

Abstract
Metatropic dysplasia is a clinical heterogeneous skeletal dysplasia characterized by short extremities, a short trunk with progressive kyphoscoliosis, and craniofacial abnormalities that include a prominent forehead, midface hypoplasia, and a squared-off jaw. Dominant mutations in the gene encoding TRPV4, a calcium permeable ion channel, were identified all 10 of a series of metatropic dysplasia cases, ranging in severity from mild to perinatal lethal. These data demonstrate that the lethal form of the disorder is dominantly inherited and suggest locus homogeneity in the disease. Electrophysiological studies demonstrated that the mutations activate the channel, indicating that the mechanism of disease may result from increased calcium in chondrocytes. Histological studies in two cases of lethal metatropic dysplasia revealed markedly disrupted endochondral ossification, with reduced numbers of hypertrophic chondrocytes and presence of islands of cartilage within the zone of primary mineralization. These data suggest that altered chondrocyte differentiation in the growth plate leads to the clinical findings in metatropic dysplasia.
Keywords: metatropic dysplasia, TRPV4, calcium channel, skeleton, chondrocyte
INTRODUCTION
Dominant mutations in the gene encoding TRPV4, a calcium permeable ion channel, have recently been identified in a spectrum of skeletal dysplasias that includes autosomal-dominant brachyolmia, spondylometaphyseal dysplasia Kozlowski type (SMDK) and nonlethal metatropic dysplasia [Rock et al., 2008; Krakow et al., 2009]. In vitro, the mutations have been shown to activate the channel constitutively, resulting in increased baseline intracellular calcium concentration as well as potentiated activation by agonists of the channel. These data suggest that the TRPV4 skeletal disorders result from an increased concentration of intracellular calcium in chondrocytes.
There is a very broad range of clinical severity among patients with metatropic dysplasia [Maroteaux et al., 1966; Kannu et al., 2007; Geneviève et al., 2008]. Common to all cases are the primary clinical findings which establish the diagnosis, including short extremities, an elongated, narrow trunk with progressive kyphoscoliosis [Leet et al., 2006], progressive joint contractures and distinct facial characteristics with a prominent forehead and squared-off jaw. Additional clinical findings, which may not be present in all cases, include odontoid hypoplasia with cervical instability and sensorineural hearing loss [Kannu et al., 2007; Geneviève et al., 2008]. Radiographic findings show dumbbell-shaped long bones with widened metaphyses, very flat and dense vertebral bodies, a halberd-shaped pelvis, and brachydactyly with markedly delayed carpal ossification. At the mildest end of the clinical spectrum, metatropic dysplasia cases resemble SMDK. Moderately severe cases exhibit more severe short stature and progressive deformities. Complications can include a small chest with some degree of respiratory compromise beginning in the neonatal period, and unrelenting kyphoscoliosis. The lethal form of metatropic dysplasia defines the most severe end of the spectrum, in which perinatal death results from cardiopulmonary compromise.
Metatropic dysplasia has also been viewed as genetically complex with both autosomal-dominant and recessive forms proposed [Beck et al., 1983]. Lethal metatropic dysplasia has been described primarily as a recessive disorder. Recognition of dominantly inherited metatropic dysplasia has been based on a preponderance of sporadic cases as well as vertical transmission of the phenotype in families. The conclusion that there are recessively inherited forms of metatropic dysplasia [Beck et al., 1983] resulted from the observation of occurrence of the disorder among multiple siblings born to unaffected parents [Kozlowski et al., 1967; Beck et al., 1983; Kannu et al., 2007; Geneviève et al., 2008]. However, recurrence can also be explained by parental germline mosaicism for a dominant mutation, a possibility further supported by the lack of increased parental consanguinity among families with recurrence. Furthermore, formal segregation analysis [Geneviève et al., 2008] does not support recessive inheritance in all instances of metatropic dysplasia and the phenotype can be viewed as a phenotypic continuum [Kannu et al., 2007].
To test the hypothesis that the full clinical spectrum of metatropic dysplasia could result from dominant mutations in TRPV4, a cohort of 10 genetically independent cases of metatropic dysplasia, ranging from mildly affected to neonatal lethal, was ascertained and analyzed. All cases were heterozygous for mutations in TRPV4, indicating dominant inheritance and a lack of significant locus heterogeneity in metatropic dysplasia, including lethal metatropic dysplasia.
