

Abstract
A long-standing literature linking endocannabinoids (ECBs) to stress, fear, and anxiety has led to growing interest in developing novel anxiolytics targeting the ECB system. Following rapid on-demand biosynthesis and degradation upon neuronal activation, the ECB N-arachidonoylethanolamide (anandamide, AEA) is actively degraded by the serine hydrolase enzyme, fatty acid amide hydrolase (FAAH). Exposure to stress rapidly mobilizes FAAH to deplete the signaling pool of AEA and increase neuronal excitability in a key anxiety-mediating region – the basolateral amygdala (BLA). Gene deletion or pharmacological inhibition of FAAH prevents stress-induced reductions in AEA and associated increases in BLA dendritic hypertrophy and anxiety-like behavior. Additionally, inhibition of FAAH facilitates long-term fear extinction and rescues deficient fear extinction in rodent models by enhancing AEA–CB1 (cannabinoid type 1) receptor signaling and synaptic plasticity in the BLA. These preclinical findings propose restoring deficient BLA AEA levels by pharmacologically inhibiting FAAH as a mechanism to therapeutically mitigate the effects of traumatic stress.
Keywords: endocannabinoid, post-traumatic stress disorder, anxiety, fear, depression, 2-AG
Cannabis, endocannabinoids, and anxiety
Cannabis is one of the most widely used drugs in the world, with historical records dating use in Eastern cultures back millennia [1]. Cannabis and its derivatives have profound effects on a wide variety of behavioral and neural functions, ranging from feeding and metabolism to pain and cognition [2]. However, epidemiological studies have indicated that the most common self-reported reason for using cannabis is rooted in its ability to reduce feelings of stress, tension, and anxiety [3]. Significant numbers of people may be self-medicating with cannabis in an attempt to reduce excessive anxiety [4,5] (even though cannabis use can also cause paranoia and heightened anxiety in certain situations and predisposed individuals, depending on the dose [6]). Furthermore, studies in controlled clinical settings confirm that administration of synthetic variants of delta-9-tetrahydrocannabinol (THC), the psychoactive constituent of cannabis, can reduce anxiety in patients with anxiety disorders [7,8]. Finally, the anxiety-reducing properties of THC extend to preclinical rodent assays and models [9], demonstrating that the anxiolytic properties of cannabinoids are well conserved across species.
Many of the neural and behavioral effects of exogenously administered cannabinoids can be traced directly to activation of the `endocannabinoid' (ECB) system [10] – a set of neurochemicals and cognate receptors densely expressed throughout the brain [11]. The discovery of the ECB system raised the possibility that ECBs could be important modulators of anxiety, and might contribute to individual differences in anxious temperament and risk for anxiety disorders. It also led to the notion that targeting components of the ECB system could represent a novel therapeutic approach to developing effective anxiolytic medications devoid of the unwanted effects of cannabis (e.g., cognitive impairment, abuse liability) [4,12] (Box 1). That is, selective alleviation of symptoms could be produced by increasing levels of endogenously activated ECBs and avoiding widespread behavioral effects caused by exogenously applied, ubiquitously activating cannabinoid receptor type 1 (CB1R) agonists.
Box 1. Developing selective targets for AEA and 2-AG.
AEA and 2-AG have differential roles in ECB signaling, with AEA providing a tonic signal that offsets excess excitability and 2-AG producing more of the phasic ECB response [55]. AEA is a relatively low efficacy agonist, whereas 2-AG has much higher agonist efficacy and induces a robust CB1R G protein mediated response [55]. Drugs that act on the catabolic enzymes for each ECB (FAAH for AEA, MAGL for 2-AG) provide one excellent approach to selectively target each ECB for therapeutic applications, as biochemical studies support the feasibility of inhibiting FAAH without altering 2-AG levels and blocking MAGL without affecting AEA [55,108]. Reflecting the earlier discovery of FAAH as the hydrolytic enzyme for AEA, FAAH inhibitors have been well studied to date and appear to influence a more limited range of behaviors than MAGL blockers [52]. This could translate into a more favor side effect profile. Chronically elevating AEA with FAAH inhibitors may also be less prone to CB1R desensitization and the associated issue of behavioral tolerance that is likely with MAGL blockade induced increases in 2-AG [109,110]. These unique properties suggest that inhibiting FAAH is a particularly amenable approach to developing ECB medications for various therapeutic indications, including anxiety disorders.
A substantial amount of research has been undertaken over the past decade to test these ideas. As a result, understanding the role of ECBs in controlling stress, fear, and anxiety has grown considerably in recent years, with some targets already having been advanced to preliminary clinical trials in patients. The broader literature on this topic has been covered in many excellent, comprehensive reviews [13] and will not be reiterated here. The focus of the current article is on one of the most rapidly moving developments in the field – the stress and anxiety-related role of a major modulatory component of the ECB system, known as fatty acid amide hydrolase (FAAH).
We first introduce the functional role of FAAH within the wider ECB system. Next, we discuss evidence from pharmacological and genetic approaches that loss/inhibition of FAAH function can both mitigate behavioral and neural sequelae of stress and promote learned reductions in fear via actions in the basolateral amygdala (BLA), a central node within the neural circuitry mediating stress and anxiety. We conclude with an outlook for the field going forward and pose some outstanding questions that remain to be addressed.
FAAH regulation of ECB signaling
ECBs are fatty acid amides and monoacylglyerols functioning as neuromodulator lipids that exhibit rapid (within seconds) on-demand biosynthesis in response to neuronal activation, and are subsequently degraded by specialized catabolic enzymes. There are two known receptors binding ECBs with high affinity – CB1R is the most densely expressed in the brain and is present at high levels in corticolimbic regions mediating anxiety, including the medial prefrontal cortex (mPFC) and hippocampus, as well as the BLA [11,14], whereas CB2R is mainly found in the periphery but also in some microglia and neuronal populations in the central nervous system (CNS) [15,16]. Unlike most neurotransmitters, however, ECBs are not stored in readily releasable pools, but instead are rapidly synthesized `on-demand' upon depolarization-induced calcium increase. Such biosynthesis occurs tonically and, under strong neuronal activation, phasically. The ECB 2-arachidonoylglycerol (2-AG) is synthesized, postsynaptically, by diacylglycerol lipase, whereas the ECB anandamide (AEA) is predominantly synthesized by N-acyl phosphatidylethanolamine phospholipase D (NAPE-PLD), also at postsynaptic sites in regions including the BLA, but also presynaptically at others (e.g., hippocampus) [17,18]. Following their synthesis, ECBs are retrogradely transported into the extracellular space to bind ECB receptors present on presynaptic terminals [19–21].
