions. We previously found that this α-β1 loop domain, which participates in formation of the γ+/β− interface (Klausberger et al., 2000), interacts with the β2 subunit N32 glycosylation site impairing receptor assembly and function (Klausberger et al., 2000; Lo W, 2013.), suggesting that the R82Q mutation disrupted primarily the α-β1-loop-mediated γ+/β− interaction.
All three mutations were predicted to cause rearrangements in other domains also (Figure 8C). While both R82Q and P83S mutations primarily produced changes among residues at the γ+/β− interface, they also produced rearrangements in the β7-β8 loop and β8 sheet (Cα RMSD of 0.07 ± 0.005 Å, and Cα RMSD of 0.02 ± 0.005 Å, respectively for residues Y213-Y220) and P83S also produced rearrangements in the β3-β4 loop (Cα RMSD of 0.06 ± 0.01 Å for residues V142-P148). Strikingly, only the P83S mutation had side-chain rearrangements among residues found at both α+/γ− and γ+/β− interfaces (Cα RMSD of 0.19 ± 0.02 Å for residues W121-K127 in the β2 sheet and β2-β3 loop) (Figure 8A and C). In addition, the P83S mutation had rearrangements of two conserved tryptophan residues (W121 and W146), which are among residues located at homologous assembly domains described as necessary for the formation of the γ2, α1, and β2 subunits interfaces (Srinivasan et al., 1999; Klausberger et al., 2000; Sarto et al., 2002). On the other hand, despite the fact that the N79S mutation had the largest Cα RMSD at the β3-β4 loop (Cα RMSD of 0.43 ± 0.04 Å for residues V142-P148), which formed part of an assembly motif between γ2 and α1 subunits (Klausberger et al., 2000; Sarto et al., 2002), this structural rearrangement did not seem as important for the stability of the receptor as it was not accompanied by additional structural changes in other loops nor in the core of the subunit. This could be due to differences in side-chain rearrangements between a buried-polar mutation at site 83 (P83S) and an exposed-polar mutation at site 79 (N79S), resulting in a somewhat smaller pocket for surrounding interactions and formation of buried polar interactions for P83S. It seems that mutations at positions 82 and 83 were less “tolerated” than at position 79, resulting in R82 and P83 contributing the most to interactions at the γ2 interfaces and the core of the subunit. Overall, both R82Q and P83S mutations caused the most disruptive rearrangements at both γ+/β− and α+/γ− interfaces and were less structurally “tolerated” than N79S. These findings are in agreement with our functional data, which demonstrated that R82Q and P83S mutations were less tolerated with marked impairment of receptor function and assembly, and in contrast, the N79S mutation was located two-three residues away and had minimal effects on GABAA receptor function and assembly.
How do the GABRG2(R82Q) and GABRG2(P83S) mutations contribute to epileptogenesis?
In contrast to the γ2 subunit N79S mutation, the P83S mutation was as detrimental to GABAA receptor function as the R82Q mutation. Heterozygous knock-in mice carrying the R82Q mutation developed absence and febrile seizures, recapitulating patients phenotype (Tan et al., 2007). Compared to a hypomorphic allele, the R82Q mutation increased seizure susceptibility indicating that it had effects in addition to haploinsufficiency (Chiu et al., 2008). A recent study also suggested that γ2 subunits haploinsufficiency could account for genesis of absence seizures in γ2R82Q/+ knock-in mice but not the increased thermal seizure susceptibility, while the R82Q mutation increased thermal seizure susceptibility, independent of genetic background (Reid et al., 2013).
