

Abstract
Many biologically and agronomically important traits are dynamic and show temporal variation. In this study, we used triticale (× Triticosecale Wittmack) as a model crop to assess the genetic dynamics underlying phenotypic plasticity of adult plant development. To this end, a large mapping population with 647 doubled haploid lines derived from four partially connected families from crosses among six parents was scored for developmental stage at three different time points. Using genome-wide association mapping, we identified main effect and epistatic quantitative trait loci (QTL) at all three time points. Interestingly, some of these QTL were identified at all time points, whereas others appear to only contribute to the genetic architecture at certain developmental stages. Our results illustrate the temporal contribution of QTL to the genetic control of adult plant development and more generally, the temporal genetic patterns of regulation that underlie dynamic traits.
Keywords: dynamic QTL, plant development, genetic architecture, triticale, complex traits, genome-wide association study (GWAS), high-density genotyping, Multiparent Advanced Generation Inter-Cross (MAGIC), multiparental populations, MPP
Many traits of biological or agronomic importance are dynamic traits that change with time (Wu and Lin 2006). Consequently, a static examination at only a single time point neglects these temporal changes and can therefore only deliver a simplified view of genetic control in time. With the availability of high-density genotypic data, approaches to unravel the genetic architecture underlying important traits have become feasible (Würschum 2012; Edwards et al. 2013) and have been applied in different crops, including maize (e.g., Buckler et al. 2009; Steinhoff et al. 2012), wheat (e.g., Reif et al. 2011a), barley (e.g., Wang et al. 2012), sugar beet (e.g., Würschum et al. 2011), and rapeseed (e.g., Snowdon et al. 2010; Würschum et al. 2013). Even for dynamic traits, however, most genomic approaches so far have been based on a single evaluation of the trait, and few studies have employed dynamic quantitative trait loci (QTL) mapping. Yan et al. (1998) performed QTL mapping for plant height in rice at several developmental stages and found that some QTL could be detected at all examined stages whereas others were only identified at some of the developmental stages. Busemeyer et al. (2013a) and Liu, et al. (2014) recently have described QTL mapping for biomass yield in triticale. They assessed three different time points and showed that the entire genetic architecture underlying biomass accumulation is under dynamic genetic control. Nevertheless, due to the low number of studies that use dynamic QTL mapping, our understanding of the genetic patterns underlying phenotypic plasticity remains rudimentary.
Adult plant development is an important trait in small grain cereals because it affects adaptation and ultimately yield. Most studies until now have investigated only the genetics underlying a single developmental stage, mainly flowering time. However, the genetic control underlying preceding or subsequent developmental stages may well be different. Winter cereals are sown in autumn and after vernalization during winter can make the transition from vegetative to generative growth, dependent on external stimuli such as photoperiod. After flowering, the grains start to fill and the plants finally ripen in summer. To enable a uniform coding of phenologically identical growth stages of crops, Lancashire et al. (1991) introduced a decimal system, the BBCH code. It is based on the well-known code for cereals developed by Zadoks et al. (1974) and is widely used by scientists and breeders alike.
Association mapping often is carried out on a diversity panel of lines but also can be used for QTL detection in multiple segregating families (Yu et al. 2008; Liu et al. 2013). Combining multiple families in a mapping population can offer the advantage of an increased QTL detection power. Triticale (× Triticosecale Wittmack) is a hexaploid crop containing the A and B genomes from wheat and the R genome from rye. QTL mapping in multiple families of triticale recently has been reported for biomass yield and plant height (Alheit et al. 2014).
The objective of this study was to investigate the genetic patterns of regulation underlying adult plant development in triticale. To this end, a large mapping population of 647 doubled haploid (DH) lines derived from four families was scored for developmental stage at three time points, roughly corresponding to the stage when awns become visible, late flowering, and very early dough development. In particular our objectives were to identify main effect QTL for the three developmental stages, assess the contribution of epistasis to the genetic architecture, and to investigate the temporal changes in the genetic control underlying phenotypic plasticity of adult plant development.
