

Abstract
Rare copy number variants (CNVs) have been established as an important cause of various neurodevelopmental disorders, including intellectual disability (ID) and epilepsy. In some cases, a second CNV may contribute to a more severe clinical presentation. Here we present two siblings and their mother who have mild ID, short stature, obesity and seizures. Array CGH studies show that each affected individual has two large, rare CNVs. The first is a deletion of chromosome 16p11.2, which has been previously associated with ID and autism. The second is a 0.9 Mb deletion of 19p13.2, which results in the deletion of a cluster of zinc finger genes. We suggest that, while the 16p11.2 deletion is likely the primary cause of the obesity and ID in this family, the 19p13.2 deletion may act as a modifier of the epilepsy phenotype, which is not a core feature of the 16p11.2 deletion syndrome. We investigate the potential role of ZNF44, a gene within the deleted region, in a cohort of patients with generalized epilepsy.
INTRODUCTION
Rare copy number variants (CNVs) have been well established as important genetic causes or risk factors for intellectual disability (ID), autism, schizophrenia and epilepsy [Mefford, et al., 2012]. A subset of CNVs, including deletions of 1q21.1, 15q13.3, 16p11.2 and 16p13, appear to be risk factors for a range of disorders [Girirajan, et al., 2010, Mefford, et al., 2009]. Deletions of 16p11.2 are one of the most common causes of neurodevelopmental disorders and have been associated with autism spectrum disorder and ID as well as early-onset obesity. In addition, 24% of individuals also have seizures [Zufferey, et al., 2012]. For each of these CNVs that are associated with a variety of phenotypes, it is thought that are additional genetic, epigenetic or environmental factors that influence phenotypic expression. One possible modifying factor is the presence of a second CNV [Girirajan, et al., 2010], referred to as the “two-hit” model. For example, in affected children with an inherited CNV tend to have a second large CNV [Girirajan, et al., 2012]. We report a family in which a mother and her two children have generalized epilepsy, developmental delays, intellectual disability and short stature. Array CGH revealed two rare, large microdeletion events in all three affected individuals, neither of which was present in the unaffected maternal grandmother. We propose that both deletions contribute to the phenotype in affected individuals and identify a potential candidate gene for epilepsy.
MATERIALS AND METHODS
Human subjects
Patient samples were obtained with informed consents following the University of Iowa, University of Washington and University of Melbourne Internal Review Board criteria and the Declaration of Helsinki criteria. Samples were de-identified as per these criteria. Clinical history and physical exam findings were obtained from review of scheduled patient visits. Stanford-Binet testing was performed in the research setting.
Array comparative genomic hybridization (CGH)
The proband was evaluated by oligonucleotide array CGH using a whole genome array (Agilent Sure Print G3 Human CGH 1 M array, Agilent Technologies, Santa Clara, CA). Data were analyzed using Genome Workbench software according to manufacturer’s instructions and all potential copy number changes were also visually inspected.
Sanger Sequencing
We carried out Sanger sequencing of all coding exons of the ZNF44 gene on chromosome 19p13.2 (primers available upon request). We evaluated 125 samples with generalized epilepsy, 44 of which were also confirmed to have mild intellectual disability. Sequence was compared to the reference genome (hg19).
RESULTS
Medical and family history
The three affected individuals in this family all presented with seizures in childhood (Figure 1). Seizure activity was most noticeable during intercurrent illnesses. EEGs for each show rare generalized interictal discharges with occasional multifocal discharges. All three affected individuals also have short stature (<3rd centile) and obesity. All have IQs (Stanford-Binet) from 60–80 (mother 80, daughter 64, son 60) and each lives in a group home setting, requiring assistance with most activities of daily living outside of dressing and feeding themselves. None of the three is able to read, and none have held a job. The maternal grandmother in the family is responsible for medical decision-making. There were no autistic features. Per mother’s report, the father was of normal stature and worked in construction. The father died at less than 50 years of age, though the exact age and cause were unknown. No further information was available from the father, and no other family members were available for questioning, examination, or analysis.
FIG. 1.