Stimulation of CB1R recruits various signaling pathways [22]. These pathways include CB1R coupling to Gi/o proteins that reduces adenylyl cyclase activity and downregulates cyclic AMP/protein kinase A signaling [23], by Gβγ-induced phospholipase C-β-mediated increases in intracellular calcium influx, and by activation of mitogen-activated protein kinases [24,25]. In addition, CB1R negatively regulates N- and P/Q-type voltage-gated calcium channels [26] and positively regulates inwardly rectifying K+ channels [26,27], as well as protein serine/threonine phosphatase 2B (calcineurin, PP2B) to change the phosphorylation state of various effector molecules [28,29]. At this time, the precise contribution of these various signaling pathways to ECB modulation of anxiety remains essentially unknown. Enriching the picture further, the actions of AEA are not restricted to CB1R, given that ECBs also act, sometimes in a non-retrograde manner, at CB2R [30], GPR55 [31], and TRPV1 (transient receptor potential vanilloid type 1) channels [32–34], as well as other G protein subtypes such as Gs and/or Gq11 [35,36]. It should be noted that not all of these actions have been demonstrated in the BLA at the present time, and this may become an important consideration for future work given differences in CB1R signaling mechanisms across brain regions [37]. Indeed, recent evidence demonstrates that diverse effects are evident even within the extended amygdala [38], whereas other parts of the amygdala, notably central amygdala subnuclei, remain largely uncharacterized.
A key mechanism governing CB1R-mediated signaling is the active degradation of released ECBs. 2-AG is degraded presynaptically by monoacylglycerol lipase (MAGL). By contrast, the catabolic fate of a number of the N-acylethanolamine (NAE) group of ECBs, including N-palmitoyl ethanolamine (PEA) [39], N-oleoyl ethanolamine (OEA), and AEA [40], was suspected to be controlled by a common enzyme as early as 1984 [41]. This enzyme was later identified as the serine hydrolase enzyme, FAAH [42–44], which was then isolated and cloned and shown to be located postsynaptically [45]. In the rodent brain, the distribution of FAAH overlaps closely with CB1R in many but not all regions. Within amygdala subnuclei, FAAH is highly expressed on pyramidal neurons in the BLA, and to a significantly lesser extent in the central nucleus (CeA) [19,20,46,47]. Although activation of CB1R in the BLA causes a decrease in both glutamatergic and GABAergic transmission, there is typically a net reduction in neuronal excitability on application of CB1R agonists, probably due to CB1R-mediated presynaptic inhibition of glutamate release [48,49]. By contrast, CB1R signaling generated by increased AEA can also produce long-term depression of inhibitory transmission in the BLA, a change in synaptic plasticity which can serve to promote excitability [50,51].
Collectively, these observations demonstrate that FAAH is functionally positioned to modulate the actions of AEA and other NAEs on BLA neurons and anxiety processes mediated by this region. By extension, manipulations that lead to alterations in FAAH activity, such as stress or administration of compounds targeting FAAH [52], would be predicted to functionally impact BLA functions, including anxiety. We consider emerging evidence in support of this hypothesis in the next section.
FAAH modulation of anxiety and stress
There is growing evidence that tonic ECB signaling in the BLA and elsewhere is mediated by AEA, whereas phasic ECB responses to robust neuronal activation are sub-served by 2-AG [53–56]. Interestingly, stress appears to produce divergent effects on AEA and 2-AG levels in the BLA – with an elevation in 2-AG levels [57], but a rapid induction of FAAH activity and a resultant decline in the pool of AEA [58–60] were reported following exposure to various types of stress. These differing directional and temporal responses probably indicate a difference in the mechanisms modulating the effects of these two ECBs and their catabolic enzymes in the BLA. These mechanisms are not currently understood, however, and it is also unclear exactly how the relative balance between changes in AEA and 2-AG impacts the response and recovery from stress.
Notwithstanding, several recent observations demonstrate that loss of AEA–CB1R signaling in the BLA can trigger behavioral and neuroendocrine responses to stress. First, presumably by removing AEA-mediated tonic inhibitory activity, CB1R antagonism increases BLA excitability [61–63], activates the hypothalamic–pituitary–adrenal (HPA) axis and increases anxiety-like behavior [58,64,65]. Second, increasing AEA by inhibiting FAAH (with URB597) reduces the HPA axis and anxiety-like response to stress [58,66]. These actions of FAAH inhibition have been localized to actions specifically within the BLA by the finding that only infusion of URB597 into the BLA, and not CeA or medial amygdala, is effective in producing these anti-stress effects [58]. Collectively, these findings support a model in which stress rapidly mobilizes FAAH, depletes the signaling pool of AEA, and increases BLA excitability to drive anxiety [67] (Figure 1).
Figure 1.
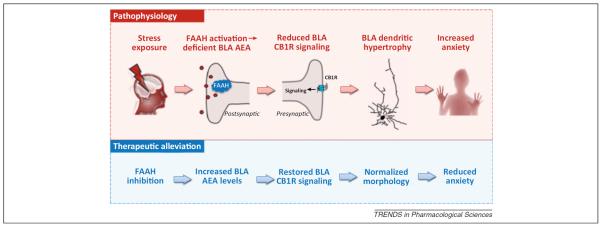
Role of FAAH in the pathophysiology and therapeutic alleviation of stress-induced anxiety. Hypothesized pathophysiological scheme by which stress activates FAAH, leading to depletion of AEA in the BLA, a reduction in CB1R signaling and dendritic hypertrophy associated with anxiety. Therapeutic alleviation of stress-induced anxiety could be achieved by inhibiting FAAH to increase BLA AEA levels to restore CB1R signaling and possibly normalize dendritic abnormal morphology. Abbreviations: FAAH, fatty acid amide hydrolase; AEA, anandamide; BLA, basolateral amygdala; CB1R, cannabinoid receptor type 1.
Disruption of FAAH and AEA may be exacerbated under conditions of chronic stress. Rodents exposed to chronic stress show sustained enhancement of FAAH activity and prolonged reductions in tissue levels of BLA AEA that persist beyond exposure [59,68,69]. Additionally, multiple studies have found that chronic stress results in an increase in dendritic arborization and spinogenesis on BLA pyramidal neurons, increasing their intrinsic excitability and afferent stimulation in a manner that correlates with heightened anxiety-like behavior [70–72]. These morphological changes are absent in mutant mice lacking FAAH [68], whereas genetic loss or repeated, pre-stress pharmacological inhibition (JNJ5003) of FAAH prevents stress-induced reductions in AEA and increases in HPA activity and anxiety-like behavior [68,73,74]. Accordingly, inhibition of FAAH can potently ameliorate multiple sequelae of chronic stress by enhancing AEA signaling and restoring BLA dysfunction (Figure 1).
