

Abstract
BACKGROUND
Neural tube defects (NTDs) are considered complex with both genetic and environmental factors implicated. To date, no major causative genes have been identified in humans despite several investigations. The first genomewide screen in NTDs (Rampersaud et al. 2005) demonstrated evidence of linkage to chromosomes 7 and 10. This screen included forty-four multiplex families and consisted of 402 microsatellite markers spaced approximately 10 cM apart. Further investigation of the genomic screen data identified a single large multiplex family, pedigree 8776, as primarily driving the linkage results on chromosome 7.
METHODS
To investigate this family more thoroughly, a high-density single nucleotide polymorphism (SNP) screen was performed. Two-point and multipoint linkage analyses were performed using both parametric and nonparametric methods.
RESULTS
For both the microsatellite and SNP markers, linkage analysis suggested the involvement of a locus or loci proximal to the telomeric regions of chromosomes 2q and 7p, with both regions having nonparametric lod* scores of ~3.0, yielding very similar evidence in favor of linkage.
CONCLUSIONS
The regions of strongest evidence for linkage map proximal to the telomeres on these two chromosomes. In addition to mutations and/or variants in a major gene, these loci may harbor a microdeletion and/or translocation; potentially, polygenic factors may also be involved. This single family may be promising for narrowing the search for NTD susceptibility genes.
Keywords: Neural tube defects (NTDs), spina bifida, genetic mapping, linkage, genome screen
Introduction
Neural Tube Defects: Clinical Impact
Birth defects are the leading cause of death for neonates in the United States; the risk of an infant being born with significant congenital abnormality in body structure or function is ~3-4%. Neural tube defects (NTDs) occur at a rate of 1 per 1000 live births in the United States (Campbell et al. 1986; Lary and Edmonds 1996), representing both the most common congenital malformation of the central nervous system and the second most common type of birth defect. Neural tube closure occurs during the first three to four weeks of development. NTDs are due to failure of closure of the neural tube during early embryogenesis, typically before a woman is aware of her pregnancy. NTDs can present clinically with large phenotypic variation. The most common presentations are spina bifida (spina bifida cystica, open spina bifida) and anencephaly. These phenotypes represent the classic definition of NTDs and whether they are caused by the same underlying genetic basis remains controversial.
NTDs can frequently be disabling. Children affected with spina bifida often undergo surgery to close the defect within 48 hr after birth, and may require frequent surgical intervention for complications such as hydrocephalus. Affected patients commonly have an impaired ability to walk and often need to use a wheelchair, and they may have little or no bowel and/or bladder control. Severely affected NTD cases may require aggressive surgical, medical, and rehabilitative care. Thus, for a child and their family affected with an NTD, the psychosocial and emotional tolls are immeasurable. These significant medical and personal costs have lead to several collaborative studies to investigate NTDs to better understand the genetic and environmental etiologies.
Evidence for genetic and environmental components
NTDs are caused by a complex interaction between genetic and environmental risk factors. Several exogenous risk factors are correlated with the development of NTDs, including maternal insulin-dependent diabetes mellitus, obesity, maternal use of anti-epileptic (valproic acid), and and maternal consumption of the mycotoxin fumonisin (Gomez 1981; Bjerkedal et al. 1982; Lindhout and Schmidt 1986; Shaw et al. 1996; Watkins et al. 1996; Marasas et al. 2004; Loeken 2005). An important environmental intervention for NTDs is periconceptional supplementation with folic acid, which has been shown to reduce the recurrence risk for NTDs by reducing the recurrence risk for NTDs by 50-70% (Milunsky et al. 1991; MRC Vitamin Study Research Group 1991; Centers for Disease Control and Prevention 1992). However, folate supplementation has not entirely eliminated the risk of NTDs, suggesting that there are additional underlying genetic factors that may contribute to the development of NTDs.
Several lines of evidence support a genetic component to NTDs (Frey and Hauser 2003). The most compelling evidence for a genetic factor is the higher recurrence risk for NTDs among offspring from parents with previous NTD pregnancies (Carter 1974; Copp and Bernfield 1994). Notably, the NTD recurrence risk for siblings is 2-5%, representing a 25-50 fold increased risk above that observed in the general population (Khoury and Wagener 1995). NTDs are also associated with established genetic syndromes including Meckel-Gruber syndrome, anterior sacral meningomyelocele, anal stenosis, trisomies 13 and 18, and other chromosomal aberrations (Hume et al. 1996; Kennedy et al. 1998; Lynch 2005). Furthermore, a plethora of mouse models with mutations leading to NTDs have been identified (Greene and Copp 2005). Overall, these observations support a genetic component to the development of NTDs.
Initial genomic screen results
Results from the first genome screen of NTDs were recently reported (Rampersaud et al. 2005). This screen included 44 multiplex families and considered 402 microsatellite markers with approximate 10 cM spacing. The genomic screen identified regions of interest on chromosomes 7 and 10, with the highest lod score of 2.45 on chromosome 7 using parametric linkage analysis. Further investigations of the genomic screen data identified a single large family 8776, a multigenerational Caucasian family with lumbosacral myelomeningocele segregating, as primarily driving the linkage results on chromosome 7. Additionally, this family revealed a region on chromosome 2 with evidence in favor of linkage (unpublished data).