Materials and Methods
Plant material, field trials, and phenotypic data
The plant material and the field trials underlying this study have been described in Busemeyer et al. (2013a). In brief, a mapping population consisting of 647 DH (Würschum et al. 2012a) triticale lines was used. The population consisted of four families (Supporting Information, Figure S1), DH06 (n = 131; parents ‘Modus’ × ‘Saka3006’), DH07 (n = 120; parents ‘Modus’ × ‘Saka3008’), EAW74 (n = 200; parents ‘HeTi117-06’ × ‘Pawo’), and EAW78 (n = 196; parents ‘HeTi117-06’ × ‘TIW671’), which have been described by Alheit et al. (2011, 2012). These four families are partially connected because families DH06 and DH07 share a common parent (‘HeTi117-06’) as well as families EAW74 and EAW78 (‘Modus’) (Figure S1). This design was chosen because the six parents are all adapted elite winter triticale lines, which obliterates the need for a NAM design (Buckler et al. 2009). Furthermore, we wanted to include six parental lines to increase the number of alleles segregating in the mapping population and at the same time wanted to study large families. Fully connected designs, for example, a diallel (Rebaï and Goffinet 1993; Blanc et al. 2006) would have required a strongly increased size of the mapping population and consequently the partially connected design was chosen.
The DH lines were grown in partially replicated designs (Williams et al. 2011), including common checks with 960 yield plots per location, at two locations in 2 yr. The plants were scored visually for their developmental stage (i.e., BBCH stage; Lancashire et al. 1991) at three time points termed DS1, DS2, and DS3, in May and June, at which time the majority of the plants were at the developmental stages: BBCH stage 49 (awns visible), BBCH 69 (late flowering), and BBCH 81 (very early dough development). Scoring was done on a plot basis, and the values represent the developmental stage of the majority of the plants within the plots. For all lines, the best linear unbiased estimates were determined across environments.
Association mapping in multiple families
The DH lines were genotyped with DArT markers, and QTL mapping was done based on the integrated consensus linkage map described by Alheit et al. (2012). For QTL mapping, an additive genetic model was chosen, and association mapping was done with a biometric model that performed well in a recent comparison of models for association mapping in multiple families and incorporates a family effect to control for differences in family means, cofactors to control the genetic background, and a marker effect across families (Liu et al. 2012; Würschum et al. 2012b):
In this model, Y is a N × 1 column vector of the best linear unbiased estimates of N DH lines, coming from F families (F = 4); l is a N × 1 column vector containing constant 1; µ is the intercept; Xq (Xc) is a N × 1 column vector containing the marker information of each DH at marker q (cofactor c); Xf is a N × F matrix whose elements are 1 or 0 according to whether or not an individual belonged to family f and Mf is a F × 1 vector of family effects; bq (bc) is the expected allele substitution effect of marker q (cofactor c); and ε is the vector of the residuals of the model.
We applied a two-step procedure for QTL detection. In the first step, stepwise multiple linear regression was used to select a set of cofactors based on the Schwarz (1978) Bayesian Criterion with a model including a family effect and cofactors. Cofactor selection was performed using Proc GLMSELECT implemented in the statistical software SAS (SAS Institute Inc, 2008). In the second step, we calculated a P value for the association of each marker with the phenotypic value for the F test with a full model (with marker effect) against a reduced model (without marker effect). The Bonferroni-Holm procedure (Holm 1979) was used to detect markers with significant (P < 0.05) main effects. QTL were declared as overlapping between the three developmental stages if they fell within an arbitrarily defined 10-cM interval surrounding the QTL. The proportion of the genotypic variance explained by the detected QTL was estimated following Utz et al. (2000) as pG = / h2. The pG explained by individual QTL (Table S2 and Table S3) was estimated as sequential partial R2 from a simultaneous fit of the detected QTL. To account for the potential presence of colinearity among the detected QTL, they were fitted in the linear model in the order of the strength of their association with the trait, i.e., the most significantly associated markers (smallest P value in the QTL scan) were fitted first in the model. Fivefold cross-validation was done as described previously (Liu et al. 2013; Würschum and Kraft 2014).