The contribution of BLA 2-AG to stress responsivity and recovery remains, in contrast to FAAH–AEA signaling, much less certain at the present time. As noted above, stress increases 2-AG in the BLA, which could suggest a protective/restorative role to counter the effects of stress-induced AEA depletion. In this context, systemically administered MAGL inhibitors have been shown to acutely reduce anxiety-like behavior [75–78], and when administered chronically, to prevent chronic stress-induced increases in anxiety-like behavior and impairments in BLA synaptic plasticity [75]. Further studies will be needed to determine the role of MAGL–2-AG signaling in moderating stress and the manner in which this mechanism interacts with the FAAH–AEA system in the BLA. Indeed, these relationships may be further complicated by sex differences in the ECB system and ECB mediation of anxiety (Box 2).
Box 2. Sex differences in ECB and FAAH modulation of anxiety.
There are known sex differences in the function of the ECB system that could impact how manipulation of CB1R and FAAH affect anxiety-related behaviors in rodents. There are some notable differences between males and females in CB1R-binding site density, including greater binding in the BLA of female rats [111] and across limbic regions in human female subjects [107]. Despite these differences, FAAH inhibitors retain anxiolytic- and antidepressant-like effects in ovariectomized female rats [112]. Interestingly, however, anxiolytic- and antidepressant-like effects produced by estradiol administration are attenuated by CB1R blockade [112], whereas estradiol administration increases AEA levels [113] or AEA signaling [114], possibly via downregulation of FAAH driven by an estrogen response element on the FAAH gene that suppresses FAAH transcription when bound by estrogen [115]. These observations suggest that FAAH–AEA signaling may be one important mechanism linking gonadal hormone status and account, at least in part, for sex differences in anxiety. Given the preponderance of diagnosed anxiety disorders in women, these emerging preclinical data will represent an important consideration of the potential clinical application of FAAH inhibitors for anxiety.
FAAH mediation of fear and extinction
In addition to being a major target and modulator of stress, the BLA is a critical node within the neural circuitry subserving learned fear behaviors. The BLA is activated during the formation, expression, and extinction of conditioned fear memories, and damage to the BLA disrupts one or more of these processes [79,80]. Implicating BLA ECB signaling in fear conditioning, several studies have shown that injecting a CB1R agonist (WIN55212-2) into the BLA enhanced the consolidation of conditioned fear (but impaired fear reconsolidation [81]), whereas CB1R antagonism/inverse agonism (AM251) had the opposite effect, impairing fear memory formation [82]. These effects on fear are not restricted to the BLA but involve a pathway between the BLA and mPFC. Responses to fear cues in mPFC neurons receiving inputs from BLA were potentiated by the CB1R agonist WIN55212-2, and CB1R blockade/inverse agonism (with AM251) in these regions disrupts fear learning and learning-related synaptic plasticity (long-term potentiation) [83–85].
Extending these findings and demonstrating a role for ECBs in fear extinction, Marsicano and colleagues reported elevated AEA and 2-AG BLA levels after extinction training in mice [86], and produced impairments in extinction learning (but not fear conditioning) by CB1R KO or systemic antagonist (SR141716A) administration [28,86] (for studies reporting similar effects, see [87–89]). Subsequent studies showed that infusing SR141716A directly into the BLA (or mPFC [90]) was sufficient to impair a cued fear extinction in rats [91,92]. Some authors have posited that these effects on extinction reflect a more general role of ECBs in promoting the long-term adaptation to aversion situations [47,93,94]. The ability of ECBs to modulate fear extinction is bidirectional. Activating CB1R via systemic or intra-hippocampal or -mPFC administration of either the CB1R agonist, WIN55212-2, the ECB reuptake blocker, AM404, or AEA itself has been shown to facilitate rodent fear extinction, in most cases without affecting conditioned fear [89,95–98] (but see [90]). Initial work has found that similar effects can be produced in humans by administering THC [99].
The ability of ECBs to facilitate fear extinction is recapitulated by selective inhibition of FAAH. Rats infused with the FAAH inhibitor, URB597, into the mPFC prior to extinction training show improved extinction retrieval when subsequently tested drug-free [90]. More recently, systemic administration of a novel, potent FAAH inhibitor, AM3506 [100], prior to extinction training augmented extinction-induced BLA AEA levels and reduced fear on extinction retrieval in a mouse model of impaired extinction [50]. Although this drug did not reduce fear during extinction training, reduced fear on extinction retrieval is not contingent upon observable facilitation of extinction learning [94,101]. The effects of AM3506 were attributed to the BLA by the finding that infusing the drug directly into the BLA promoted extinction, and the demonstration that intra-BLA infusion of SR141716A blocked the pro-extinction effect of systemically delivered AM3506 [50]. Thus, these data show that CEA–CB1R signalling in the BLA is both necessary and sufficient for FAAH inhibitors to facilitate fear extinction. Interestingly, there is preliminary evidence that analogous effects may result from genetically driven variation in FAAH in human subjects (Box 3).
Box 3. FAAH gene variation and risk for anxiety disorders.
Preclinical findings linking FAAH activity to stress and anxiety in rodents is supported by emerging evidence that gene variation in human FAAH may moderate risk for anxiety disorders by regulating amygdala processing of threat. A common (~25% in those of Caucasian ancestry [116]) single nucleotide polymorphism (C385A; rs324420) that results in the conversion of a conserved proline residue to threonine (P129T) in the amino acid sequence of the FAAH gene is associated with reduced expression of FAAH in lymphocytes and elevations in circulating levels of AEA [117,118]. Studies using BOLD functional magnetic resonance imaging have found that people with this gene variant had significantly less amygdala responses to threat, in the form of fearful faces, and trait anxiety levels [119] (but see [120]). Furthermore, in parallel with increased amygdala plasticity produced by FAAH inhibitors in mice [50], the same gene variant predicted more rapid habituation of amygdala responses to repeated threat [50]. These preliminary observations are in line with preclinical evidence that FAAH modulates amygdala responses to stress and fear extinction encoding, and lend support for a model in which increased FAAH works to mitigate the effects of stress.
The mechanisms underlying the pro-extinction effects of BLA–FAAH inhibition remain to be elucidated. One notable finding is that a CB1R-dependent form of synaptic plasticity in BLA [51,86], long-term depression of inhibitory GABAergic transmission (LTDi), is enhanced by FAAH KO [51] and by the FAAH inhibitor AM3506 at doses that promote extinction [50]. This suggests a scheme in which AEA released during extinction relieves a tonic inhibitory brake on BLA output neurons [50,51,86] necessary for the encoding of extinction memories [102] (Figure 2). However, it would be premature to discount the involvement of other mechanisms, including more direct effects on glutamatergic transmission, and this remains a key question for future work.