MATERIALS AND METHODS
Subjects
Ten cases of metatropic dysplasia were assessed under an IRB approved protocol and informed consent was obtained for all subjects. All of the cases were ascertained through the International Skeletal Dysplasia Registry at Cedars-Sinai Medical Center. The diagnosis of metatropic dysplasia was made based on clinical and radiographic information (see text).
Mutation Analysis
Genomic DNA was extracted from whole blood samples or cheek swabs by standard protocols using a Qiagen (Valencia, CA) QIAamp DNA Mini Kit. The coding exons of TRPV4 (Ensembl, ENST00000261740) were amplified from 50 ng of genomic DNA using a touchdown PCR reaction with an annealing temperature at 60°C for 5 cycles, 58°C for another 5 cycles, and 54°C for the last 35 cycles. Oligonucleotide sequences have been described previously [Rock et al., 2008]. Bidirectional sequence analysis was performed (MC Laboratories, San Francisco, CA) and the resulting sequences were compared to the reference sequence for TRPV4, with nucleotide numbering starting from the A of the ATG initiation codon. For mutations resulting from insertion and/or deletion events, PCR fragments were cloned into plasmid DNA (TOPO TA Cloning Kit for Sequencing, Invitrogen, Carlsbad, CA). Twenty clones were selected for each mutation, plasmid DNA was isolated (QIAamp DNA Mini Kit, Qiagen) and sequence analysis was performed as described above.
Histology
In case R09-035 the leg bones were dissected from the surrounding soft tissue at the time of necropsy and bisected longitudinally. A representative longitudinal 0.5-cm thick section of tissue was taken and fixed in 10% phosphate-buffered formalin (Fisher Diagnostics, Kalamazoo, MI), decalcified using Decalcifier I (Surgipath Medical Industries, Richmond, IL), and embedded in paraffin. Five-micron thick paraffin sections were cut and stained with hematoxylin and eosin. Decalcified sections were also similarly prepared from bone samples from case R08-023 and examined.
Electrophysiology of Normal and Mutant TRPV4 in HEK293 cells
Human embryonic kidney cells (HEK293T) were grown in DMEM containing 10% (v/v) human serum, 2 mM L-glutamine, 2 U/ml penicillin, and 2 mg/ml streptomycin at 37°C in a humidity-controlled incubator with 10% CO2. As previously described, cells were transiently transfected with a human TRPV4 (accession number NP_671737) vector, cloned as a BamHI fragment into the BcII-site of the pCAGGS/IRES-GFP vector. Mutagenesis to introduce the I331F and P799L mutations, and HEK cell transfections were performed as previously described [Rock et al., 2007; Krakow et al., 2009].
Whole-cell membrane currents were measured with an EPC-10 (HEKA Elektronik, Lambrecht, Germany) using ruptured patches sampled at 20 kHz and filtered at 2.9 kHz as previously described [Rock et al., 2007; Krakow et al., 2009] with and without treatment with the TRPV4-specific activating phorbal ester, 4α-phorbol 12,13, didecanoate (4α-PDD). Similar extracellular solution and pipette solution were used as described in [Krakow et al., 2009]. Intracellular [Ca2+]i was measured in Fura-2-loaded cells with a monochromator-based imaging system [Vriens et al., 2005, 2007]. Ca2+ influxes were monitored by Cell™ imaging software (Olympus, Essex, UK) using OLYMPUS IX81 (Olympus) with standard extracellular solution similar to that described for electrophysiology.
RESULTS
Ten genetically independent metatropic dysplasia cases were studied, comprising the full range of clinical and radiographic severity (Figs. 1 and 2 and Table I). Based on the severity of the spinal abnormalities, the degree of metaphyseal flaring, and the presence of respiratory compromise, each of the five nonlethal cases was assigned as mild, moderate, or severe. Mild cases had little or no scoliosis, mild platyspondyly, mild metaphyseal widening, and carpal ossification delay. Moderately severe cases had scoliosis, platyspondyly with irregular end plates, widened irregular metaphyses, and marked epiphyseal delay. Severe cases of nonlethal metatropic dysplasia exhibited severe scoliosis, wafer-thin vertebral bodies with wedging, dumbbell-shaped long bones with metaphyseal irregularity and significant epiphyseal delay. The five lethal metatropic dysplasia cases were separated according to whether they died in the immediate neonatal period (two cases) or prior to 12 months of age (infantile-lethal).