For the detection of epistasis the model was extended and included the main effects of the two loci q and q’, Xqbq and Xq’bq’, and the interaction effect of the marker pair under consideration Xqq’bqq’. For the significance level for the epistatic QTL we used an α-level of 0.05 and followed the suggestion of Holland et al. (2002) dividing the α-level by the number of possible independent pairwise interactions between chromosome regions, assuming two separate regions per chromosome (P < 5.3e-5). The circular plots illustrating the epistatic interactions were created with Circos (Krzywinski et al. 2009).
Results
For all four families and developmental stages there were only small differences between the phenotypic values of the parental lines (Figure 1). The parents are all adapted elite winter triticale lines and the differences between families were marginal compared with the variation within families. In each of the four families DH lines transgressed the respective parents for all three time points and consistently, we observed highly significant (P < 0.01) genotypic variances () and in addition genotype-by-environment interaction variances () for developmental stage at all three time points (DS1–DS3) (Table S1). The heritabilities in the triticale mapping population with 647 genotypes were high with 0.90 for DS1, 0.85 for DS2, and 0.72 for DS3. The phenotypic correlations between developmental stage at the three time points were 0.84 for DS1−DS2, 0.74 for DS2−DS3, and 0.69 for DS1−DS3 (all significant at P < 0.01). Taken together, the phenotypic data capture the developmental progression over time and the data set is therefore well suited to study the underlying genetic dynamics.
Figure 1.

Histograms of the phenotypic values for developmental stage (BBCH) shown for the entire population and for each of the four families at three time points (DS1−DS3). The arrowheads indicate the parents of the families.
Employing association mapping in multiple families, we identified 29, 14, and 14 markers significantly associated with developmental stage at DS1, DS2, and DS3, respectively (Figure 2, Table 1, and Table S2). Interestingly, we identified eight markers that were found to be associated with developmental stage at all three time points, whereas the other markers were only detected at one or two of the time points (Figure 3). These markers explained 73.9% of the genotypic variance for DS1, 52.1% for DS2, and 57.4% for DS3. The proportion of genotypic variance (pG) explained by single markers ranged from 0.1 to 13.0% for DS1, from 0.1 to 18.3% for DS2, and from 0.2 to 17.4% for DS3 (Table S2). Major QTL explaining more than 3% of the genotypic variance were identified on chromosomes 6A, 5R, and 6R for DS1; on 4R and 5R for DS2; and on 2A, 6A, and 5R for DS3. The six parental lines showed differences in their allele composition at the detected QTL, despite their rather similar developmental stage at each of the three time points (Table S2). Consistent with the finding of time point-specific as well as overlapping QTL, we observed temporal dynamics of the contribution of these QTL regions to adult plant phenotypic development (Figure 4). Some QTL regions contributed to a similar extent throughout development, whereas for others the contribution to the genotypic variance was temporally defined. In line with these findings, we also detected QTL significantly associated with the developmental progression from one time point to another (Table S3 and Figure S2).
Figure 2.

Quantitative trait loci for developmental stage at three time points (DS1−DS3). The dashed horizontal line indicates the significance threshold and significantly associated markers are shown in red.
Table 1. Results of QTL mapping and fivefold cross-validation for developmental stage (DS) at three time points.
| DS1 | DS2 | DS3 | |
|---|---|---|---|
| QTLDS | 29 | 14 | 14 |
| pG-DS | 73.9 | 52.1 | 57.4 |
| QTLES | 21.8 | 12.2 | 10.5 |
| pG-ES | 68.4 | 57.7 | 64.3 |
| pG-TS | 42.9 | 35.2 | 40.6 |
| Relative bias | 37.3 | 39.0 | 36.9 |
Number of detected quantitative trait loci (QTLDS), proportion of genotypic variance (%) explained by the detected QTL across all families in the data set (pG-DS), number of QTL (QTLES), and proportion of genotypic variance averaged over estimation sets (pG-ES) and averaged over test sets (pG-TS), and relative bias (%) in the estimation of pG.
Figure 3.