Figure 2.

Putative mechanism mediating fatty acid amide hydrolase (FAAH) inhibitor effects on fear extinction. Following formation of a fear memory, systemic administration of a FAAH inhibitor increases anandamide (AEA) levels in the basolateral amygdala (BLA) and increases cannabinoid receptor type 1 (CB1R) signaling to enhance attenuation of GABAergic transmission via long-term depression of inhibitory transmission (LTDi) [50]. Lesser inhibition may remove a brake on the activity/plasticity of BLA neurons [102] recruited to encode extinction, allowing FAAH to gate the formation of extinction memories.
Another important question for future studies will be whether FAAH inhibitors work to normalize impairments in fear extinction that are known to be produced by environmental insults such as exposure to abused drugs and stress [97,103]. Indeed, several authors have posited utility of FAAH inhibitors for drug and alcohol addiction [104], and the effects related to stress are particularly pertinent given the protective effects of boosting ECBs (by inhibiting FAAH) for stress-induced anxiety-related behaviors discussed above. Interestingly in this context, deficient fear extinction caused by a restraint/swim stressor was recently shown to be rescued by intra-BLA (or hippocampal, but not mPFC) injection of a CB1R agonist (WIN55212-2) immediately after stress [105,106]. Demonstrating similar effects with selective FAAH inhibitors would support the utility of these compounds across a range of extinction-impaired settings.
Concluding remarks
Preclinical studies offer strong support for a major role of FAAH, via effects on BLA AEA–CB1R signaling, in modulating stress-induced anxiety and fear extinction. Moreover, recent clinical reports have demonstrated decreased peripheral levels of AEA and increases in brain CB1R-binding in patients with anxiety disorders, such as post-traumatic stress disorder [107]. Taken together, these findings encourage the development of novel anxiolytics based around restoring deficient AEA levels by pharmacologically inhibiting FAAH. Encouragingly, preliminary clinical trials with selective FAAH inhibitors, including PF04457845 and URB597, are either underway (www.clinicaltrials.gov/ct2/show/NCT01665573) or being planned. The `on-demand' nature of ECB release makes it a particularly attractive target for drug development because FAAH inhibitors would selectively augment CB1R signaling in neural circuits where AEA was recruited [52]. This refined mechanism of action would avoid the widespread activity of THC or CB1R agonism and, as a result, is expected to produce fewer clinically unwanted side effects and be less liable to CB1R downregulation after repeated dosing. Although this prediction awaits thorough clinical investigation, and some important questions also remain to be addressed (Box 4), the field is at an exciting juncture and has genuine promise for advancing our understanding and treatment of anxiety disorders.
Box 4. Outstanding questions.
What role does the FAAH–AEA–CB1R signaling system play in modulating the fear and anxiety-related functions of corticolimbic regions beyond the BLA, including the CeA, mPFC, and hippo-campus?
How does 2-AG-related ECB signaling contribute to stress and extinction? And do 2-AG and AEA work in tandem or opposition to modulate these behaviors?
The ECB system is perturbed by various environmental insults other than stress, including chronic alcohol exposure and traumatic brain injury. Does loss of FAAH function contribute to the fear- and anxiety-related abnormalities [103,121] associated with these insults?
AEA regulates a wide range of behaviors in addition to stress and anxiety [2]. To what extent does amygdala FAAH contribute to behaviors, such as reward seeking, that are known to be subserved by the amygdala?
What are the key upstream and downstream molecular mechanisms regulating FAAH activity? Elucidating these mechanisms could suggest new (e.g., pharmacological) approaches to modulating FAAH activity.
Does treatment with FAAH inhibitors facilitate extinction in human subjects and/or normalize impaired extinction in patients with anxiety disorders?
Acknowledgments
O.G-C. and A.H. are supported by the NIAAA Intramural Research Program and Department of Defense in the Center for Neuroscience and Regenerative Medicine. M.N.H. is the recipient of a Tier II Canada Research Chair and operating funds from the Canadian Institutes of Health Research.
References
- 1.Aggarwal SK, et al. Medicinal use of cannabis in the United States: historical perspectives, current trends, and future directions. J. Opioid Manag. 2009;5:153–168. doi: 10.5055/jom.2009.0016. [DOI] [PubMed] [Google Scholar]
- 2.Katona I, Freund TF. Multiple functions of endocannabinoid signaling in the brain. Annu. Rev. Neurosci. 2012;35:529–558. doi: 10.1146/annurev-neuro-062111-150420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheung JT, et al. Anxiety and mood disorders and cannabis use. Am. J. Drug Alcohol Abuse. 2010;36:118–122. doi: 10.3109/00952991003713784. [DOI] [PubMed] [Google Scholar]
- 4.Van Dam NT, et al. Characteristics of clinically anxious versus non-anxious regular, heavy marijuana users. Addict. Behav. 2012;37:1217–1223. doi: 10.1016/j.addbeh.2012.05.021. [DOI] [PubMed] [Google Scholar]
- 5.Agosti V, et al. Rates of psychiatric comorbidity among U.S. residents with lifetime cannabis dependence. Am. J. Drug Alcohol Abuse. 2002;28:643–652. doi: 10.1081/ada-120015873. [DOI] [PubMed] [Google Scholar]
- 6.Thomas H. Psychiatric symptoms in cannabis users. Br. J. Psychiatry. 1993;163:141–149. doi: 10.1192/bjp.163.2.141. [DOI] [PubMed] [Google Scholar]
- 7.Fabre LF, McLendon D. The efficacy and safety of nabilone (a synthetic cannabinoid) in the treatment of anxiety. J. Clin. Pharmacol. 1981;21:377S–382S. doi: 10.1002/j.1552-4604.1981.tb02617.x. [DOI] [PubMed] [Google Scholar]
- 8.Fraser GA. The use of a synthetic cannabinoid in the management of treatment-resistant nightmares in posttraumatic stress disorder (PTSD) CNS Neurosci. Ther. 2009;15:84–88. doi: 10.1111/j.1755-5949.2008.00071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moreira FA, Wotjak CT. Cannabinoids and anxiety. Curr. Top. Behav. Neurosci. 2010;2:429–450. doi: 10.1007/7854_2009_16. [DOI] [PubMed] [Google Scholar]
- 10.Mechoulam R, Parker LA. The endocannabinoid system and the brain. Annu. Rev. Psychol. 2013;64:21–47. doi: 10.1146/annurev-psych-113011-143739. [DOI] [PubMed] [Google Scholar]