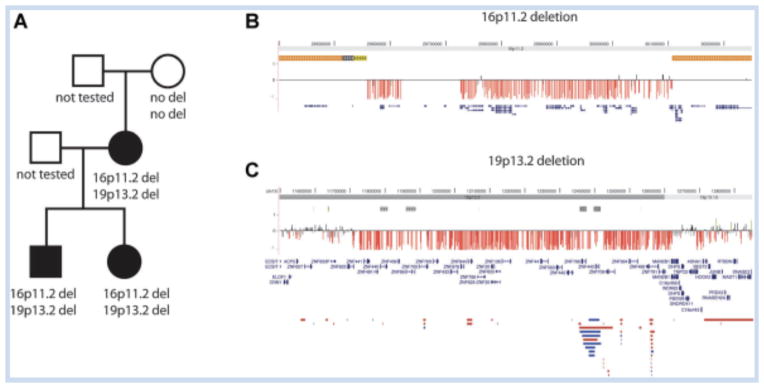
A: Pedigree of family discussed in the text. Black shading indicates intellectual disability and generalized epilepsy. B and C: Two deletions identified in the proband (data shown), sibling and mother (not shown). Solid horizontal bars at the bottom indicated CNVs in controls from the Database of Genomic Variants (http://projects.tcag.ca/variation/). Blue bars represent duplications, red bars represent deletions and brown bars indicate that both deletions and duplications have been seen.
Array CGH results
Array CGH in the proband revealed two large deletions: one on chromosome 16p11.2 (chr16: 29.5–30.2 Mb, hg19 coordinates) and one at 19p13.2 (chr19: 11.7–12.6 Mb). Follow-up studies in the affected sibling and affected mother revealed that both deletions are present in all three affected family members. Neither is present in the unaffected maternal grandmother. The father and maternal grandfather were not available for testing.
Sanger sequencing results: ZNF44
Because of the rarity of 19p13.2 deletions reported in the literature and the paucity of common copy number variants in the region, we wondered whether one or more genes in the region might contribute to epilepsy susceptibility. Because it is expressed in the brain, and because there are no CNVs involving the gene, we selected ZNF44 for sequencing in 125 individuals with GGE, some of which also have intellectual disability. We identified four rare nonsynonymous single nucleotide variants in three patients, each of which is present in <1% of control exomes; we did not detect any variants that are completely absent in controls (Table 1).
Table 1.
single nucleotide variants in ZNF44 identified in 125 samples
| Patient ID | Genomic Position (hg19) | cDNA Position | Ref. Allele | Alt. Allele | SNP ID | AA Change | Frequency in EVS | PolyPhen |
|---|---|---|---|---|---|---|---|---|
| T19969 | chr19:12386800 | 245 | G | C | rs137984148 | Arg 82 Pro | 47/10711 | 0.005 |
| T19969 | chr19:12384411 | 803 | G | A | rs142288021 | Thr 268 Ile | 48/10710 | 1.00 |
| T16233 | chr19:12383893 | 1321 | C | T | -- | His 441 Tyr | 4/10754 | 0.966 |
| EP-093 | chr19:12384105 | 1109 | G | A | rs144293271 | Gly 370 Glu | 20/10738 | 0.962 |
EVS, Exome Variant Server (http://evs.gs.washington.edu/EVS/)
DISCUSSION
We report a family with three affected individuals who have a history of developmental delays, ID, generalized epilepsy, short stature and obesity. We identified two rare deletions in all affected individuals that likely contribute to their clinical presentation.
Deletions of 16p11.2 have been associated with a range of phenotypes, including autism spectrum disorders, developmental delay and obesity [Bijlsma, et al., 2009, Kumar, et al., 2008, Shinawi, et al., 2010, Walters, et al., 2010, Weiss, et al., 2008]. Mean IQ for deletion carriers is 76, on average 32 points lower than the IQ in non-carrier relatives [Zufferey, et al., 2012]. In addition, individuals with 16p11.2 deletions appear to have an increased risk of developing seizures. In three studies, three of 14 [Bijlsma, et al., 2009] two of 18 [Rosenfeld, et al., 2009] and five of 16 [Shinawi, et al., 2010] patients had seizures or abnormal EEG findings. In a recent, large study of 285 deletion carriers, 24% suffered from seizures [Zufferey, et al., 2012]. On the other hand, the deletion is not a common finding in cohorts of patients who present with epilepsy as a primary diagnosis. For example, the 16p11.2 deletion was reported in only one of 1234 patients with GGE [de Kovel, et al., 2010], and was not seen in 517 patients with various epilepsy syndromes [Mefford, et al., 2010] or 315 patients with epileptic encephalopathies [Mefford, et al., 2011], although the reciprocal duplication was present in1/517 [Mefford, et al., 2010] and 2/315 [Mefford, et al., 2011] patients. The reciprocal duplication has also been reported in a child with West syndrome [Bedoyan, et al., 2010]. All three affected individuals in the family studied here have mild ID and require assistance with activities of daily living. In addition they all suffer from generalized seizures and obesity. Given the phenotypes associated with the 16p11.2 deletion, it is highly likely that the deletion is a significant contributor to both of those phenotypes.