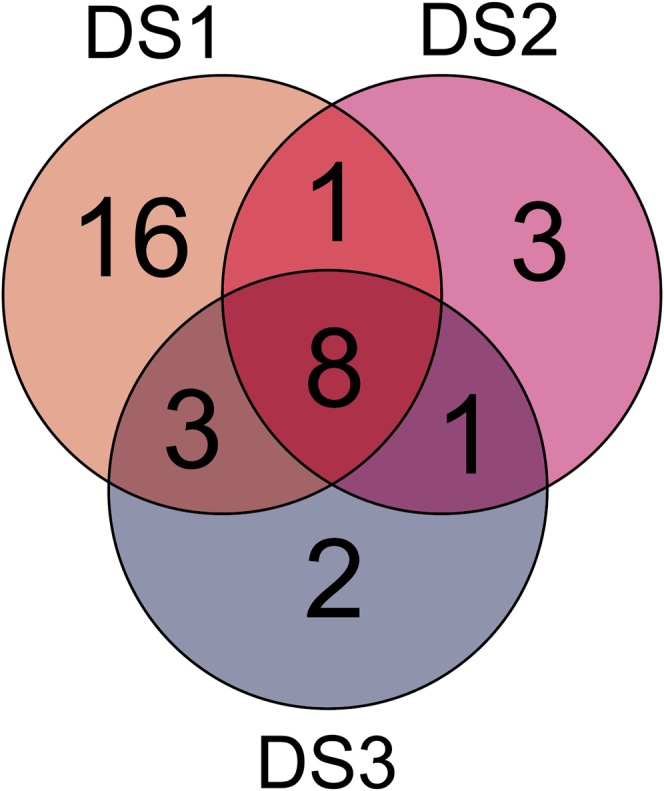
Venn diagram for developmental stage quantitative trait loci detected at three time points (DS1−DS3).
Figure 4.

Temporal development of the contribution of the quantitative trait loci detected for any of the three time points to the proportion of explained genotypic variance for developmental stage (DS).
We employed a fivefold cross-validation approach to obtain asymptotically unbiased estimates of the proportion of genotypic variance explained by the detected markers. The cross-validated pG still ranged between 35.2% for DS2 to 42.9% for DS1 (Table 1). The obtained QTL frequency distributions revealed that most major QTL were identified in the majority of the runs whereas some other markers were only identified in few of the runs (Figure S3).
We next performed a full two-dimensional scan for epistatic QTL at each of the three developmental stages, which revealed two epistatic interactions for DS1 and DS3 and four for DS2 (Figure 5). Of these, one epistatic QTL involving chromosomes 1B and 5R was identified at all three time points. The pG of these epistatic QTL ranged between 0.0 and 3.5%.
Figure 5.

Epistatic quantitative trait loci for developmental stage at three time points (DS1−DS3).
Discussion
The progression of adult plant development affects traits such as flowering time or ripening and thus has consequences for many agronomically important traits, including adaptation and grain yield. The developmental stage of a plant is a plastic trait that undergoes dynamic temporal changes. Nevertheless, attempts to decipher the underlying genetics have thus far concentrated on single time points, mainly flowering time. In this study, we used a large mapping population and three time points to investigate the temporal dynamics of the genetic architecture during adult plant development.
Phenotypic progression of adult plant development
Developmental stage was assessed in the mapping population at three time points when the majority of the plants were at the BBCH stages 49, 69, and 81, corresponding to the stages of awns becoming visible, late flowering, and very early dough development, respectively (Lancashire et al. 1991). The heritability was high for all three time points but highest for DS1, the first evaluated time point for which also the highest ratio of genotypic to genotype-by-environment interaction variance was observed (Table 1). This may indicate a stronger contribution of genotype-by-environment interactions at the later developmental stages.
The parents of the families were very similar in their developmental stage at each of the three time points (Figure 1). Nevertheless, we observed trangressive segregation, suggesting that the parents have all been adapted to the Central European environment by breeding but achieve this by different underlying genetics. The DH lines transgressing their respective parents suggest that the parents carry in part complementary QTL alleles that are newly combined in the progeny (Figure 1). The observed trait distributions indicate that adult plant development is a complex trait controlled by multiple loci and that different alleles for these loci are present in elite triticale germplasm. This allelic diversity is the foundation for the phenotypic plasticity that can be used in breeding programs. The data set under study thus represents an excellent resource to unravel the genetic basis of adult plant development in triticale as a representative for small grain cereals.