- 11.Di Marzo V, De Petrocellis L. Why do cannabinoid receptors have more than one endogenous ligand? Philos. Trans. R. Soc. Lond. B: Biol. Sci. 2012;367:3216–3228. doi: 10.1098/rstb.2011.0382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buckner JD, et al. Problematic alcohol and cannabis use among young adults: the roles of depression and discomfort and distress tolerance. Addict. Behav. 2007;32:1957–1963. doi: 10.1016/j.addbeh.2006.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Micale V, et al. Endocannabinoid system and mood disorders: priming a target for new therapies. Pharmacol. Ther. 2012;138:18–37. doi: 10.1016/j.pharmthera.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 14.Mackie K. Distribution of cannabinoid receptors in the central and peripheral nervous system. Handb. Exp. Pharmacol. 2005;168:299–325. doi: 10.1007/3-540-26573-2_10. [DOI] [PubMed] [Google Scholar]
- 15.Gong JP, et al. Cannabinoid CB2 receptors: immunohistochemical localization in rat brain. Brain Res. 2006;1071:10–23. doi: 10.1016/j.brainres.2005.11.035. [DOI] [PubMed] [Google Scholar]
- 16.Xi ZX, et al. Brain cannabinoid CB2 receptors modulate cocaine's actions in mice. Nat. Neurosci. 2011;14:1160–1166. doi: 10.1038/nn.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nyilas R, et al. Enzymatic machinery for endocannabinoid biosynthesis associated with calcium stores in glutamatergic axon terminals. J. Neurosci. 2008;28:1058–1063. doi: 10.1523/JNEUROSCI.5102-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Egertova M, et al. Localization of N-acyl phosphatidyl-ethanolamine phospholipase D (NAPE-PLD) expression in mouse brain: a new perspective on N-acylethanolamines as neural signaling molecules. J. Comp. Neurol. 2008;506:604–615. doi: 10.1002/cne.21568. [DOI] [PubMed] [Google Scholar]
- 19.Katona I, et al. Distribution of CB1 cannabinoid receptors in the amygdala and their role in the control of GABAergic transmission. J. Neurosci. 2001;21:9506–9518. doi: 10.1523/JNEUROSCI.21-23-09506.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gulyas AI, et al. Segregation of two endocannabinoid-hydrolyzing enzymes into pre- and postsynaptic compartments in the rat hippocampus, cerebellum and amygdala. Eur. J. Neurosci. 2004;20:441–458. doi: 10.1111/j.1460-9568.2004.03428.x. [DOI] [PubMed] [Google Scholar]
- 21.McDonald AJ, Mascagni F. Localization of the CB1 type cannabinoid receptor in the rat basolateral amygdala: high concentrations in a subpopulation of cholecystokinin-containing interneurons. Neuroscience. 2001;107:641–652. doi: 10.1016/s0306-4522(01)00380-3. [DOI] [PubMed] [Google Scholar]
- 22.Ramikie TS, Patel S. Endocannabinoid signaling in the amygdala: anatomy, synaptic signaling, behavior, and adaptations to stress. Neuroscience. 2012;204:38–52. doi: 10.1016/j.neuroscience.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Azad SC, et al. Activation of CB1 specifically located on GABAergic interneurons inhibits LTD in the lateral amygdala. Learn. Mem. 2008;15:143–152. doi: 10.1101/lm.741908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rhee MH, et al. Cannabinoid receptor activation differentially regulates the various adenylyl cyclase isozymes. J. Neurochem. 1998;71:1525–1534. doi: 10.1046/j.1471-4159.1998.71041525.x. [DOI] [PubMed] [Google Scholar]
- 25.Guo J, Ikeda SR. Endocannabinoids modulate N-type calcium channels and G-protein-coupled inwardly rectifying potassium channels via CB1 cannabinoid receptors heterologously expressed in mammalian neurons. Mol. Pharmacol. 2004;65:665–674. doi: 10.1124/mol.65.3.665. [DOI] [PubMed] [Google Scholar]
- 26.Mato S, et al. Role of the cyclic-AMP/PKA cascade and of P/Q-type Ca2+ channels in endocannabinoid-mediated long-term depression in the nucleus accumbens. Neuropharmacology. 2008;54:87–94. doi: 10.1016/j.neuropharm.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 27.Twitchell W, et al. Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J. Neurophysiol. 1997;78:43–50. doi: 10.1152/jn.1997.78.1.43. [DOI] [PubMed] [Google Scholar]
- 28.Cannich A, et al. CB1 cannabinoid receptors modulate kinase and phosphatase activity during extinction of conditioned fear in mice. Learn. Mem. 2004;11:625–632. doi: 10.1101/lm.77904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heifets BD, et al. Interneuron activity controls endocannabinoid-mediated presynaptic plasticity through calcineurin. Proc. Natl. Acad. Sci. U.S.A. 2008;105:10250–10255. doi: 10.1073/pnas.0711880105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mechoulam R, et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- 31.Ryberg E, et al. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007;152:1092–1101. doi: 10.1038/sj.bjp.0707460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zygmunt PM, et al. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–457. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]
- 33.Smart D, et al. The endogenous lipid anandamide is a full agonist at the human vanilloid receptor (hVR1) Br. J. Pharmacol. 2000;129:227–230. doi: 10.1038/sj.bjp.0703050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Melck D, et al. Unsaturated long-chain N-acyl-vanillyl-amides (N-AVAMs): vanilloid receptor ligands that inhibit anandamide-facilitated transport and bind to CB1 cannabinoid receptors. Biochem. Biophys. Res. Commun. 1999;262:275–284. doi: 10.1006/bbrc.1999.1105. [DOI] [PubMed] [Google Scholar]
- 35.Howlett AC. Efficacy in CB1 receptor-mediated signal transduction. Br. J. Pharmacol. 2004;142:1209–1218. doi: 10.1038/sj.bjp.0705881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Glass M, Felder CC. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: evidence for a Gs linkage to the CB1 receptor. J. Neurosci. 1997;17:5327–5333. doi: 10.1523/JNEUROSCI.17-14-05327.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bosier B, et al. Functionally selective cannabinoid receptor signalling: therapeutic implications and opportunities. Biochem. Pharmacol. 2010;80:1–12. doi: 10.1016/j.bcp.2010.02.013. [DOI] [PubMed] [Google Scholar]
- 38.Puente N, et al. Polymodal activation of the endocannabinoid system in the extended amygdala. Nat. Neurosci. 2011;14:1542–1547. doi: 10.1038/nn.2974. [DOI] [PubMed] [Google Scholar]