There is controversy concerning whether mutant γ2(R82Q) subunits have dominant negative effects on partnering α1 and β2 subunits (Hales et al., 2005; Kang et al., 2006; Eugene et al., 2007; Frugier et al., 2007). Here we determined the effects of mutant γ2 subunits on surface expression of α1 and β2 subunits using flow cytometry, which is more quantifiable and sensitive. Surface α1 levels were significantly reduced by all three mutant subunits when coexpressed with β2γ2S subunits, but not when coexpressed with β2γ2L subunits. The dominant negative effects may have been caused by the formation of dimers between mutant γ2 subunits and α1 or β2 subunits, preventing formation of αβ receptors and trapping partnering subunits in the ER. It could also have been caused by internalization of receptors containing these mutations (Chaumont et al., 2013), which would result in subunit degradation through the endosome/lysosome pathway. We did not observe a significant decrease of surface β2 subunits, probably because there were increased αβ receptors on the cell surface, which would increase surface β2 subunit levels. Compared to γ2L subunits, γ2S subunits can be trafficked to the cell surface in the absence of α and β subunits (Kofuji et al., 1991; Connolly et al., 1999; Boileau et al., 2010), probably by forming trafficking-competent homopentamers. With excess γ2 subunits, unassembled γ2L subunits are retained in the ER, while γ2S subunits assemble into pentamers and are trafficked to the cell surface. As unassembled γ2L subunits can still interact with α1 and β2 subunits, excessive wildtype γ2L subunits could produce dominant negative effects on partnering subunits similar to those caused by trafficking-incompetent mutant subunits. This could have contributed to the failure to find significant dominant-negative effects of mutant γ2L subunits.
In summary, while R82Q and P83S mutations decreased surface α1β2γ2 receptors and increased surface α1β2 receptors, they also decreased the amount of surface receptors through slight dominant negative effects, both of which would be expected to contribute to the epileptogenesis.
Implications for future treatments
Trafficking-deficient mutant proteins have been shown to be rescued by lower temperature and molecular or pharmacological chaperones (Denning et al., 1992; Thomas et al., 2003; Varga et al., 2008; Guo et al., 2012; Saxena et al., 2012; Cestele et al., 2013). Here we found that decreased incubation temperature (30°C) increased surface and total levels of wildtype and mutant γ2L subunits. Interestingly, the surface α1 subunit levels were increased only slightly and insignificantly. This demonstrated that the biogenesis of GABAA receptor subunits was complex and inefficient. Many misfolded or unassembled subunits were degraded, which were rescued by lower temperature. However, the assembly of GABAA receptor subunits was still slow at 30°C. Trafficking of mutant γ2L(R82Q) and γ2L(P83S) subunits was not further improved further compared to wildtype γ2L subunits, and the dramatic increase of surface γ2L subunits could be partially caused by increased γ2L homopentamers formed at the low temperature .
Compared to temperature-induced rescue, which affects multiple proteins, specific pharmacological chaperones may be favored. It was reported that GABAA receptor ligands could promote receptor trafficking as ligand chaperones (Eshaq et al., 2010). However, we did not find significant chaperone effects of either GABA or diazepam on wildtype or mutant receptors (data not shown). With more thorough drug screening, chemicals with specific chaperone effects on GABAA receptors may be identified and developed for future treatment of GEs.
Highlights.
R82Q and P83S are epilepsy-associated mutations that reduce surface GABAA receptors
γ2(R82Q) and γ2(P83S) subunits are poorly assembled, trapped in the ER and degraded
γ2(R82Q) and γ2(P83S) subunits reduce the trafficking of α1 and β2 subunits
N79S is likely a benign rare or susceptibility variant with only small effects
Lower temperature increased surface and total levels of γ2 subunits
Acknowledgements
Confocal microscope experiments were performed in part through the use of the VUMC Cell Imaging Shared Resource (supported by NIH grants CA68485, DK20593, DK58404, HD15052, DK59637 and EY08126).
Flow Cytometry experiments were performed in the VMC Flow Cytometry Shared Resource. The VMC Flow Cytometry Shared Resource is supported by the Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404).
This work was supported by NIH R01 NS 33300 to RLM.