FIG. 1.

Radiographs of nonlethal metatropic dysplasia cases. Case numbers are shown below each radiograph. Arrows identify some of the characteristic findings in metatropic dysplasia including marked platyspondyly with dense, wafer-like vertebral bodies (A–D) and metaphyseal abnormalities of the metacarpals (I).
FIG. 2.

Two cases of neonatal lethal metatropic dysplasia. (A, B) Both infants have short limbs and small chest cavities. (C, D) Note the nodular appearance of the right ear of R09-35 in B. The left ear was similarly enlarged.
TABLE I.
Metatropic Dysplasia Cases and Mutations
| Patient | Clinical severity | TRPV4 exon | Nucleotide substitution | Predicted amino acid change | Protein domain |
|---|---|---|---|---|---|
| R94-316 | Lethal, neonatal | 2 | c.C366 >T | p.T89I | Cytoplasmic (NH2 end) |
| R99-441 | Lethal, infantile | 4 | c.A590 >G | p.K197R | ANK2, vanilloid receptor |
| R00-067a | Severe | 6 | c.A991 >T | p.I331F | ANK5 |
| R97-288 | Mild | 6 | c.999_1010del | p.D333_E337delinsE | ANK5 |
| R94-199 | Lethal, infantile | 8 | c.1412_1414del | p.F471del | TM1 |
| R08-023 | Lethal, neonatal | 11 | c.C1812 >G | p.I604M | Cytoplasmic (TM4–TM5) |
| R08-325 | Mild | 12 | c.C1851 >A | p.F617L | TM5 |
| R09-035 | Lethal, neonatal | 12 | c.T1853 >C | p.L618P | TM5 |
| R01-187 | Mild | 15 | c.G2389 >A | p.E797K | Cytoplasmic (COOH end) |
| R91-149 | Moderate | 15 | c.C2396 >T | p.P799L | Cytoplasmic (COOH end) |
| R08-078 | Moderate | 15 | c.C2396 >T | p.P799L | Cytoplasmic (COOH end) |
| R03-386a | Moderate | 15 | c.C2396 >T | p.P799L | Cytoplasmic (COOH end) |
Previously published [Krakow et al., 2009].
As seen in the two cases previously studied [Krakow et al., 2009], all five nonlethal metatropic dysplasia cases were heterozygous for sequence changes in TRPV4 (Table I and Fig. 3). Two of the cases with mild metatropic dysplasia, R01-187 and R08-325, were each heterozygous for point mutations predicting the amino acid substitutions p.E797K in the carboxyl-terminal tail of the protein and p.F617L in transmembrane segment 5(TM5), respectively. A third mild case, R97-288, was heterozygous for an in-frame deletion–insertion (c.999_1010del), predicting a p.D333_E337delinsE change in ankyrin repeat 5, toward the amino-terminal end of the molecule. Parental DNA samples were not available for these cases, but all of the sequence alterations are predicted to alter highly evolutionarily conserved residues in the protein. Furthermore, none of the sequence changes was found among at least 200 alleles from ethnically matched control individuals, indicating that they are not common polymorphisms in the population.
FIG. 3.
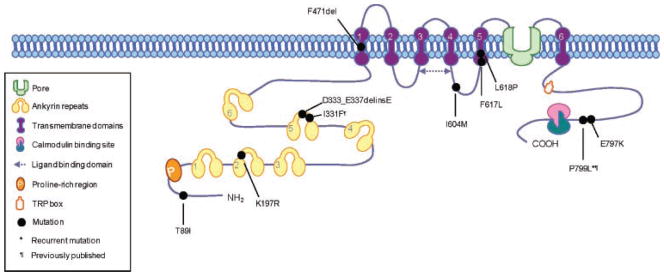
Model of the TRPV4 molecule showing the locations of the implied sequence changes in the protein relative to the recognized protein domains.
The two cases with moderately severe metatropic dysplasia, R08-078 and R91-149, were each heterozygous for the same single-nucleotide substitution c.C2396>T in exon 15, predicted to lead to the amino acid substitution p.P799L in the carboxyl-terminal cytoplasmic tail of the protein. This substitution was previously seen in another patient with moderately severe metatropic dysplasia, R03-386, which was determined to be de novo based on absence of the change in DNA from the unaffected parents [Krakow et al., 2009].