- 39.Facci L, et al. Mast cells express a peripheral cannabinoid receptor with differential sensitivity to anandamide and palmitoylethanolamide. Proc. Natl. Acad. Sci. U.S.A. 1995;92:3376–3380. doi: 10.1073/pnas.92.8.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Devane WA, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 41.Natarajan V, et al. Catabolism of N-acylethanolamine phospholipids by dog brain preparations. J. Neurochem. 1984;42:1613–1619. doi: 10.1111/j.1471-4159.1984.tb12750.x. [DOI] [PubMed] [Google Scholar]
- 42.Ueda N, et al. Partial purification and characterization of the porcine brain enzyme hydrolyzing and synthesizing anandamide. J. Biol. Chem. 1995;270:23823–23827. doi: 10.1074/jbc.270.40.23823. [DOI] [PubMed] [Google Scholar]
- 43.Desarnaud F, et al. Anandamide amidohydrolase activity in rat brain microsomes. Identification and partial characterization. J. Biol. Chem. 1995;270:6030–6035. doi: 10.1074/jbc.270.11.6030. [DOI] [PubMed] [Google Scholar]
- 44.Cravatt BF, et al. Chemical characterization of a family of brain lipids that induce sleep. Science. 1995;268:1506–1509. doi: 10.1126/science.7770779. [DOI] [PubMed] [Google Scholar]
- 45.Cravatt BF, et al. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 46.Tsou K, et al. Fatty acid amide hydrolase is located preferentially in large neurons in the rat central nervous system as revealed by immunohistochemistry. Neurosci. Lett. 1998;254:137–140. doi: 10.1016/s0304-3940(98)00700-9. [DOI] [PubMed] [Google Scholar]
- 47.Kamprath K, et al. Short-term adaptation of conditioned fear responses through endocannabinoid signaling in the central amygdala. Neuropsychopharmacology. 2011;36:652–663. doi: 10.1038/npp.2010.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Azad SC, et al. Activation of the cannabinoid receptor type 1 decreases glutamatergic and GABAergic synaptic transmission in the lateral amygdala of the mouse. Learn. Mem. 2003;10:116–128. doi: 10.1101/lm.53303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Domenici MR, et al. Cannabinoid receptor type 1 located on presynaptic terminals of principal neurons in the forebrain controls glutamatergic synaptic transmission. J. Neurosci. 2006;26:5794–5799. doi: 10.1523/JNEUROSCI.0372-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gunduz-Cinar O, et al. Convergent translational evidence of a role for anandamide in amygdala-mediated fear extinction, threat processing and stress-reactivity. Mol. Psychiatry. 2013;18:813–823. doi: 10.1038/mp.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Azad SC, et al. Circuitry for associative plasticity in the amygdala involves endocannabinoid signaling. J. Neurosci. 2004;24:9953–9961. doi: 10.1523/JNEUROSCI.2134-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blankman JL, Cravatt BF. Chemical probes of endocannabinoid metabolism. Pharmacol. Rev. 2013;65:849–871. doi: 10.1124/pr.112.006387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gorzalka BB, et al. Regulation of endocannabinoid signaling by stress: implications for stress-related affective disorders. Neurosci. Biobehav. Rev. 2008;32:1152–1160. doi: 10.1016/j.neubiorev.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 54.Kim J, Alger BE. Reduction in endocannabinoid tone is a homeostatic mechanism for specific inhibitory synapses. Nat. Neurosci. 2010;13:592–600. doi: 10.1038/nn.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ahn K, et al. Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem. Rev. 2008;108:1687–1707. doi: 10.1021/cr0782067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jourdan T, et al. Endocannabinoid-activated Nlrp3 inflammasome in infiltrating macrophages mediates β-cell loss in type 2 diabetes. Nat. Med. 2013;19:1132–1140. doi: 10.1038/nm.3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Patel S, et al. Repeated homotypic stress elevates 2-arachidonoylglycerol levels and enhances short-term endocannabinoid signaling at inhibitory synapses in basolateral amygdala. Neuropsychopharmacology. 2009;34:2699–2709. doi: 10.1038/npp.2009.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hill MN, et al. Suppression of amygdalar endocannabinoid signaling by stress contributes to activation of the hypothalamic–pituitary–adrenal axis. Neuropsychopharmacology. 2009;34:2733–2745. doi: 10.1038/npp.2009.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rademacher DJ, et al. Effects of acute and repeated restraint stress on endocannabinoid content in the amygdala, ventral striatum, and medial prefrontal cortex in mice. Neuropharmacology. 2008;54:108–116. doi: 10.1016/j.neuropharm.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 60.Patel S, et al. Inhibition of restraint stress-induced neural and behavioural activation by endogenous cannabinoid signalling. Eur. J. Neurosci. 2005;21:1057–1069. doi: 10.1111/j.1460-9568.2005.03916.x. [DOI] [PubMed] [Google Scholar]
- 61.Patel S, et al. Synergistic interactions between cannabinoids and environmental stress in the activation of the central amygdala. Neuropsychopharmacology. 2005;30:497–507. doi: 10.1038/sj.npp.1300535. [DOI] [PubMed] [Google Scholar]
- 62.Meye FJ, et al. Neutral antagonism at the cannabinoid 1 receptor: a safer treatment for obesity. Mol. Psychiatry. 2013 doi: 10.1038/mp.2012.145. http://dx.doi.org/10.1038/mp.2012.145. [DOI] [PubMed]
- 63.Newsom RJ, et al. Cannabinoid receptor type 1 antagonism significantly modulates basal and loud noise induced neural and hypothalamic–pituitary–adrenal axis responses in male Sprague–Dawley rats. Neuroscience. 2012;204:64–73. doi: 10.1016/j.neuroscience.2011.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dono LM, Currie PJ. The cannabinoid receptor CB1 inverse agonist AM251 potentiates the anxiogenic activity of urocortin I in the basolateral amygdala. Neuropharmacology. 2012;62:192–199. doi: 10.1016/j.neuropharm.2011.06.019. [DOI] [PubMed] [Google Scholar]
- 65.Ganon-Elazar E, Akirav I. Cannabinoid receptor activation in the basolateral amygdala blocks the effects of stress on the conditioning and extinction of inhibitory avoidance. J. Neurosci. 2009;29:11078–11088. doi: 10.1523/JNEUROSCI.1223-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Haller J, et al. Interactions between environmental aversiveness and the anxiolytic effects of enhanced cannabinoid signaling by FAAH inhibition in rats. Psychopharmacology (Berl.) 2009;204:607–616. doi: 10.1007/s00213-009-1494-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hill MN, et al. Functional interactions between stress and the endocannabinoid system: from synaptic signaling to behavioral output. J. Neurosci. 2010;30:14980–14986. doi: 10.1523/JNEUROSCI.4283-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hill MN, et al. Disruption of fatty acid amide hydrolase activity prevents the effects of chronic stress on anxiety and amygdalar microstructure. Mol. Psychiatry. 2013 doi: 10.1038/mp.2012.90. http://dx.doi.org/10.1038/mp.2012.90. [DOI] [PMC free article] [PubMed]