Detection of QTL regions for developmental stage
The progression of developmental stage in small grain cereals is likely affected by a number of pathways and thus potentially also by the major QTL underlying plant height (Rht genes), vernalization (Vrn genes), or photoperiod (Ppd). The semi-dwarf Reduced height (Rht)-1 homeologous loci from wheat are located on the group 4 chromosomes of the different genomes (Wilhelm et al. 2013). In the mapping population underlying this study, the major plant height gene Ddw1 (Korzun et al. 1996; Börner et al. 1999), located on the rye chromosome 5R has been shown to segregate and to exhibit a strong effect on plant height (Alheit et al. 2014). Photoperiod (Ppd) homeologs are located on group 2 chromosomes and in wheat Ppd-D1 has been shown to possess the strongest effect on photoperiod insensitivity and thus the transition from vegetative to generative growth (Beales et al. 2007). The homeologs of the three Vernalization (Vrn) genes are located on chromosomes 5 (Vrn1), 4 or 5 (Vrn2), and 7 (Vrn3), respectively (Yan et al. 2003, 2004, 2006). We identified QTL for all three time points which cross-validated still explained a considerable proportion of genotypic variance of approximately 40% (Table 1). The identified major QTL for the different time points, explaining more than 3% of the genotypic variance, were located on chromosomes 2A, 6A, 4R, 5R, and 6R (Table S2). The major QTL on chromosome 2A identified to affect plant development might correspond to a Ppd homeolog and the QTL on 5B to Vrn-1, and both may be identified due to an accelerated transition to the generative phase by certain alleles at these loci. The other identified major QTL do not appear to correspond to any of the abovementioned major genes and therefore may represent other pathways affecting plant development. We have previously performed a QTL mapping in this population for plant height (Alheit et al. 2014) and 9 of the 12 plant height QTL regions were identified in this study to affect developmental stage. Although these QTL were not major QTL, this finding illustrates the interrelations between plant height and adult plant development. As discussed previously, the evaluation of the allele composition at the detected QTL for the six parental lines indeed revealed differences among them consistent with the observed strong variation within the families.
Epistasis refers to interactions between two or more loci in the genome and has recently been shown to contribute to the genetic control of complex traits in both triticale parents, wheat and rye (Li et al. 2011; Reif et al. 2011b). In addition to main effect QTL, we also identified few epistatic QTL for all three time points in development (Figure 5). In general, however, identified epistatic interactions do not allow to infer the biological mechanisms of the gene interactions (Carlborg and Haley 2004). The limited number of detected epistatic QTL may be misleading because the power to detect them depends more on population size than the power for the detection of main effect QTL. Thus, a higher number of epistatic QTL may be involved but remained undetected despite the comparably large size of the mapping population. Our results thus illustrate that epistatic interactions are a component of the genetic architecture underlying adult plant development in triticale.
Temporal dynamics underlying the genetic control
Chromosomal regions harboring QTL were identified for all three time points but more for DS1 than for the later two time points (Figure 2 and 3). DS1 corresponded to the developmental stage when the awns became visible. This may indicate that development until that stage is shaped predominantly by QTL with effects large enough to be detected by QTL mapping, whereas the subsequent development might require more fine-tuning orchestrated by smaller effect QTL that escape detection. Interestingly, some QTL were detected at all three time points, whereas others were only identified at one or two time points. As illustrated by the Manhattan plots (Figure 3), this temporal contribution of QTL to adult plant development was not simply an artifact of these QTL being just above or below the significance threshold at the different time points; rather, it shows that some QTL are active and contribute to developmental progression during a longer period of time encompassing several developmental stages whereas other QTL act in a more timely restricted manner. This conclusion was further substantiated by the varying contribution of the identified QTL regions to the genotypic variance (Figure 4). An alternative approach to assess the genetics underlying dynamic traits is termed functional mapping, where genetic differences in growth trajectories during trait development are used for QTL mapping (Ma et al. 2002; Zhao et al. 2005; Wu and Lin 2006). This, however, requires a much higher number of time points as used in this study, but as illustrated by our results warrants future research for adult plant development in cereals.