REFERENCES
- Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nature methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audenaert D, et al. A novel GABRG2 mutation associated with febrile seizures. Neurology. 2006;67:687–90. doi: 10.1212/01.wnl.0000230145.73496.a2. [DOI] [PubMed] [Google Scholar]
- Baumann SW, et al. Forced subunit assembly in alpha1beta2gamma2 GABAA receptors. Insight into the absolute arrangement. The Journal of biological chemistry. 2002;277:46020–5. doi: 10.1074/jbc.M207663200. [DOI] [PubMed] [Google Scholar]
- Bianchi MT, et al. Two different mechanisms of disinhibition produced by GABAA receptor mutations linked to epilepsy in humans. J Neurosci. 2002;22:5321–7. doi: 10.1523/JNEUROSCI.22-13-05321.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boileau AJ, et al. The short splice variant of the gamma 2 subunit acts as an external modulator of GABA(A) receptor function. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:4895–903. doi: 10.1523/JNEUROSCI.5039-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cestele S, et al. Nonfunctional NaV1.1 familial hemiplegic migraine mutant transformed into gain of function by partial rescue of folding defects. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:17546–51. doi: 10.1073/pnas.1309827110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaumont S, et al. Agonist-dependent endocytosis of GABAA receptors revealed by a gamma2(R43Q) epilepsy mutation. The Journal of biological chemistry. 2013 doi: 10.1074/jbc.M113.470807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu C, et al. Developmental impact of a familial GABAA receptor epilepsy mutation. Annals of neurology. 2008;64:284–93. doi: 10.1002/ana.21440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly CN, et al. Assembly and cell surface expression of heteromeric and homomeric gamma-aminobutyric acid type A receptors. J Biol Chem. 1996;271:89–96. doi: 10.1074/jbc.271.1.89. [DOI] [PubMed] [Google Scholar]
- Connolly CN, et al. Subcellular localization and endocytosis of homomeric gamma2 subunit splice variants of gamma-aminobutyric acid type A receptors. Molecular and cellular neurosciences. 1999;13:259–71. doi: 10.1006/mcne.1999.0746. [DOI] [PubMed] [Google Scholar]
- Denning GM, et al. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–4. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- Eshaq RS, et al. GABA acts as a ligand chaperone in the early secretory pathway to promote cell surface expression of GABAA receptors. Brain research. 2010;1346:1–13. doi: 10.1016/j.brainres.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugene E, et al. GABA(A) receptor gamma 2 subunit mutations linked to human epileptic syndromes differentially affect phasic and tonic inhibition. J Neurosci. 2007;27:14108–16. doi: 10.1523/JNEUROSCI.2618-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frugier G, et al. A gamma 2(R43Q) mutation, linked to epilepsy in humans, alters GABAA receptor assembly and modifies subunit composition on the cell surface. J Biol Chem. 2007;282:3819–28. doi: 10.1074/jbc.M608910200. [DOI] [PubMed] [Google Scholar]
- Gallagher MJ, et al. The GABAA receptor alpha1 subunit epilepsy mutation A322D inhibits transmembrane helix formation and causes proteasomal degradation. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:12999–3004. doi: 10.1073/pnas.0700163104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrie GH, et al. Assembly of GABAA receptors composed of alpha1 and beta2 subunits in both cultured neurons and fibroblasts. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1997;17:6587–96. doi: 10.1523/JNEUROSCI.17-17-06587.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, et al. A422T mutation in HERG potassium channel retained in ER is rescurable by pharmacologic or molecular chaperones. Biochemical and biophysical research communications. 2012;422:305–10. doi: 10.1016/j.bbrc.2012.04.153. [DOI] [PubMed] [Google Scholar]