The two infantile-lethal cases were each heterozygous for sequence changes corresponding to different domains in the protein (Table I and Fig. 3). Case R99-441 was heterozygous for a point mutation in exon 4, predicting the amino acid substitution p.K197R in the second ankyrin repeat at the amino-terminal end of the molecule. Proband R94-199 was heterozygous for a 3 bp deletion, removing the codon for F471 in the first transmembrane domain of the protein. Parental DNA samples were not available for these two cases. These sequence changes were not found among over 200 control chromosomes, and both sequence changes imply alteration of amino acid residues that are highly conserved among vertebrates.
Point mutations predicting single amino acid substitutions in TRPV4 were found in all three neonatal lethal cases studied (Table I and Fig. 3). The p.T89I substitution implied by the sequence change found in proband R94-316 was the most amino-terminal change found to date, occurring in a domain of unknown significance. In case R08-023, the p.I604M substitution occurs in the cytoplasmic loop between TM4 and TM5. Case R09-035 was heterozygous for a single-nucleotide substitution predicting a p.L618P substitution in TM5. As in the other cases, all sequence changes are predicted to alter highly evolutionarily conserved amino acid residues and none of the changes was found among at least 200 control alleles.
To determine the effect of two of the metatropic dysplasia mutations on TRPV4 activity, normal and mutant human TRPV4 were expressed in HEK cells [Rock et al., 2008; Krakow et al., 2009]. In comparison to wild-type TRPV4, the I331F and P799L mutations yielded much larger basal currents which represent constitutive open channels without any additional activation (Fig. 4A–D). Similar results were obtained from Ca2+ imaging experiments. Figure 4E–H shows the increase in the intracellular Ca2+ concentration, [Ca2+]i, after application of the TRPV4 selective agonist 4α-PDD which cause much higher responses than in the wild-type TRPV4-transfected cells. Note that the basal [Ca2+]i before 4α-PDD application was much higher that in the control channels (data not shown, but see traces before 4α-PDD application). Supporting information Figures 1 and 2 (supporting information Figs. 1 and 2 may be found in the online version of this article) show the effects for both [Ca2+]i changes and current modulation for the three different modes of activation: 4α-PDD agonist, cell swelling, and application of arachidonic acid (AA). For both mutations significantly higher [Ca2+]i responses were obtained with 4α-PDD and cell swelling, but not with AA. The same results were obtained in whole current clamp measurements. Both mutants I331F and P799L showed a dramatic increase in the activity of the constitutive open channels (Fig. 4A–D), but they also had increased activity following stimulation with all three protocols, for example, including AA (see supporting information Fig. 2G–I which may be found in the online version of this article). These data are very similar to those generated for mutations in TRPV4 that produce autosomal-dominant brachyolmia and SMDK [Rock et al., 2008; Krakow et al., 2009].
FIG. 4.

Basal currents through TRPV4 in metatropic dysplasia (A–C) and effects of stimulation with 2 μM 4α-PDD in WT TRPV4 (E), I331F (F), P799L (G) on the increase in the intracellular Ca2+ concentration [Ca2+]i. A voltage step protocol was used activating TRPV4 channels consisting of 40 mV steps from −80 to +200 mV. Note that the constitutive channel activity of the mutant channels was higher than that of the wild-type channel (histogram in panel D). Panels E–G show the Ca2+ imaging data for activation of TRPV4 with 2 μM 4α-PDD. Note the increased basal Ca2+ level before the agonist stimulation. H: Histogram showing average increases in fluorescence ratio in response to 4α-PDD. Error bars represent means ±SE. Asterisk (*) indicates significant differences when compared with cells expressing wild-type TRPV4 (one-sided Student’s t-test, P <0.01).