In this study of family 8776, the microsatellite genomic screen was followed up with a whole-genome high-density SNP screen to confirm the previously identified region on chromosomes 7 and 2 and narrow the minimum candidate interval for each. In addition, the SNP screen could identify previously unidentified regions of linkage. The utility of SNPs over microsatellites for genomewide screens has been validated, with studies demonstrating increases in information content with SNP screens being dependent on family structure, previous marker density, and other factors (Evans and Cardon 2004; Middleton et al. 2004; John et al. 2004; Schaid et al. 2004). However, this increased density comes at a cost: simulation-based studies have lead to concern regarding inter-marker linkage disequilibrium (LD) in dense SNP-genomic screens. These simulations demonstrated that dense SNP data can cause false positive elevation of multipoint lod scores, especially in the presence of missing parental genotypes (Huang et al. 2004; Boyles et al. 2005).
Materials and Methods
Clinical data collection and power analysis
NTD family 8776 was identified by self-referral through the NTD Collaborative Study. First-degree relatives and relatives connecting related affected individuals were ascertained in this extended family. A detailed family history (Melvin et al. 1998) was obtained and medical records including operative reports and pre-surgical x-ray films were collected for review of diagnosis by a neurosurgeon (TMG). Of the affected individuals in the pedigree, three were diagnosed with lumbosacral myelomeningocele and a fourth individual was diagnosed with congenital dermoid cyst with tethered cord.
Certified phlebotomists obtained blood samples from affected individuals and related family members at medical centers, clinics, or by visiting participants’ homes. Some participants sent in mailer kits with their enclosed samples. This study was conducted under the oversight of Duke University Medical Center Institutional Review Board and informed consent was obtained from all participants.
Power analysis of family 8776, under the broad phenotypic classification, was performed for the microsatellites screen by using the program SIMLINK (Boehnke 1986; Ploughman and Boehnke 1989). The assumptions for the power studies included an autosomal dominant inheritance pattern with low penetrance (affecteds-only) model, a disease allele frequency of 0.001, and a marker with 75% heterozygosity linked at 5% recombination with the disease allele. These power studies generated an average parameric lod score for family 8776 at a recombination fraction θ = 0.05 of 1.063 ± 0.02 and a maximum lod score of 2.515. These power estimations are based on 2-point parametric analysis with a specified dominant mode of inheritance; therefore this analysis may underestimate power to detect linkage when using a high-density SNP approach.
Genotyping methods
Blood samples were collected on 4 affected individuals and 27 unaffected individuals in family 8776. DNA was extracted from whole blood using the Puregene system (Gentra Systems, Minneapolis, MN). Whole genome SNP typing was performed using the Affymetrix 10K SNP Chip (Affymetrix, Santa Clara, CA). In brief, the 10K SNP Chip requires only 250 ng of genomic DNA and yields genotypes for ~10,644 SNPs from the SNP Consortium database with an average heterozygosity of 0.37, and covering on average ~1 SNP every 210 kb. Microarrays were scanned by using the GeneChip Scanner 3000 according to the manufacturer’s protocol (Affymetrix). Data acquisition was performed by using the GeneChip GCOS software.
Whole genome linkage analyses
The integrity of the family’s pedigree structure was confirmed as previously reported (Rampersaud et al. 2005) using data generated from the genomewide microsatellite screen with the program RELPAIR (Boehnke and Cox, 1997; Epstein et. al., 2000).
All marker information from the genomic screen was databased using the PEDIGENE system (Haynes et al. 1995). Data were evaluated for Mendelian inconsistencies using PedCheck (O’Connell and Weeks, 1998). Markers were retained in the map files for linkage analysis if they demonstrated at least 85% efficiency. Allele frequencies for the SNP markers were estimated based on 126 unrelated Caucasian individuals that were genotyped on the 10K SNP chip. Map order for the SNP markers was generated from sequence data provided by Translational Genomics (Phoenix, AZ). To account for potential inter-marker LD in the dense SNP screen, LD Select was used (Carlson et al. 2004) with pairwise r2 > 0.16 (Boyles et al. 2005) to generate map files that contained only tagging SNPs.
Linkage analysis was performed using the program Allegro (Gudbjartsson et al. 2000) for both 2-point and multipoint analysis. Because the exact inheritance pattern of NTDs is unknown, different analytic approaches were applied to these data, including parametric (assuming dominant inheritance and a disease allele frequency of 0.001) and nonparametric analysis. For the nonparametric analysis, an identity-by-descent relative pair sharing method lod* was assessed between all pairs of affected individuals using Spairs (Whittemore and Halpern 1994) and the exponential model (Kong and Cox, 1997) as implemented in Allegro. Since the unaffected individuals in the pedigree 8776 could represent asymptomatic gene carriers or have undiagnosed NTDs, a low penetrance affecteds-only parametric model was applied such that unaffected individuals contributed no information to the lod score other than to infer missing genotypic data on unsampled individuals. Size limitations in Allegro’s computational capability allowed inclusion of only 24 individuals from family 8776 in the linkage analyses, so the least informative individuals were eliminated from the analysis. Phenotypic criteria included both broad (all 6 affected individuals) and narrow (only 5 affected individuals with lumbosacral myelomeningocele), with the narrow phenotypic definition excluding individual 100, who has a congenital dermoid cyst. The 2 affected individuals 102 and 1075 who were not sampled were included in the analyses, because the genotypic information on their siblings and/or parents using maximum-likelihood estimation techniques can help to infer the affected individual’s missing genotypes.