- Gurba KN, et al. GABRB3 mutation, G32R, associated with childhood absence epilepsy alters alpha1beta3gamma2L gamma-aminobutyric acid type A (GABAA) receptor expression and channel gating. The Journal of biological chemistry. 2012;287:12083–97. doi: 10.1074/jbc.M111.332528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales TG, et al. The epilepsy mutation, gamma2(R43Q) disrupts a highly conserved inter-subunit contact site, perturbing the biogenesis of GABAA receptors. Mol Cell Neurosci. 2005;29:120–7. doi: 10.1016/j.mcn.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Hernandez CC, et al. THE GABRA6 MUTATION, R46W, ASSOCIATED WITH CHILDHOOD ABSENCE EPILEPSY ALTERS {alpha}6{beta}2{gamma}2 and {alpha}6{beta}2{delta} GABAA RECEPTOR CHANNEL GATING AND EXPRESSION. J Physiol. 2011 doi: 10.1113/jphysiol.2011.218883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbs RE, Gouaux E. Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature. 2011;474:54–60. doi: 10.1038/nature10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, Macdonald RL. The GABAA receptor gamma2 subunit R43Q mutation linked to childhood absence epilepsy and febrile seizures causes retention of alpha1beta2gamma2S receptors in the endoplasmic reticulum. J Neurosci. 2004;24:8672–7. doi: 10.1523/JNEUROSCI.2717-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, et al. Slow degradation and aggregation in vitro of mutant GABAA receptor gamma2(Q351X) subunits associated with epilepsy. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:13895–905. doi: 10.1523/JNEUROSCI.2320-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, et al. Why does fever trigger febrile seizures? GABAA receptor gamma2 subunit mutations associated with idiopathic generalized epilepsies have temperature-dependent trafficking deficiencies. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26:2590–7. doi: 10.1523/JNEUROSCI.4243-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, et al. Detection and binding properties of GABA(A) receptor assembly intermediates. The Journal of biological chemistry. 2001a;276:16024–32. doi: 10.1074/jbc.M009508200. [DOI] [PubMed] [Google Scholar]
- Klausberger T, et al. GABA(A) receptor assembly. Identification and structure of gamma(2) sequences forming the intersubunit contacts with alpha(1) and beta(3) subunits. The Journal of biological chemistry. 2000;275:8921–8. doi: 10.1074/jbc.275.12.8921. [DOI] [PubMed] [Google Scholar]
- Klausberger T, et al. Alternate use of distinct intersubunit contacts controls GABAA receptor assembly and stoichiometry. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2001b;21:9124–33. doi: 10.1523/JNEUROSCI.21-23-09124.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofuji P, et al. Generation of two forms of the gamma-aminobutyric acid A receptor gamma 2-subunit in mice by alternative splicing. Journal of neurochemistry. 1991;56:713–5. doi: 10.1111/j.1471-4159.1991.tb08209.x. [DOI] [PubMed] [Google Scholar]
- Lachance-Touchette P, et al. Novel alpha1 and gamma2 GABAA receptor subunit mutations in families with idiopathic generalized epilepsy. The European journal of neuroscience. 2011;34:237–49. doi: 10.1111/j.1460-9568.2011.07767.x. [DOI] [PubMed] [Google Scholar]
- Lauck F, et al. RosettaBackrub--a web server for flexible backbone protein structure modeling and design. Nucleic acids research. 2010;38:W569–75. doi: 10.1093/nar/gkq369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo W LA, Hernandez CC, Gurba KN, Macdonald RL. Co-expression of γ2 Subunits Hinders Processing of N-Linked Glycans Attached to the N104 Glycosylation Sites of GABAA Receptor β2 Subunits. Neurochem Res. 2013 doi: 10.1007/s11064-013-1187-9. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo WY, et al. A conserved Cys-loop receptor aspartate residue in the M3-M4 cytoplasmic loop is required for GABAA receptor assembly. J Biol Chem. 2008;283:29740–52. doi: 10.1074/jbc.M802856200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo WY, et al. Glycosylation of {beta}2 subunits regulates GABAA receptor biogenesis and channel gating. J Biol Chem. 2010;285:31348–61. doi: 10.1074/jbc.M110.151449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald RL, Kang JQ. Molecular pathology of genetic epilepsies associated with GABAA receptor subunit mutations. Epilepsy Curr. 2009;9:18–23. doi: 10.1111/j.1535-7511.2008.01278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini C, et al. Childhood absence epilepsy and febrile seizures: a family with a GABA(A) receptor mutation. Brain. 2003;126:230–40. doi: 10.1093/brain/awg018. [DOI] [PubMed] [Google Scholar]