The three affected individuals in this family also carry a 0.9 Mb deletion encompassing 26 genes on chromosome 19p13.2. All of the deleted genes except for MAN2B1, which is partially deleted, are zinc finger genes. Constitutional deletions of 19p13.2 are rarely reported. In the DECIPHER database (http://decipher.sanger.ac.uk/) there are no patients with deletions encompassing the same region that we report, though there are two patients with large, partially overlapping deletions (cases 260269 with no available phenotype information and1857 with ID, behavioral problems and dysmorphic features). There is one report of a child with infantile spasms who has a de novo 0.7 Mb deletion of 19p13 that includes the CACNA1A gene [Auvin, et al., 2009], just distal to the deletion in our patients. The authors hypothesize that deletion of the CACNA1A gene may be responsible for the epilepsy phenotype in their patient. The CACNA1A gene is not included within the deleted region in our family, though it is possible that there are regulatory effects that might influence the expression of this gene.
A two-hit model for severe developmental delay has been proposed for deletions at another locus on chromosome 16, 16p12.1 [Girirajan, et al., 2010]. In patients with the 16p12 deletion, probands who had a second CNV >500 kb were more severely affected. We hypothesize that the second large deletion on 19p13.2 in all affected individuals is contributing the severity and nature of the phenotype. Because generalized epilepsy is not a primary phenotype associated with deletions of 16p11.2, we investigated whether genes within the 19p13.2 deletion might be involved in epilepsy susceptibility. ZNF44 encodes a zinc finger protein also known as gonadotropin-inducible transcription factor (GIOT-2). In yeast two-hybrid screens, ZNF44 binds CSNK2B, the beta subunit of the ubiquitous serine/threonine kinase CK2, which is abundant in developing nervous tissue [Blanquet, 2000, Lehner, et al., 2004]. In addition, there are no CNVs involving this gene in control studies to date. We therefore considered this gene a reasonable candidate and performed sequence analysis of all coding exons in patients with generalized epilepsy with or without ID. While we identified four variants that are rare (<1% frequency) in the general population, we did not identify any variants that are absent from control studies. While ZNF44 is a reasonable candidate, it is possible that other genes in the region are involved as well.
Taken together, these data suggest that the combination of a recurrent CNV at 16p11.2 and a rare CNV at 19p13.2 is responsible for the clinical phenotype in this family and suggest that deletions encompassing ZNF44 and/or rare variants in ZNF44 may contribute to the risk of epilepsy in the general population. Ongoing studies genotyping large cohorts of epilepsy patients (http://www.epgp.org/epi4k/ and others) will help clarify the contribution of genes within both the 16p11.2 and 19p13 loci to epilepsy, and may suggest novel diagnostic and therapeutic targets.
References
- Auvin S, Holder-Espinasse M, Lamblin MD, Andrieux J. Array-CGH detection of a de novo 0.7-Mb deletion in 19p13.13 including CACNA1A associated with mental retardation and epilepsy with infantile spasms. Epilepsia. 2009;50:2501–3. doi: 10.1111/j.1528-1167.2009.02189.x. [DOI] [PubMed] [Google Scholar]
- Bedoyan JK, Kumar RA, Sudi J, Silverstein F, Ackley T, Iyer RK, Christian SL, Martin DM. Duplication 16p11.2 in a child with infantile seizure disorder. Am J Med Genet A. 2010;152A:1567–74. doi: 10.1002/ajmg.a.33415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijlsma EK, Gijsbers AC, Schuurs-Hoeijmakers JH, van Haeringen A, Fransen van de Putte DE, Anderlid BM, Lundin J, Lapunzina P, Perez Jurado LA, Delle Chiaie B, Loeys B, Menten B, Oostra A, Verhelst H, Amor DJ, Bruno DL, van Essen AJ, Hordijk R, Sikkema-Raddatz B, Verbruggen KT, Jongmans MC, Pfundt R, Reeser HM, Breuning MH, Ruivenkamp CA. Extending the phenotype of recurrent rearrangements of 16p11.2: Deletions in mentally retarded patients without autism and in normal individuals. Eur J Med Genet. 2009;52:77–87. doi: 10.1016/j.ejmg.2009.03.006. [DOI] [PubMed] [Google Scholar]