To confirm segregation of a disease-linked haplotypes within regions of interest, haplotype analysis was performed using both visual inspection and a statistical approach with Simwalk (Weeks et al., 1995).
Sequencing of Candidate Genes
Several genes mapping to the regions of interest were identified using an internally developed bioinformatics tool Genominator2 that operates via Ensembl (Xu et al., 2002). Primers were designed using Primer 3 (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi) on genomic sequence from UCSC’s database (http://genome.ucsc.edu/). Primers were designed to flank the exons and include the splicing region to account for splice variants. PCR products covered all the exons and respective intronic boundaries. PCR product was purified using Sephadex G-50 fine (Amersham Biosciences, Piscataway, NJ) and QuickStep2 SOPE Resin (Edge BioSystems, Gaithersburg, MD). Samples were sequenced using the BigDye Terminator v3.1 Cycle Sequencing Ready Reaction kit (Applied Biosystems, Foster City, CA) and purified using Sephadex G-50 fine. Both forward and reverse strands were sequenced. The sequence data was analyzed to evaluate for putative polymorphisms using ABI 3100 Data Collection Software Version 1.01 and ABI Sequencing Analysis 3.7. All sequences for candidate genes MEOX-2 and FZD7 were visually inspected with this software to evaluate noteworthy variants such as a premature termination codon, a frameshift mutation(s) or a missense mutation(s). The sequence data were compared to unaffected parents to ascertain if a variant was transmitted or a de novo mutation.
Results
Genetic mapping
Linkage Results for Family 8776
The individuals included in the linkage analyses using Allegro are indicated in Figure 1. The regions of interest were defined by a one lod unit drop support interval from the peak lod to approximate 95% confidence intervals. Table 1 summarizes the linkage findings for the SNP data based on the results from Allegro multipoint lod scores > 2.5 and with two-point lod scores ≥ 1.8; plots for these regions are shown in Figure 2. The strongest evidence for linkage occurred near the telomeres of chromosomes 7 (7p21.1-pter) and 2 (2q33.1-q35). Specifically, the maximum multipoint nonparametric lod* score was 3.01 on chromosome 2 in the region spanning rs1379007-rs1992235. For chromosome 7, the maximum multipoint nonparametric lod* score was 2.99 in the region spanning rs2353788-rs1368215. These results provide an approximate 10% increase in information content over the maximum multipoint scores in the microsatellite genomic screen.
Figure 1.
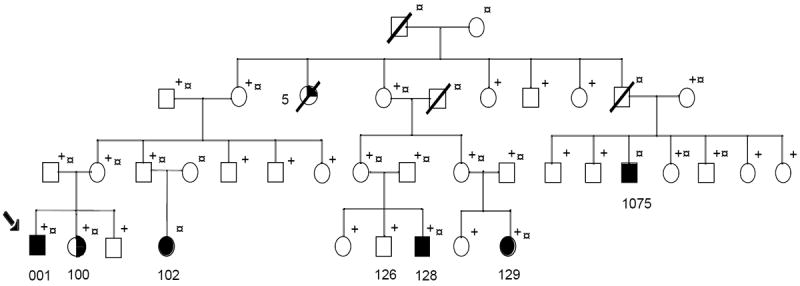
Pedigree diagram of family 8776: Condensed version of extended family 8776. Fully shaded symbols represent individuals affected with lumbosacral myelomeningocele. The half shaded circle for individual 100 represents congenital dermoid cyst. The quarter shaded circle denotes the 5 individuals who are affected by history alone. + indicates sampled (DNA) individuals. § refers to individuals included in linkage analysis. Individuals 102 and 1075 are reported to have lumbosacral myelomeningocele; however, the medical records and radiographic studies are not currently available to confirm this diagnosis.
Table 1.
Multipoint LOD scores (> 2.5) for regions of interest on chromosomes 2 and 7.
| Parametric | Nonparametric | ||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Cytogenetic Band | Marker | Region Spans | One LOD unit (CI) (Total distance, cM) | Broad n = 6 | Narrow n = 5 | Broad n = 6 | Narrow n = 5 |
| Chromosome 2 2q33.1-q35 | SNPs | rs1437872-rs2888386 | 196.5 – 213.6 (17.1 cM) | 2.69 | 2.39 | 3.01 | 2.41 |
|
| |||||||
| Microsat-telite | D2S2978-D2S1363 | 186.0 – 229.2 (43.2 cM) | 2.32 | 2.02 | 2.64 | 2.04 | |
|
| |||||||
| Chromosome 7 7p21.1-pter | SNPs | rs2389831- 7pter | 0 – 31.68 (31.7 cM) | 2.66 | 2.36 | 2.99 | 2.38 |
|
| |||||||
| Microsat. | D7S3051-7pter | 0 – 32.34 (32.3 cM) | 2.31 | 2.02 | 2.63 | 2.03 | |
n represents the number of affected individuals included in each analysis.
Figure 2.