Bone samples were taken at autopsy from two of the lethal metatropic dysplasia cases (R08-023 and R09-035) with TRPV4 mutations. As illustrated for case R09-035 in Figure 5A,B, sections cut through the femur and tibia showed that they had extremely short diaphyses. The ends of these bones and the adjacent patella were enlarged with abundant abnormally thick cartilage and the cortices were composed of bone matrix (Fig. 5A,B). Microscopic examination of the bones from this fetus revealed extensive nodular proliferation of immature cartilage at the ends of the tibia and femur, associated with abnormal bone formation. The patella was composed predominantly of abnormal nodular cartilaginous tissue (Fig. 5B). Analysis of the cartilage growth plates in both cases showed absence of well-organized columns of pre-hypertrophic and hypertrophic chondrocytes in any of the bones examined (Fig. 5C,D). In case R09-035 there were irregular nodules of cartilage within the abnormal primary spongiosa (Fig. 5C) and in case R08-023, irregular tongues of hypertrophic chondrocytes were observed invading the primary spongiosa (Fig. 5D). Nodules of cartilage appeared to develop from areas of proliferating mesenchymal stromal cells in which small foci of immature chondrocytes were present along with fully differentiated chondrocytes, suggesting that mesenchymal stromal cells progressed through the typical cell differentiation stages of endochondral ossification (Fig. 5E). However, other islands of abnormal cells were composed of immature woven bone with no maturing chondrocytes and were surrounded by spindle-shaped mesenchymal cells. It appeared that bone may have originated directly from mesenchymal precursors with no intermediate chondrogenesis (Fig. 5E). Similar but less extensive abnormal chondrogenesis was also observed in bone samples from the second case (data not shown). The cortical bone of the diaphysis had numerous porosities and a trabecularized appearance. Some trabecular structures were composed of lamellar matrix and others consisted of abnormally large islands of immature of woven bone (Fig. 5F). Osteoclasts were present within the metaphyseal and cortical bone, but their numbers appeared to be reduced in comparison to control.
FIG. 5.

Morphology of the bones in metatropic dysplasia cases R08-023 and R09-035. A: Gross appearance of sectioned lower limb and histology of case R09-035. The femur and tibia are shortened with very little diaphyseal bone (arrow). There is extensive nodular overgrowth of disordered cartilage in the patella and at the ends of both long bones, which contain brown appearing bone marrow and bone. P, patella; DF, distal femur; PF, proximal femur; T, tibia. B: Low magnification of an H&E stained, paraffin-embedded whole section cut through the knee joint of case R09-035 and mounted on a 7.5 cm × 5 cm glass slide showing lack of growth plates, the nodular nature of the cartilaginous proliferation, and cortical bone (arrow) of the short femoral diaphyses. P, patella; F, femur; T, tibia; BM, bone marrow. C: Section showing the growth plate in case R09-035 illustrating the short hypertrophic zone and cartilage within the primary spongiosa (arrow). D: Growth plate from case R08-023 showing irregular primary spongiosa with trapped cartilage nodules (arrowhead) and a poorly organized hypertrophic zone (thin arrow). PS, primary spongiosa; H, hypertrophic zone. E: Nodules of cartilage (arrowheads) and woven bone (arrows) in case R09-035 arising in islands of mesenchymal cells. M, mesenchymal cell. F: H&E stained diaphyseal cortical bone from case R09-035 showing islands of immature woven bone (arrows) surrounded by normal lamellar bone.
A striking finding was that the tracheal and bronchial cartilage rings were increased in volume due to nodular cartilaginous proliferation and there appeared to be an increase in the number of cartilage elements within the bronchial walls (data not shown). Occasional islands of immature woven bone were seen within the trachea at the level of the thyroid gland. These bony islands were surrounded by stromal mesenchymal cells similar to those observed at the ends of the tibia and femur (Fig. 6).
FIG. 6.

Tracheal cartilage in case R09-035. A: The cartilage in the trachea at the level of the thyroid gland is thickened by irregular nodules of cartilage separated by strands of mesenchymal cells. The adjacent tracheal mucosa (arrow) is unremarkable. B: Trabeculae of immature woven bone (arrows) are present within the foci of mesenchymal cell proliferation.
DISCUSSION
All cases of metatropic dysplasia studied to date, including 10 cases described herein and 2 previously published cases [Krakow et al., 2009], have been heterozygous for sequence changes in TRPV4. A total of seven cases of nonlethal metatropic dysplasia have now been studied, providing evidence of locus homogeneity in nonlethal forms of the disease and suggesting that recessive forms, if they exist, are rare. The mutations alter amino acid residues that are distributed throughout the protein, making it difficult to predict a particular phenotype from the genotype based on the domain of the protein involved. However, three cases with the same mutation, predicting the amino acid substitution p.P799L in the carboxyl-terminal tail of the molecule, all had a moderately severe form of the disorder, indicating that there may be consistency from case to case for the same mutation. The I331F and P799L substitutions activated the TRPV4 channel much as the autosomal-dominant brachyolmia and SMDK mutations do [Rock et al., 2008; Krakow et al., 2009], suggesting that all of the TRPV4 mutations produce disease by similar mechanisms. No clear relationship has emerged between the phenotypes produced by the mutations and the domain of the molecule involved, but it is likely that the degree of activation of TRPV4 is correlated with phenotypic severity.