- Blanquet PR. Casein kinase 2 as a potentially important enzyme in the nervous system. Progress in neurobiology. 2000;60:211–46. doi: 10.1016/s0301-0082(99)00026-x. [DOI] [PubMed] [Google Scholar]
- de Kovel CG, Trucks H, Helbig I, Mefford HC, Baker C, Leu C, Kluck C, Muhle H, von Spiczak S, Ostertag P, Obermeier T, Kleefuss-Lie AA, Hallmann K, Steffens M, Gaus V, Klein KM, Hamer HM, Rosenow F, Brilstra EH, Trenite DK, Swinkels ME, Weber YG, Unterberger I, Zimprich F, Urak L, Feucht M, Fuchs K, Moller RS, Hjalgrim H, De Jonghe P, Suls A, Ruckert IM, Wichmann HE, Franke A, Schreiber S, Nurnberg P, Elger CE, Lerche H, Stephani U, Koeleman BP, Lindhout D, Eichler EE, Sander T. Recurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsies. Brain. 2010;133:23–32. doi: 10.1093/brain/awp262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girirajan S, Eichler EE. Phenotypic variability and genetic susceptibility to genomic disorders. Hum Mol Genet. 2010;19:R176–87. doi: 10.1093/hmg/ddq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girirajan S, Rosenfeld JA, Coe BP, Parikh S, Friedman N, Goldstein A, Filipink RA, McConnell JS, Angle B, Meschino WS, Nezarati MM, Asamoah A, Jackson KE, Gowans GC, Martin JA, Carmany EP, Stockton DW, Schnur RE, Penney LS, Martin DM, Raskin S, Leppig K, Thiese H, Smith R, Aberg E, Niyazov DM, Escobar LF, El-Khechen D, Johnson KD, Lebel RR, Siefkas K, Ball S, Shur N, McGuire M, Brasington CK, Spence JE, Martin LS, Clericuzio C, Ballif BC, Shaffer LG, Eichler EE. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. The New England journal of medicine. 2012;367:1321–31. doi: 10.1056/NEJMoa1200395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girirajan S, Rosenfeld JA, Cooper GM, Antonacci F, Siswara P, Itsara A, Vives L, Walsh T, McCarthy SE, Baker C, Mefford HC, Kidd JM, Browning SR, Browning BL, Dickel DE, Levy DL, Ballif BC, Platky K, Farber DM, Gowans GC, Wetherbee JJ, Asamoah A, Weaver DD, Mark PR, Dickerson J, Garg BP, Ellingwood SA, Smith R, Banks VC, Smith W, McDonald MT, Hoo JJ, French BN, Hudson C, Johnson JP, Ozmore JR, Moeschler JB, Surti U, Escobar LF, El-Khechen D, Gorski JL, Kussmann J, Salbert B, Lacassie Y, Biser A, McDonald-McGinn DM, Zackai EH, Deardorff MA, Shaikh TH, Haan E, Friend KL, Fichera M, Romano C, Gecz J, DeLisi LE, Sebat J, King MC, Shaffer LG, Eichler EE. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat Genet. 2010;42:203–9. doi: 10.1038/ng.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar RA, Kara Mohamed S, Sudi J, Conrad DF, Brune C, Badner JA, Gilliam TC, Nowak NJ, Cook EH, Jr, Dobyns WB, Christian SL. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet. 2008;17:628–38. doi: 10.1093/hmg/ddm376. [DOI] [PubMed] [Google Scholar]
- Lehner B, Semple JI, Brown SE, Counsell D, Campbell RD, Sanderson CM. Analysis of a high-throughput yeast two-hybrid system and its use to predict the function of intracellular proteins encoded within the human MHC class III region. Genomics. 2004;83:153–67. doi: 10.1016/s0888-7543(03)00235-0. [DOI] [PubMed] [Google Scholar]