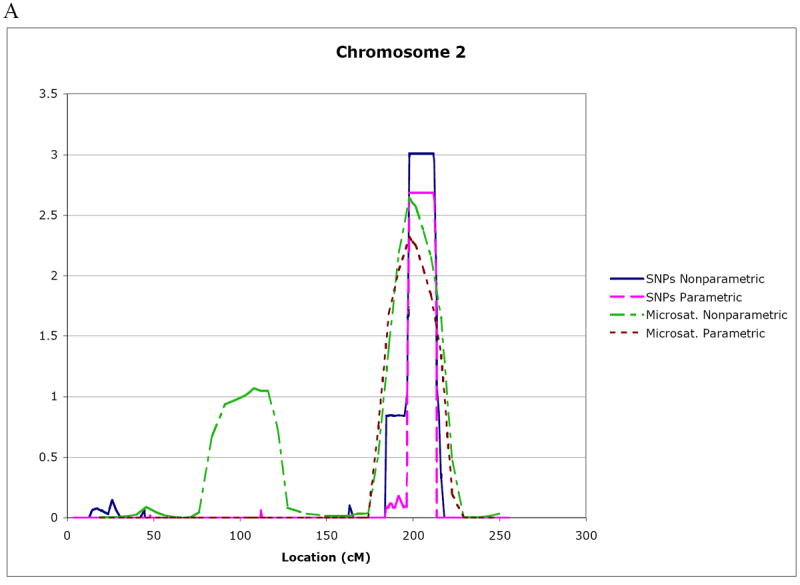
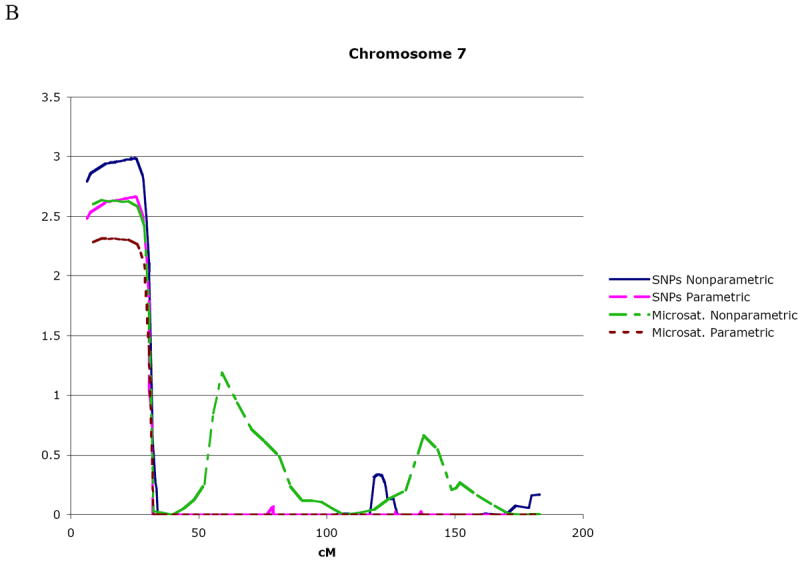
Multipoint lod score curves for regions of interest on chromosome 2 (A) and chromosome 7 (B). Both SNP and microsatellite marker linkage results using parametric and nonparametric linkage methods are plotted. The total genetic distance covered by the analyzed markers is given on the x axis in cM.
When individual 100, who has a closed NTD, was included as affected (under the broad phenotype), her genotypic information increased the overall lod score under both parametric and nonparametric analyses.
Haplotyping
Preliminary haplotyping using Simwalk (Weeks et al. 1995) and hand-haplotyping confirmed the linkage regions identified in Table 1. The haplotype results are consistent with autosomal dominant transmission with reduced penetrance. Individual 100, who has the closed NTD variant of congenital dermoid cyst, also carried the disease-associated haplotypes on chromosomes 2 and 7. Visual inspection of marker data for chromosome 2 expanded the candidate interval to span markers D2S1776 to D2S1363 approximating a 55.2 cM region. For chromosome 7, marker coverage of 7pter extended only to the middle of 7p22.3; as a result, the flanking marker for the 7pter end of the disease-associated haplotype could not be determined. Thus, the minimum candidate interval for chromosome 7 spans from D7S3051 to 7pter approximating a 32.3 cM region. The presumed unaffected individual 126 carried the chromosome 7 disease-associated haplotype and showed a crossover between D7S2201 and D7S641. If individual 126 is truly “unaffected,” the crossover could reduce the minimum candidate interval to 17.4 cM.
Candidate genes
Several biologically plausible candidate genes reside in the regions of interest defined by haplotyping results. Using Ensembl (http://www.ensembl.org/index.html), a list of candidate genes for chromosomes 2 and 7 was assembled for mutational analysis. For chromosome 2, the frizzled 7 receptor (FZD7) gene that maps to 2q33.1 was sequenced. Members of the frizzled gene family encode 7-transmembrane domain proteins that are receptors for Wnt signaling proteins. The highly conserved Wnt-frizzled signal non-canonical transduction pathways have been shown in several animal models to be involved in convergent extension, the process that leads to elongation of the neural plate during neural tube development (Wallingford and Harland, 2002; Lawrence and Morel, 2003; Copp et al., 2003). Human mesenchyme homeobox 2 (MEOX 2), located at 7p21.3-p22.1, is a homeobox gene involved in formation, patterning, and differentiation of somites. The mouse homolog Meox-2 is required for migration of limb bud and null Meox-2 mice have decreased limb muscle mass and demonstrate downregulation of Pax3 (Mankoo et al., 2003). Both genes evaluated (FZD7 and MEOX-2) did not show any novel genetic polymorphisms.