Furthermore, we observed that for some chromosomal regions identified as QTL at more than one time point, the sign of the additive effect changed between time points (e.g., QTL on chromosome 6A) (Table S2). This was mirrored by QTL detected for the same regions for the developmental progression between the stages when the change in sign of the QTL effects occurred (Table S2, Table S3, and Figure S2). This finding may indicate that certain QTL alleles accelerate or decelerate developmental progression in a developmental stage-specific manner. Such information could be useful in applied plant breeding to tailor varieties which, for example, have an extended grain filling phase to maximize yield potential. Taken together, the plasticity of adult plant development in triticale is mirrored by dynamic genetic patterns of regulation.
In this study we used a large triticale mapping population, phenotyped at different time points, to investigate the genetic control underlying adult plant development. Our results show that both main and epistatic QTL contribute to the genetic architecture of developmental stage at all three time points. By considering more than just a single time point in development we demonstrate that development during the reproductive phase in triticale is controlled by dynamic patterns of genetic regulation. This confirms that a temporal assessment, as facilitated for example by precision phenotyping platforms (Busemeyer et al. 2013b), is of paramount importance to study the changes in the genetic control underlying dynamic traits. In a wider context, our discoveries on the dynamic genetic control underlying triticale development are likely to have broad relevance to other crops as well as other plant species in general, as adult plant development likely shares a similar dynamic genetic pattern of regulation.
Supplementary Material
Acknowledgments
This research was funded by the German Federal Ministry of Education and Research (BMBF) under the promotional reference 0315414. This publication only reflects the views of the authors. We acknowledge the handling of the funding by the Project Management Organization Jülich (PtJ). We thank Christiane Maus, Agnes Rölfing-Finze, Hans Häge, Jacek Till, and Justus von Kittlitz for outstanding work in the field.
Footnotes
Communicating editor: A. Charcosset
Literature Cited
- Alheit K. V., Reif J. C., Maurer H. P., Hahn V., Weissmann E. A., et al. , 2011. Detection of segregation distortion loci in triticale (× Triticosecale Wittmack) based on a high-density DArT marker consensus genetic linkage map. BMC Genomics 12: 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alheit K. V., Maurer H. P., Reif J. C., Tucker M. R., Hahn V., et al. , 2012. Genome-wide evaluation of genetic diversity and linkage disequilibrium in winter and spring triticale (× Triticosecale Wittmack). BMC Genomics 13: 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alheit K. V., Busemeyer L., Liu W., Maurer H. P., Gowda M., et al. , 2014. Multiple-line cross QTL mapping for biomass yield and plant height in triticale (× Triticosecale Wittmack). Theor. Appl. Genet. 127: 251–260 [DOI] [PubMed] [Google Scholar]
- Beales J., Turner A., Griffiths S., Snape J. W., Laurie D. A., 2007. A pseudo-response regulator is missexpressed in the photoperiod insensitive Ppd-D1a mutant of wheat (Triticum aestivum L.). Theor. Appl. Genet. 115: 721–733 [DOI] [PubMed] [Google Scholar]
- Blanc G., Charcosset A., Mangin B., Gallais A., Moreau L., 2006. Connected populations for detecting quantitative trait loci and testing for epistasis: an application in maize. Theor. Appl. Genet. 113: 206–224 [DOI] [PubMed] [Google Scholar]
- Börner A., Korzun V., Voylokov A. V., Weber W. E., 1999. Detection of quantitative trait loci on chromosome 5R of rye (Secale cereale L.). Theor. Appl. Genet. 98: 1087–1090 [Google Scholar]
- Buckler E. S., Holland J. B., Bradbury P. J., Acharya C. B., Brown P. J., et al. , 2009. The genetic architecture of maize flowering time. Science 325: 714–718 [DOI] [PubMed] [Google Scholar]