- Mefford HC, Batshaw ML, Hoffman EP. Genomics, intellectual disability, and autism. The New England journal of medicine. 2012;366:733–43. doi: 10.1056/NEJMra1114194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford HC, Eichler EE. Duplication hotspots, rare genomic disorders, and common disease. Curr Opin Genet Dev. 2009;19:196–204. doi: 10.1016/j.gde.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford HC, Muhle H, Ostertag P, von Spiczak S, Buysse K, Baker C, Franke A, Malafosse A, Genton P, Thomas P, Gurnett CA, Schreiber S, Bassuk AG, Guipponi M, Stephani U, Helbig I, Eichler EE. Genome-wide copy number variation in epilepsy: novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genetics. 2010;6:e1000962. doi: 10.1371/journal.pgen.1000962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford HC, Yendle SC, Hsu C, Cook J, Geraghty E, McMahon JM, Eeg-Olofsson O, Sadleir LG, Gill D, Ben-Zeev B, Lerman-Sagie T, Mackay M, Freeman JL, Andermann E, Pelakanos JT, Andrews I, Wallace G, Eichler EE, Berkovic SF, Scheffer IE. Rare copy number variants are an important cause of epileptic encephalopathies. Annals of neurology. 2011;70:974–85. doi: 10.1002/ana.22645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld JA, Coppinger J, Bejjani BA, Girirajan S, Eichler EE, Shaffer LG, Ballif BC. Speech delays and behavioural problems are the predominant features in individuals with developmental delays and 16p11.2 microdeletions and microduplications. J Neurodevelop Disord. 2009;2:26–38. doi: 10.1007/s11689-009-9037-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinawi M, Liu P, Kang SH, Shen J, Belmont JW, Scott DA, Probst FJ, Craigen WJ, Graham BH, Pursley A, Clark G, Lee J, Proud M, Stocco A, Rodriguez DL, Kozel BA, Sparagana S, Roeder ER, McGrew SG, Kurczynski TW, Allison LJ, Amato S, Savage S, Patel A, Stankiewicz P, Beaudet AL, Cheung SW, Lupski JR. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J Med Genet. 2010;47:332–41. doi: 10.1136/jmg.2009.073015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters RG, Jacquemont S, Valsesia A, de Smith AJ, Martinet D, Andersson J, Falchi M, Chen F, Andrieux J, Lobbens S, Delobel B, Stutzmann F, El-Sayed Moustafa JS, Chevre JC, Lecoeur C, Vatin V, Bouquillon S, Buxton JL, Boute O, Holder-Espinasse M, Cuisset JM, Lemaitre MP, Ambresin AE, Brioschi A, Gaillard M, Giusti V, Fellmann F, Ferrarini A, Hadjikhani N, Campion D, Guilmatre A, Goldenberg A, Calmels N, Mandel JL, Le Caignec C, David A, Isidor B, Cordier MP, Dupuis-Girod S, Labalme A, Sanlaville D, Beri-Dexheimer M, Jonveaux P, Leheup B, Ounap K, Bochukova EG, Henning E, Keogh J, Ellis RJ, Macdermot KD, van Haelst MM, Vincent-Delorme C, Plessis G, Touraine R, Philippe A, Malan V, Mathieu-Dramard M, Chiesa J, Blaumeiser B, Kooy RF, Caiazzo R, Pigeyre M, Balkau B, Sladek R, Bergmann S, Mooser V, Waterworth D, Reymond A, Vollenweider P, Waeber G, Kurg A, Palta P, Esko T, Metspalu A, Nelis M, Elliott P, Hartikainen AL, McCarthy MI, Peltonen L, Carlsson L, Jacobson P, Sjostrom L, Huang N, Hurles ME, O’Rahilly S, Farooqi IS, Mannik K, Jarvelin MR, Pattou F, Meyre D, Walley AJ, Coin LJ, Blakemore AI, Froguel P, Beckmann JS. A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature. 2010;463:671–5. doi: 10.1038/nature08727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H, Ferreira MA, Green T, Platt OS, Ruderfer DM, Walsh CA, Altshuler D, Chakravarti A, Tanzi RE, Stefansson K, Santangelo SL, Gusella JF, Sklar P, Wu BL, Daly MJ. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358:667–75. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- Zufferey F, Sherr EH, Beckmann ND, Hanson E, Maillard AM, Hippolyte L, Mace A, Ferrari C, Kutalik Z, Andrieux J, Aylward E, Barker M, Bernier R, Bouquillon S, Conus P, Delobel B, Faucett WA, Goin-Kochel RP, Grant E, Harewood L, Hunter JV, Lebon S, Ledbetter DH, Martin CL, Mannik K, Martinet D, Mukherjee P, Ramocki MB, Spence SJ, Steinman KJ, Tjernagel J, Spiro JE, Reymond A, Beckmann JS, Chung WK, Jacquemont S. A 600 kb deletion syndrome at 16p11.2 leads to energy imbalance and neuropsychiatric disorders. Journal of medical genetics. 2012;49:660–8. doi: 10.1136/jmedgenet-2012-101203. [DOI] [PMC free article] [PubMed] [Google Scholar]