Discussion
For some complex diseases, traditional linkage analysis approaches in rare families with Mendelian or near-Mendelian inheritance patterns has identified susceptibility loci. For example, investigators who studied rare multigenerational families with early-onset fully penetrant Alzheimer’s disease identified linkage to chromosome 14 and subsequently identified the amyloid precursor protein (APP) gene (Tanzi and Bertram, 2001). Later investigations identified a second locus and the genetic risk gene presenilin 1 (PSEN 1) (Tanzi and Bertram, 2001). Other examples using single large families to identify linkage regions that influence disease susceptibility include familial focal segmental glomerulosclerosis (Winn et al. 2005), coronary artery disease (Wang et al., 2003), dementia (Ashley-Koch et al., 2005), tonic-clonic seizure (Puranam et al., 2005) sensoineural hearing loss (Blanton et al., 2002; Tlili et al., 2005), among others. This focused approach of genetic analysis is advantageous for minimizing genetic heterogeneity, thereby increasing the investigator’s ability to identify areas of allele sharing in large multiplex families.
Using a high-density SNP genomic screen, linkage of Family 8776 was confirmed for the previously reported locus at 7p21.1- pter. Additionally, a locus at 2q33.1-q35 yielded increased evidence in favor of linkage over that from the earlier microsatellite genomic screen (Rampersaud et al., 2005). These two regions of interest exhibit similar statistical support for an NTD susceptibility locus, which may reflect multiple distinct genetic effects. Haplotype analysis verified these regions of interest and demonstrated that the high-risk haplotypes segregate in a pattern consistent with autosomal dominant inheritance and reduced penetrance, or alternatively, an environmental factor that triggers disease presentation. As with any genetic linkage study, these results could represent a spurious finding.
An avenue for further investigation involves more precise phenotypic definition in the presumptively unaffected individuals. For instance, determining whether these unaffected individuals have subtle dysraphic changes such as spina bifida occulta may stimulate broadening of the NTD phenotype, increase the power for future linkage analyses, enhance the ability to minimize a candidate interval, and identify individuals at high risk to conceive a pregnancy with an NTD. Alternatively, the failure to identify radiographic changes in presumptively unaffected “transmitting” individuals may reflect a non-penetrant allele or a susceptibility locus in the absence of an eliciting environmental insult. Studies to elucidate the phenotype involve radiographic confirmation by x-ray and/or magnetic resonance imaging and may be difficult to obtain in a research setting due to increased risk to the subject. Any detected differences in affected individuals’ NTD phenotype from family 8776 may potentially be explained by pleiotropic effects of a common underlying gene, different genetic backgrounds containing a modifier gene, and/or different environmental exposures at a critical developmental time point. Two individuals in family 8776 demonstrate the importance of accurate phenotypic definition.
For instance, when individual 100, who has an NTD variant of congenital dermoid cyst, was included in the analyses as affected, the lod scores for the 7p and 2q regions were increased. Individual 100 also carries the same high-risk haplotype as other affected individuals with lumbosacral myelomeningocele in the family.
And, haplotype analysis also revealed that individual 126, who appears phenotypically unaffected, to be recombinant for the disease-associated chromosome 7 haplotype. Since individual 126 is a minor, radiographic studies are not available for clarification of the phenotype. If individual 126 demonstrated no evidence for a radiographic abnormality, the 7p interval could potentially be narrowed by 14.9 cM to a 17.4 cM region between D7S641-7pter. However, this crossover for the unaffected individual should be interrupted cautiously because she may harbor a subtle dysraphic variant which would lead to a change in her affection status, thereby modifying the minimum candidate interval.
Several promising candidate genes reside in the regions of interest for chromosomes 2 and 7. FZD7, CASP8, CASP10, PAX3, and a cluster of HOXD genes map to the region of interest on 2q. MEOX2 and TWIST1 genes map to 7p. Sequence data for FZD7 and MEOX2 genes did not reveal any potential causal variants for this family; however, the promoter regions for these genes have yet to be evaluated. In addition to evaluation of candidate genes, family 8776 is also under investigation for potential chromosomal abnormalities using both comparative genomic hybridization (CGH)-arrays and fluorescent in situ hybridization (FISH).
Overall, the highest linkage results for family 8776 map near the telomeric regions of chromosomes 2 and 7. Telomeric regions typically are difficult to map due to limited marker density, and in fact, the chromosome 7 region of interest could be narrowed with an increased marker density. Regions proximal to the telomeres tend to have a high degree of interchromosomal recombination, so it is plausible that a microdeletion and/or translocation event has taken place in one or both of these regions. Chromosomal abnormalities are associated with NTDs, and reports suggest 2-16% of non-syndromic NTDs have detectable cytogenetic abnormalities (Lynch, 2005). The 2q region of interest for family 8776 includes the PAX3 gene, for which an interstitial de novo microdeletion of 2q35-q36.2 has been previously reported for two patients with Waardenburg syndrome type III and myelomeningocele (OMIM 148820) (Nye et al., 1998). PAX3 is a member of the Paired Box transcription factor gene family that has been extensively studied for its role in early development. Pax3 is expressed in the mouse neural tube and mutant Pax3 mice (Splotch) embryos have neural tube defects, including spina bifida and exencephaly, (the equivalent of human anencephaly).