- Busemeyer L., Ruckelshausen A., Möller K., Melchinger A. E., Alheit K. V., et al. , 2013a Precision phenotyping of biomass accumulation in triticale reveals temporal genetic patterns of regulation. Sci. Rep. 3: 2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busemeyer L., Mentrup D., Möller K., Wunder E., Alheit K. V., et al. , 2013b Breedvision - A multi-sensor platform for non-destructive field-based phenotyping in plant breeding. Sensors (Switzerland) 13: 2830–2847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlborg Ö., Haley C. S., 2004. Epistasis: too often neglected in complex trait studies? Nat. Rev. Genet. 5: 618–625 [DOI] [PubMed] [Google Scholar]
- Edwards D., Batley J., Snowdon R., 2013. Assessing complex crop genomes with next-generation sequencing. Theor. Appl. Genet. 126: 1–11 [DOI] [PubMed] [Google Scholar]
- Holland J. B., Portyanko V. A., Hoffmann D. L., Lee M., 2002. Genomic regions controlling vernalization and photoperiod responses in oat. Theor. Appl. Genet. 105: 113–126 [DOI] [PubMed] [Google Scholar]
- Holm S., 1979. A simple sequentially rejective Bonferroni test orocedure. Scand. J. Stat. 6: 65–70 [Google Scholar]
- Korzun V., Melz G., Börner A., 1996. RFLP mapping of the dwarfing (Ddw1) and hairy peduncle (Hp) genes on chromosome 5 of rye (Secale cereale L.). Theor. Appl. Genet. 92: 1073–1077 [DOI] [PubMed] [Google Scholar]
- Krzywinski M., Schein J., Birol I., Connors J., Gascoyne R., et al. , 2009. Circos: an information aesthetic for comparative genomics. Genome Res. 19: 1639–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancashire P. D., Bleiholder H., van Boom T. D., Langelüddeke P., Stauss R., et al. , 1991. A uniform decimal code for growth stages of crops and weeds. Ann. Appl. Biol. 119: 561–601 [Google Scholar]
- Li Y., Böck A., Haseneyer G., Korzun V., Wilde P., et al. , 2011. Association analysis of frost tolerance in rye using candidate genes and phenotypic data from controlled, semi-controlled, and field phenotyping platforms. BMC Plant Biol. 11: 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W., Reif J. C., Ranc N., Porta G. D., Würschum T., 2012. Comparison of biometrical approaches for QTL detection in multiple segregating families. Theor. Appl. Genet. 125: 987–998 [DOI] [PubMed] [Google Scholar]
- Liu W., Maurer H. P., Reif J. C., Melchinger A. E., Utz H. F., et al. , 2013. Optimum design of family structure and allocation of resources in association mapping with lines from multiple crosses. Heredity 110: 71–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W., Gowda M., Reif J. C., Hahn V., Ruckelshausen A., et al. , 2014. Genetic dynamics underlying phenotypic development of biomass yield in triticale. BMC Genomics 15: 458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C.-X., Casella G., Wu R., 2002. Functional mapping of quantitative trait loci underlying the character process: a theoretical framework. Genetics 161: 1751–1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebaï A., Goffinet B., 1993. Power of tests for QTL detection using replicated progenies derived from a diallel cross. Theor. Appl. Genet. 86: 1014–1022 [DOI] [PubMed] [Google Scholar]
- Reif J. C., Gowda M., Maurer H. P., Longin C. F. H., Korzun V., et al. , 2011a Association mapping for quality traits in soft winter wheat. Theor. Appl. Genet. 122: 961–970 [DOI] [PubMed] [Google Scholar]
- Reif J. C., Maurer H. P., Korzun V., Ebmeyer E., Miedaner T., et al. , 2011b Mapping QTLs with main and epistatic effects underlying grain yield and heading time in soft winter wheat. Theor. Appl. Genet. 123: 283–292 [DOI] [PubMed] [Google Scholar]
- SAS Institute Inc , 2008. SAS User’s Guide, version 9.2. SAS Institute, Cary, NC [Google Scholar]
- Schwarz G., 1978. Estimating the dimension of a model. Ann. Stat. 6: 461–464 [Google Scholar]
- Snowdon R. J., Wittkop B., Rezaidad A., Hasan M., Lipsa F., et al. , 2010. Regional association analysis delineates a sequenced chromosome region influencing antinutritive seed meal compounds in oilseed rape. Genome 53: 917–928 [DOI] [PubMed] [Google Scholar]