- 69.Reich CG, et al. Differential effects of chronic unpredictable stress on hippocampal CB1 receptors in male and female rats. Behav. Brain Res. 2009;203:264–269. doi: 10.1016/j.bbr.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vyas A, et al. Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J. Neurosci. 2002;22:6810–6818. doi: 10.1523/JNEUROSCI.22-15-06810.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mitra R, et al. Stress duration modulates the spatiotemporal patterns of spine formation in the basolateral amygdala. Proc. Natl. Acad. Sci. U.S.A. 2005;102:9371–9376. doi: 10.1073/pnas.0504011102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Padival M, et al. Effects of repeated stress on excitatory drive of basal amygdala neurons in vivo. Neuropsychopharmacology. 2013;38:1748–1762. doi: 10.1038/npp.2013.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rossi S, et al. Preservation of striatal cannabinoid CB1 receptor function correlates with the antianxiety effects of fatty acid amide hydrolase inhibition. Mol. Pharmacol. 2010;78:260–268. doi: 10.1124/mol.110.064196. [DOI] [PubMed] [Google Scholar]
- 74.Hill MN, et al. Endogenous cannabinoid signaling is essential for stress adaptation. Proc. Natl. Acad. Sci. U.S.A. 2010;107:9406–9411. doi: 10.1073/pnas.0914661107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sumislawski JJ, et al. Reversible gating of endocannabinoid plasticity in the amygdala by chronic stress: a potential role for monoacylglycerol lipase inhibition in the prevention of stress-induced behavioral adaptation. Neuropsychopharmacology. 2011;36:2750–2761. doi: 10.1038/npp.2011.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kinsey SG, et al. Inhibition of endocannabinoid catabolic enzymes elicits anxiolytic-like effects in the marble burying assay. Pharmacol. Biochem. Behav. 2011;98:21–27. doi: 10.1016/j.pbb.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Busquets-Garcia A, et al. Differential role of anandamide and 2-arachidonoylglycerol in memory and anxiety-like responses. Biol. Psychiatry. 2011;70:479–486. doi: 10.1016/j.biopsych.2011.04.022. [DOI] [PubMed] [Google Scholar]
- 78.Sciolino NR, et al. Enhancement of endocannabinoid signaling with JZL184, an inhibitor of the 2-arachidonoylglycerol hydrolyzing enzyme monoacylglycerol lipase, produces anxiolytic effects under conditions of high environmental aversiveness in rats. Pharmacol. Res. 2011;64:226–234. doi: 10.1016/j.phrs.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Orsini CA, Maren S. Neural and cellular mechanisms of fear and extinction memory formation. Neurosci. Biobehav. Rev. 2012;36:1773–1802. doi: 10.1016/j.neubiorev.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Herry C, et al. Neuronal circuits of fear extinction. Eur. J. Neurosci. 2010;31:599–612. doi: 10.1111/j.1460-9568.2010.07101.x. [DOI] [PubMed] [Google Scholar]
- 81.Lin HC, et al. Effects of intra-amygdala infusion of CB1 receptor agonists on the reconsolidation of fear-potentiated startle. Learn. Mem. 2006;13:316–321. doi: 10.1101/lm.217006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Campolongo P, et al. Endocannabinoids in the rat basolateral amygdala enhance memory consolidation and enable glucocorticoid modulation of memory. Proc. Natl. Acad. Sci. U.S.A. 2009;106:4888–4893. doi: 10.1073/pnas.0900835106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Laviolette SR, Grace AA. Cannabinoids potentiate emotional learning plasticity in neurons of the medial prefrontal cortex through basolateral amygdala inputs. J. Neurosci. 2006;26:6458–6468. doi: 10.1523/JNEUROSCI.0707-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tan H, et al. Cannabinoid transmission in the basolateral amygdala modulates fear memory formation via functional inputs to the prelimbic cortex. J. Neurosci. 2011;31:5300–5312. doi: 10.1523/JNEUROSCI.4718-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tan H, et al. Integrated cannabinoid CB1 receptor transmission within the amygdala–prefrontal cortical pathway modulates neuronal plasticity and emotional memory encoding. Cereb. Cortex. 2010;20:1486–1496. doi: 10.1093/cercor/bhp210. [DOI] [PubMed] [Google Scholar]
- 86.Marsicano G, et al. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- 87.Suzuki A, et al. Memory reconsolidation and extinction have distinct temporal and biochemical signatures. J. Neurosci. 2004;24:4787–4795. doi: 10.1523/JNEUROSCI.5491-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chhatwal JP, et al. Functional interactions between endocannabinoid and CCK neurotransmitter systems may be critical for extinction learning. Neuropsychopharmacology. 2009;34:509–521. doi: 10.1038/npp.2008.97. [DOI] [PubMed] [Google Scholar]
- 89.Pamplona FA, et al. The cannabinoid receptor agonist WIN 55,212-2 facilitates the extinction of contextual fear memory and spatial memory in rats. Psychopharmacology (Berl.) 2006;188:641–649. doi: 10.1007/s00213-006-0514-0. [DOI] [PubMed] [Google Scholar]
- 90.Lin HC, et al. The role of prefrontal cortex CB1 receptors in the modulation of fear memory. Cereb. Cortex. 2009;19:165–175. doi: 10.1093/cercor/bhn075. [DOI] [PubMed] [Google Scholar]
- 91.Roche M, et al. The effect of CB1 receptor antagonism in the right basolateral amygdala on conditioned fear and associated analgesia in rats. Eur. J. Neurosci. 2007;26:2643–2653. doi: 10.1111/j.1460-9568.2007.05861.x. [DOI] [PubMed] [Google Scholar]
- 92.Finn DP, et al. Evidence for differential modulation of conditioned aversion and fear-conditioned analgesia by CB1 receptors. Eur. J. Neurosci. 2004;20:848–852. doi: 10.1111/j.1460-9568.2004.03509.x. [DOI] [PubMed] [Google Scholar]