Focusing on rare Mendelian-like families for linkage studies can identify susceptibility loci that may later be shown to alter risk in the non-Mendelian forms of the disease. Family 8776, as one of the largest reported NTD families, may represent an important resource for narrowing the search for NTD candidate genes since it provides a unique opportunity for identification of a single major locus. However, evaluation of this one family may or may not be generalizable to all NTDs, so any putative high-risk haplotypes, genetic variant(s) and/or chromosomal abnormalities may or may not be unique to this family. Consequently, any results identified in family 8776 must be characterized in other families to determine generalizability.
Acknowledgments
We thank all of the patients and family members who participated in this study for their generous contributions of time, energy, and biological samples. We also thank Bei Zhao, Kristen Deak, and Carol Haynes for their contribution to this project.
This work was supported by grants from the National Institutes of Health (HD39948, HD33400, NS39818, ES11375, ES011961, NS26630).
Footnotes
Presented at the “NTD and Beyond 2005” meeting held at Palm Springs, CA on September 10-13, 2005.
Literature Cited
- Ashley-Koch AE, Shao Y, Rimmler JB, Gaskell PC, Welsh-Bohmer KA, Jackson CE, Scott WK, Haines JL, Pericak-Vance MA. An autosomal genomic screen for dementia in an extended Amish family. Neurosci Lett. 2005;379:199–204. doi: 10.1016/j.neulet.2004.12.065. [DOI] [PubMed] [Google Scholar]
- Bjerkedal T, Czeizel A, Goujard J, Kallen B, Mastroiacova P, Nevin N, Oakley G, Jr, Robert E. Valproic acid and spina bifida. Lancet. 1982;2:1096. doi: 10.1016/s0140-6736(82)90018-6. [DOI] [PubMed] [Google Scholar]
- Blanton SH, Liang CY, Cai MW, Pandya A, Du LL, Landa B, Mummalanni S, Li KS, Chen ZY, Qin XN, Liu YF, Balkany T, nance WE, Liu XZ. A novel locus for autosomal dominant non-syndromic deafness (DFNA41) maps to chromosome 12q24-qter. J Med Genet. 2002;39:567–570. doi: 10.1136/jmg.39.8.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehnke M. Estimating the power of a proposed linkage study: a practical computer simulation approach. Am J Hum Genet. 1986;39:513–527. [PMC free article] [PubMed] [Google Scholar]
- Boehnke M, Cox NJ. Accurate inference of relationships in sib-pair linkage studies. Am J Hum Genet. 1997;61:423–9. doi: 10.1086/514862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyles AL, Scott WK, Martin ER, Schmidt S, Li YJ, Ashley-Koch A, Bass MP, Schmidt M, Pericak-Vance MA, Speer MC, Hauser ER. Linkage disequilibrium inflates type I error rates in multipoint linkage analysis when parental genotypes are missing. Hum Hered. 2005;59:220–227. doi: 10.1159/000087122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell LR, Dayton DH, Sohal GS. Neural tube defects: a review of human and animal studies on the etiology of neural tube defects. Teratology. 1986;34:171–187. doi: 10.1002/tera.1420340206. [DOI] [PubMed] [Google Scholar]
- Carlson CS, Eberle MA, Rieder MJ, Yi Q, Kruglyak L, Nickerson DA. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am J Hum Genet. 2004;74:106–120. doi: 10.1086/381000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter CO. Clues to the aetiology of neural tube malformations. Dev Med Child Neurol. 1974;16:3–15. doi: 10.1111/j.1469-8749.1974.tb03442.x. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention. Recommendations for the use of folic acid to reduce the number of cases of spina bifida and other neural tube defects. Morbidity and Mortality Weekly. 1992;41:1–7. [PubMed] [Google Scholar]
- Copp AJ, Bernfield M. Etiology and pathogenesis of human neural tube defects: insights from mouse models. Curr Opin Pediatr. 1994;6:624–631. doi: 10.1097/00008480-199412000-00002. [DOI] [PubMed] [Google Scholar]
- Copp AJ, Greene ND, Murdoch JN. The genetic basis of mammalian neurulation. Nat Rev Genet. 2003;4:784–793. doi: 10.1038/nrg1181. [DOI] [PubMed] [Google Scholar]
- Epstein MP, Duren WL, Boehnke M. Improved inference of relationship for pairs of individuals. Am J Hum Genet. 2000;67:1219–31. doi: 10.1016/s0002-9297(07)62952-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans DM, Cardon LR. Guidelines for genotyping in genomewide linkage studies: single-nucleotide-polymorphism maps versus microsatellite maps. Am J Hum Genet. 2004;75:687–692. doi: 10.1086/424696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey L, Hauser WA. Epidemiology of neural tube defects. Epilepsia. 2003;44(Suppl 3):4–13. doi: 10.1046/j.1528-1157.44.s3.2.x. [DOI] [PubMed] [Google Scholar]