- Steinhoff J., Liu W., Reif J. C., Della Porta G., Ranc N., et al. , 2012. Detection of QTL for flowering time in multiple families of elite maize. Theor. Appl. Genet. 125: 1539–1551 [DOI] [PubMed] [Google Scholar]
- Utz H. F., Melchinger A. E., Schön C.-C., 2000. Bias and sampling error of the estimated proportion of genotypic variance explained by quantitative trait loci determined from experimental data in maize using cross validation and validation with independent samples. Genetics 154: 1839–1849 [PMC free article] [PubMed] [Google Scholar]
- Wang M., Jiang N., Jia T., Leach L., Cockram J., et al. , 2012. Genome-wide association mapping of agronomic and morphologic traits in highly structured populations of barley cultivars. Theor. Appl. Genet. 124: 233–246 [DOI] [PubMed] [Google Scholar]
- Wilhelm E. P., Mackay I. J., Saville R. J., Korolev A. V., Balfourier F., et al. , 2013. Haplotype dictionary for the Rht-1 loci in wheat. Theor. Appl. Genet. 126: 1733–1747 [DOI] [PubMed] [Google Scholar]
- Williams E., Piepho H.-P., Whitaker D., 2011. Augmented p-rep designs. Biom. J. 53: 19–27 [DOI] [PubMed] [Google Scholar]
- Wu R., Lin M., 2006. Functional mapping—how to map and study the genetic architecture of dynamic camplex traits. Nat. Rev. Genet. 7: 229–237 [DOI] [PubMed] [Google Scholar]
- Würschum T., 2012. Mapping QTL for agronomic traits in breeding populations. Theor. Appl. Genet. 125: 201–210 [DOI] [PubMed] [Google Scholar]
- Würschum T., Kraft T., 2014. Cross-validation in association mapping and its relevance for the estimation of QTL parameters of complex traits. Heredity 112: 463–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Würschum T., Maurer H. P., Schulz B., Möhring J., Reif J. C., 2011. Genome-wide association mapping reveals epistasis and genetic interaction networks in sugar beet. Theor. Appl. Genet. 123: 109–118 [DOI] [PubMed] [Google Scholar]
- Würschum T., Tucker M. R., Reif J. C., Maurer H. P., 2012a Improved efficiency of doubled haploid generation in hexaploid triticale by in vitro chromosome doubling. BMC Plant Biol. 12: 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Würschum T., Liu W., Gowda M., Maurer H. P., Fischer S., et al. , 2012b Comparison of biometrical models for joint linkage association mapping. Heredity 108: 332–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Würschum T., Maurer H. P., Dreyer F., Reif J. C., 2013. Effect of inter- and intragenic epistasis on the heritability of oil content in rapeseed (Brassica napus L.). Theor. Appl. Genet. 126: 435–441 [DOI] [PubMed] [Google Scholar]
- Yan J., Zhu J., He C., Benmoussa M., Wu P., 1998. Molecular dissection of developmental behaviour of plant height in rice (Oryza sativa L.). Genetics 150: 1257–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L., Loukoianov A., Tranquilli G., Helguera M., Fahima T., et al. , 2003. Positional cloning of the wheat vernalization gene VRN1. Proc. Natl. Acad. Sci. USA 100: 6263–6268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L., Loukoianov A., Blechl A., Tranquilli G., Ramakrishna W., et al. , 2004. The wheat VRN2 gene is a flowering repressor down-regulated by vernalization. Science 303: 1640–1644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L., Fu D., Li C., Blechl A., Tranquilli G., et al. , 2006. The wheat and barley vernalization gene VRN3 is an orthologue of FT. Proc. Natl. Acad. Sci. USA 103: 19581–19586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J., Holland J. B., McMullen M. D., Buckler E. S., 2008. Genetic design and statistical power of nested association mapping in maize. Genetics 178: 539–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadoks J. C., Chang T. T., Konzak C. F., 1974. A decimal code for the growth stages of cereals. Weed Res. 14: 415–421 [Google Scholar]
- Zhao W., Chen Y. Q., Casella G., Cheverud J. M., Wu R., 2005. A non-stationary model for functional mapping of complex traits. Bioinformatics 21: 2469–2477 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.