- 93.Plendl W, Wotjak CT. Dissociation of within- and between-session extinction of conditioned fear. J. Neurosci. 2010;30:4990–4998. doi: 10.1523/JNEUROSCI.6038-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kamprath K, et al. Cannabinoid CB1 receptor mediates fear extinction via habituation-like processes. J. Neurosci. 2006;26:6677–6686. doi: 10.1523/JNEUROSCI.0153-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pamplona FA, et al. Short- and long-term effects of cannabinoids on the extinction of contextual fear memory in rats. Neurobiol. Learn. Mem. 2008;90:290–293. doi: 10.1016/j.nlm.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 96.Chhatwal JP, et al. Enhancing cannabinoid neurotransmission augments the extinction of conditioned fear. Neuropsychopharmacology. 2005;30:516–524. doi: 10.1038/sj.npp.1300655. [DOI] [PubMed] [Google Scholar]
- 97.Izquierdo A, et al. Brief uncontrollable stress causes dendritic retraction in infralimbic cortex and resistance to fear extinction in mice. J. Neurosci. 2006;26:5733–5738. doi: 10.1523/JNEUROSCI.0474-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.de Oliveira Alvares L, et al. Opposite action of hippocampal CB1 receptors in memory reconsolidation and extinction. Neuroscience. 2008;154:1648–1655. doi: 10.1016/j.neuroscience.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 99.Rabinak CA, et al. Cannabinoid facilitation of fear extinction memory recall in humans. Neuropharmacology. 2013;64:396–402. doi: 10.1016/j.neuropharm.2012.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Godlewski G, et al. Inhibitor of fatty acid amide hydrolase normalizes cardiovascular function in hypertension without adverse metabolic effects. Chem Biol. 2010;17:1256–1266. doi: 10.1016/j.chembiol.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Camp MC, et al. Genetic strain differences in learned fear inhibition associated with variation in neuroendocrine, autonomic, and amygdala dendritic phenotypes. Neuropsychopharmacology. 2012;37:1534–1547. doi: 10.1038/npp.2011.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Herry C, et al. Switching on and off fear by distinct neuronal circuits. Nature. 2008;454:600–606. doi: 10.1038/nature07166. [DOI] [PubMed] [Google Scholar]
- 103.Holmes A, et al. Chronic alcohol remodels prefrontal neurons and disrupts NMDAR-mediated fear extinction encoding. Nat. Neurosci. 2012;15:1359–1361. doi: 10.1038/nn.3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Panlilio LV, et al. Inhibition of FAAH and activation of PPAR: new approaches to the treatment of cognitive dysfunction and drug addiction. Pharmacol. Ther. 2013;138:84–102. doi: 10.1016/j.pharmthera.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ganon-Elazar E, Akirav I. Cannabinoids prevent the development of behavioral and endocrine alterations in a rat model of intense stress. Neuropsychopharmacology. 2009;37:456–466. doi: 10.1038/npp.2011.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ganon-Elazar E, Akirav I. Cannabinoids and traumatic stress modulation of contextual fear extinction and GR expression in the amygdala–hippocampal–prefrontal circuit. Psychoneuroendocrinology. 2013;38:1675–1687. doi: 10.1016/j.psyneuen.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 107.Neumeister A, et al. Elevated brain cannabinoid CB1 receptor availability in post-traumatic stress disorder: a positron emission tomography study. Mol. Psychiatry. 2013;18:1034–1040. doi: 10.1038/mp.2013.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Long JZ, et al. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat. Chem. Biol. 2009;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lichtman AH, et al. Pharmacological activity of fatty acid amides is regulated, but not mediated, by fatty acid amide hydrolase in vivo. J. Pharmacol. Exp. Ther. 2002;302:73–79. doi: 10.1124/jpet.302.1.73. [DOI] [PubMed] [Google Scholar]
- 110.Schlosburg JE, et al. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat. Neurosci. 2010;13:1113–1119. doi: 10.1038/nn.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Riebe CJ, et al. Estrogenic regulation of limbic cannabinoid receptor binding. Psychoneuroendocrinology. 2010;35:1265–1269. doi: 10.1016/j.psyneuen.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hill MN, et al. Estrogen recruits the endocannabinoid system to modulate emotionality. Psychoneuroendocrinology. 2007;32:350–357. doi: 10.1016/j.psyneuen.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 113.Scorticati C, et al. The inhibitory effect of anandamide on luteinizing hormone-releasing hormone secretion is reversed by estrogen. Proc. Natl. Acad. Sci. U.S.A. 2004;101:11891–11896. doi: 10.1073/pnas.0404366101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Huang GZ, Woolley CS. Estradiol acutely suppresses inhibition in the hippocampus through a sex-specific endocannabinoid and mGluR-dependent mechanism. Neuron. 2012;74:801–808. doi: 10.1016/j.neuron.2012.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Waleh NS, et al. Transcriptional regulation of the mouse fatty acid amide hydrolase gene. Gene. 2002;291:203–210. doi: 10.1016/s0378-1119(02)00598-x. [DOI] [PubMed] [Google Scholar]
- 116.Flanagan JM, et al. The fatty acid amide hydrolase 385 A/A (P129T) variant: haplotype analysis of an ancient missense mutation and validation of risk for drug addiction. Hum. Genet. 2006;120:581–588. doi: 10.1007/s00439-006-0250-x. [DOI] [PubMed] [Google Scholar]
- 117.Sipe JC, et al. Biomarkers of endocannabinoid system activation in severe obesity. PLoS ONE. 2010;5:e8792. doi: 10.1371/journal.pone.0008792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chiang KP, et al. Reduced cellular expression and activity of the P129T mutant of human fatty acid amide hydrolase: evidence for a link between defects in the endocannabinoid system and problem drug use. Hum. Mol. Genet. 2004;13:2113–2119. doi: 10.1093/hmg/ddh216. [DOI] [PubMed] [Google Scholar]
- 119.Hariri AR, et al. Divergent effects of genetic variation in endocannabinoid signaling on human threat- and reward-related brain function. Biol. Psychiatry. 2009;66:9–16. doi: 10.1016/j.biopsych.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Conzelmann A, et al. A polymorphism in the gene of the endocannabinoid-degrading enzyme FAAH (FAAH C385A) is associated with emotional–motivational reactivity. Psycho-pharmacology (Berl.) 2012;224:573–579. doi: 10.1007/s00213-012-2785-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Depoy L, et al. Chronic alcohol produces neuroadaptations to prime dorsal striatal learning. Proc. Natl. Acad. Sci. U.S.A. 2013;110:14783–14788. doi: 10.1073/pnas.1308198110. [DOI] [PMC free article] [PubMed] [Google Scholar]