- Gomez MR. Possible teratogenicity of valproic acid. J Pediatr. 1981;98:508–509. doi: 10.1016/s0022-3476(81)80743-3. [DOI] [PubMed] [Google Scholar]
- Greene ND, Copp AJ. Mouse models of neural tube defects: investigating preventive mechanisms. Am J Med Genet C Semin Med Genet. 2005;135:31–41. doi: 10.1002/ajmg.c.30051. [DOI] [PubMed] [Google Scholar]
- Gudbjartsson DF, Jonasson K, Frigge ML, Kong A. Allegro, a new computer program for multipoint linkage analysis. Nat Genet. 2000;25:12–13. doi: 10.1038/75514. [DOI] [PubMed] [Google Scholar]
- Haynes C, Speer MC, Peedin M, Roses AD, Haines JL, Vance JM, Pericak-Vance MA. PEDIGENE: A comprehensive data management system to facilitate efficient and rapid disease gene mapping. Am J Hum Genet. 1995;57:A193. [Google Scholar]
- Huang Q, Shete S, Amos CI. Ignoring linkage disequilibrium among tightly linked markers induces false-positive evidence of linkage for affected sib pair analysis. Am J Hum Genet. 2004;75:1106–1112. doi: 10.1086/426000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume RF, Drugan A, Reichler A, Lampinen J, Martin LS, Johnson MP, Evans MI. Aneuploidy among prenatally detected neural tube defects. Am J Med Genet. 1996;61:171–173. doi: 10.1002/(SICI)1096-8628(19960111)61:2<171::AID-AJMG14>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- John S, Shephard N, Liu G, Zeggini E, Cao M, Chen W, Vasavda N, Mills T, Barton A, Hinks A, Eyre S, Jones KW, Ollier W, Silman A, Gibson N, Worthington J, Kennedy GC. Whole-genome scan, in a complex disease, using 11,245 single-nucleotide polymorphisms: comparison with microsatellites. Am J Hum Genet. 2004;75:54–64. doi: 10.1086/422195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy D, Chitayat D, Winsor EJT, Silver M, Toi A. Prenatally diagnosed neural tube defects: Ultrasound, chromosome, and autopsy or postnatal findings in 212 cases. Am J Med Genet. 1998;77:317–321. doi: 10.1002/(sici)1096-8628(19980526)77:4<317::aid-ajmg13>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Khoury MJ, Wagener DK. Epidemiological evaluation of the use of genetics to improve the predictive value of disease risk factors. Am J Hum Genet. 1995;56:835–844. [PMC free article] [PubMed] [Google Scholar]
- Kong A, Cox NJ. Allele-sharing models: LOD scores and accurate linkage tests. Am J Hum Genet. 1997;61:1179–1188. doi: 10.1086/301592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lary JM, Edmonds LD. Prevalence of spina bifida at birth-United States, 1983-1990: a comparison of two surveillance systems. MMWR Morb Mortal Wkly Rep. 1996;45:15–26. [PubMed] [Google Scholar]
- Lawrence N, Morel V. Dorsal closure and convergent extension: two polarised morphogenetic movements controlled by similar mechanisms? Mech Dev. 2003;120:1385–1393. doi: 10.1016/j.mod.2003.07.004. [DOI] [PubMed] [Google Scholar]
- Lindhout D, Schmidt D. In-utero exposure to valproate and neural tube defects (letter) Lancet. 1986;1:1392–1393. doi: 10.1016/s0140-6736(86)91711-3. [DOI] [PubMed] [Google Scholar]
- Loeken MR. Current perspectives on the causes of neural tube defects resulting from diabetic pregnancy. American Journal of Medical Genetics Part C-Seminars in Medical Genetics. 2005;135C:77–87. doi: 10.1002/ajmg.c.30056. [DOI] [PubMed] [Google Scholar]
- Lynch SA. Non-multifactorial neural tube defects. Am J Med Genet C Semin Med Genet. 2005;135:69–76. doi: 10.1002/ajmg.c.30055. [DOI] [PubMed] [Google Scholar]
- Mankoo BS, Skuntz S, Harrigan I, Grigorieva E, Candia A, Wright CV, Arnheiter H, Pachnis V. The concerted action of Meox homeobox genes is required upstream of genetic pathways essential for the formation, patterning and differentiation of somites. Development. 2003;130:4655–4664. doi: 10.1242/dev.00687. [DOI] [PubMed] [Google Scholar]
- Marasas WF, Riley RT, Hendricks KA, Stevens VL, Sadler TW, Gelineau-van Waes J, Missmer SA, Cabrera J, Torres O, Gelderblom WC, Allegood J, Martinez C, Maddox J, Miller JD, Starr L, Sullards MC, Roman AV, Voss KA, Wang E, Merrill AH., Jr Fumonisins disrupt sphingolipid metabolism, folate transport, and neural tube development in embryo culture and in vivo: a potential risk factor for human neural tube defects among populations consuming fumonisin-contaminated maize. J Nutr. 2004;134:711–716. doi: 10.1093/jn/134.4.711. [DOI] [PubMed] [Google Scholar]
- Melvin EC, Scott WK, Speer MC, Wolpert C, Pericak-Vance MA. Development of a standardized family history questionnaire for research purposes. Journal of Genetic Counseling. 1998;7:475. [Google Scholar]