- Migita K, et al. Properties of a novel GABAA receptor gamma2 subunit mutation associated with seizures. Journal of pharmacological sciences. 2013;121:84–7. doi: 10.1254/jphs.12222sc. [DOI] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome research. 2001;11:863–74. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid CA, et al. Mechanisms of human inherited epilepsies. Prog Neurobiol. 2009;87:41–57. doi: 10.1016/j.pneurobio.2008.09.016. [DOI] [PubMed] [Google Scholar]
- Reid CA, et al. Multiple molecular mechanisms for a single GABAA mutation in epilepsy. Neurology. 2013 doi: 10.1212/WNL.0b013e3182872867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancar F, Czajkowski C. A GABAA receptor mutation linked to human epilepsy (gamma2R43Q) impairs cell surface expression of alphabetagamma receptors. J Biol Chem. 2004;279:47034–9. doi: 10.1074/jbc.M403388200. [DOI] [PubMed] [Google Scholar]
- Sander JW. The epidemiology of epilepsy revisited. Curr Opin Neurol. 2003;16:165–70. doi: 10.1097/01.wco.0000063766.15877.8e. [DOI] [PubMed] [Google Scholar]
- Sarto I, et al. Homologous sites of GABA(A) receptor alpha(1), beta(3) and gamma(2) subunits are important for assembly. Neuropharmacology. 2002;43:482–91. doi: 10.1016/s0028-3908(02)00160-0. [DOI] [PubMed] [Google Scholar]
- Saxena A, et al. Human heat shock protein 105/110 kDa (Hsp105/110) regulates biogenesis and quality control of misfolded cystic fibrosis transmembrane conductance regulator at multiple levels. The Journal of biological chemistry. 2012;287:19158–70. doi: 10.1074/jbc.M111.297580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwede T, et al. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–5. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, et al. Mutational analysis of GABRG2 in a Japanese cohort with childhood epilepsies. Journal of human genetics. 2010;55:375–8. doi: 10.1038/jhg.2010.47. [DOI] [PubMed] [Google Scholar]
- Srinivasan S, et al. Two invariant tryptophans on the alpha1 subunit define domains necessary for GABA(A) receptor assembly. The Journal of biological chemistry. 1999;274:26633–8. doi: 10.1074/jbc.274.38.26633. [DOI] [PubMed] [Google Scholar]
- Steinlein OK. Genetic mechanisms that underlie epilepsy. Nat Rev Neurosci. 2004;5:400–8. doi: 10.1038/nrn1388. [DOI] [PubMed] [Google Scholar]
- Tan HO, et al. Reduced cortical inhibition in a mouse model of familial childhood absence epilepsy. Proc Natl Acad Sci U S A. 2007;104:17536–41. doi: 10.1073/pnas.0708440104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D, et al. Defective protein trafficking in hERG-associated hereditary long QT syndrome (LQT2): molecular mechanisms and restoration of intracellular protein processing. Cardiovascular research. 2003;60:235–41. doi: 10.1016/j.cardiores.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Tian M, Macdonald RL. The intronic GABRG2 mutation, IVS6+2T->G, associated with childhood absence epilepsy altered subunit mRNA intron splicing, activated nonsense-mediated decay, and produced a stable truncated gamma2 subunit. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:5937–52. doi: 10.1523/JNEUROSCI.5332-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd E, et al. GABAA Receptor Biogenesis is Impaired by the γ2 Subunit Febrile Seizure-Associated Mutation, GABRG2(R177G). doi: 10.1016/j.nbd.2014.05.013. submitted. [DOI] [PubMed] [Google Scholar]
- Tretter V, et al. Stoichiometry and assembly of a recombinant GABAA receptor subtype. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1997a;17:2728–37. doi: 10.1523/JNEUROSCI.17-08-02728.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretter V, et al. Stoichiometry and assembly of a recombinant GABAA receptor subtype. J Neurosci. 1997b;17:2728–37. doi: 10.1523/JNEUROSCI.17-08-02728.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga K, et al. Enhanced cell-surface stability of rescued DeltaF508 cystic fibrosis transmembrane conductance regulator (CFTR) by pharmacological chaperones. The Biochemical journal. 2008;410:555–64. doi: 10.1042/BJ20071420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace RH, et al. Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat Genet. 2001;28:49–52. doi: 10.1038/ng0501-49. [DOI] [PubMed] [Google Scholar]