- Middleton FA, Pato MT, Gentile KL, Morley CP, Zhao X, Eisener AF, Brown A, Petryshen TL, Kirby AN, Medeiros H, Carvalho C, Macedo A, Dourado A, Coelho I, Valente J, Soares MJ, Ferreira CP, Lei M, Azevedo MH, Kennedy JL, Daly MJ, Sklar P, Pato CN. Genomewide linkage analysis of bipolar disorder by use of a high-density single-nucleotide-polymorphism (SNP) genotyping assay: a comparison with microsatellite marker assays and finding of significant linkage to chromosome 6q22. Am J Hum Genet. 2004;74:886–897. doi: 10.1086/420775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milunsky A, Jick H, Jick SS, Bruell CL, MacLaughlin DS, Rothman KJ, Willett W. Multivitamin/folic acid supplementation in early pregnancy reduces the prevalence of neural tube defects. JAMA. 1991;262:2847–2852. doi: 10.1001/jama.262.20.2847. [DOI] [PubMed] [Google Scholar]
- MRC Vitamin Study Research Group. Prevention of neural tube defects: Results of the Medical Research Council Vitamin Study. Lancet. 1991;338:131–137. [PubMed] [Google Scholar]
- Nye JS, Balkin N, Lucas H, Knepper P, McLone D, Charrow J. Meningomyelocele and Waardenburg syndrome (type 3) in patients with interstitial deletion of 2q35 and the PAX3 gene: possible digenic inheritance of a neural tube defect. Am J Med Genet. 1998;75:401–408. [PubMed] [Google Scholar]
- O’Connell JR, Weeks DE. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploughman LM, Boehnke M. Estimating the power of a proposed linkage study for a complex genetic trait. Am J Hum Genet. 1989;44:543–551. [PMC free article] [PubMed] [Google Scholar]
- Puranam RS, Jain S, Kleindienst AM, Saxena S, Kim MK, Kelly CB, Padma MV, Andrews I, Elston RC, Tiwari HK, McNamara JO. A locus for generalized tonic-clonic seizure susceptibility maps to chromosome 10q25-q26. Ann Neurol. 2005;58:449–458. doi: 10.1002/ana.20598. [DOI] [PubMed] [Google Scholar]
- Rampersaud E, Bassuk AG, Enterline DS, George TM, Siegel DG, Melvin EC, Aben J, Allen J, Aylsworth A, Brei T, Bodurtha J, Buran C, Floyd LE, Hammock P, Iskandar B, Ito J, Kessler JA, Lasarsky N, Mack P, Mackey J, McLone D, Meeropol E, Mehltretter L, Mitchell LE, Oakes WJ, Nye JS, Powell C, Sawin K, Stevenson R, Walker M, West SG, Worley G, Gilbert JR, Speer MC. Whole genomewide linkage screen for neural tube defects reveals regions of interest on chromosomes 7 and 10. J Med Genet. 2005;42:940–946. doi: 10.1136/jmg.2005.031658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaid DJ, Guenther JC, Christensen GB, Hebbring S, Rosenow C, Hilker CA, McDonnell SK, Cunningham JM, Slager SL, Blute ML, Thibodeau SN. Comparison of microsatellites versus single-nucleotide polymorphisms in a genome linkage screen for prostate cancer-susceptibility Loci. Am J Hum Genet. 2004;75:948–965. doi: 10.1086/425870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw GM, Velie EM, Schaffer D. Risk of neural tube defect-affected pregnancies among obese women. JAMA. 1996;275:1093–1096. doi: 10.1001/jama.1996.03530380035028. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Bertram L. New frontiers in Alzheimer’s disease genetics. Neuron. 2001;32:181–184. doi: 10.1016/s0896-6273(01)00476-7. [DOI] [PubMed] [Google Scholar]
- Tlili A, Mannikko M, Charfedine I, Lahmar I, Benzina Z, Ben Amor M, Driss N, Ala-Kokko L, Drira M, Masmoudi S, Ayadi H. A Novel Autosomal Recessive Non-Syndromic Deafness Locus, DFNB66, Maps to Chromosome 6p21.2-22.3 in a Large Tunisian Consanguineous Family. Hum Hered. 2005;60:123–128. doi: 10.1159/000088974. [DOI] [PubMed] [Google Scholar]
- Wallingford JB, Harland RM. Neural tube closure requires Dishevelled-dependent convergent extension of the midline. Development. 2002;129:5815–5825. doi: 10.1242/dev.00123. [DOI] [PubMed] [Google Scholar]
- Wang L, Fan C, Topol SE, Topol EJ, Wang Q. Mutation of MEF2A in an inherited disorder with features of coronary artery disease. Science. 2003;302:1578–1581. doi: 10.1126/science.1088477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins ML, Scanlon KS, Mulinare J, Muin JK. Is maternal obesity a risk factor for anencephaly and spina bifida? Epidemiology. 1996;7:507–512. [PubMed] [Google Scholar]
- Weeks DE, Sobel E, O’Connell JR, Lange K. Computer programs for multilocus haplotyping of general pedigrees. Am J Hum Genet. 1995;56:1506–1507. [PMC free article] [PubMed] [Google Scholar]
- Whittemore AS, Halpern J. A class of tests for linkage using affected pedigree members. Biometrics. 1994;50:118–127. [PubMed] [Google Scholar]
- Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, Daskalakis N, Kwan SY, Ebersviller S, Burchette JL, Pericak-Vance MA, Howell DN, Vance JM, Rosenberg PB. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308:1801–1804. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- Xu H, Vance JM, Stenger JE. Integrating genomic laboratory data with whole genome annotation using Endembl-DAS. Am J Hum Genet. 2002;71:393–1301A. [Google Scholar]