All of five cases of lethal metatropic dysplasia were also heterozygous for TRPV4 mutations. These data demonstrate that lethal metatropic dysplasia is primarily a dominant disorder and that activation of the channel exerts its phenotypic effect independent of the presence of the normal allele. Implicit in these findings is that recurrence among siblings with unaffected parents results from parental germline mosaicism, an inference that remains to be proven experimentally. Consequently, the empiric recurrence risk for parents who have had one child with lethal metatropic dysplasia is likely to be well below the 25% figure associated with recessive disorders.
Histologic analysis of bones from the two cases of perinatal lethal metatropic dysplasia, each with a different TRPV4 mutation, has provided a consistent picture of the morphologic consequences of the mutations and suggests abnormalities in differentiation of mesenchymal progenitors within the bones and in chondrocyte proliferation and differentiation. Vertebrate long bones are formed initially as cartilage anlagen or moulds. They lengthen at both ends by proliferation of chondrocytes, which differentiate into columns of pre-hypertrophic and hypertrophic chondrocytes in the middle of the moulds. Endochondral ossification begins with in-growth of blood vessels into the hypertrophic cartilage, which gets resorbed and replaced by bone. Eventually, growth plates form at both ends of the bones and a medullary cavity is formed [Karsenty et al., 2009]. In both cases of lethal metatropic dysplasia studied, proliferating chondrocytes did not align properly with hypertrophic chondrocytes to form typical columns. Instead, there was disordered proliferation of chondrocytes with formation of nodules of cartilage that varied in size, shape, and degree of maturation. These nodules caused enlargement of the ends of the long bones examined and filled the areas where growth plates typically would have been present. There were hypertrophic chondrocytes in their centers and proliferating chondrocytes at their peripheries and many were surrounded by bone, indicating that that some of the hypertrophic cartilage was replaced by bone, as happens during endochondral ossification. Additionally, these islands of proliferating mesenchymal cells also appeared to give rise directly to osteoblasts, which formed irregularly shaped trabeculae of woven bone between the cartilage nodules. This suggests the possibility that TRPV4 could have a role in bone that prevents mesenchymal cell differentiation directly into osteoblasts in the developing long bone anlagen.
The role of TPRV4 in bone and cartilage has been investigated recently. Osteoblasts and osteoclasts express TRPV4 [Mizoguchi et al., 2008], and Masuyama et al. [2008] showed that a TRPV4 knock-out mouse model had increased bone mass due to a defect in the terminal differentiation of osteoclasts. In the absence of Trpv4, calcium oscillations declined leading to an effect on NFATc-1-regulated transcription that affected normal terminal differentiation and activity of osteoclasts; expression of NFATC-1 is required in osteoclast precursors for their differentiation. Loss of osteoclast activity or formation during skeletal development leads to increased bone mass. Analysis of bone in the R09-035 case showed a diminished number of osteoclasts, but it is not clear what the effect of heterozygosity for activating mutations in TRPV4 is on osteoclast differentiation.
Trpv4 was identified originally as a channel protein activated by hypotonic cell swelling [Xu et al., 2003; Chen et al., 2007; Loukin et al., 2009], but it can also be activated by other influences, including temperature and acidic pH [Clark et al., 2008; Damann et al., 2008]. Activation of normal Trpv4 in the ATDC5 chondrocyte cell model was shown to increase the steady-state levels of Sox9 mRNA and protein and derepression of Sox6 [Muramatsu et al., 2007]. Sox9 is a transcription factor that initiates the commitment of mesenchymal cells into chondrocytes and is critical for the early stages of chondrogenesis [Akiyama et al., 2002]. Sox9 also plays a critical role in endochondral ossification by directing the expression of cartilage-specific extracellular matrix molecules, including the cartilage collagens [Lefebvre et al., 1997; Bridgewater et al., 1998; Zhang et al., 2003]. Analysis of a Sox9 knock-in mutant mouse [Akiyama et al., 2004] showed that overexpression of Sox9 disrupts the orderly formation of chondrocytes in the growth plate and delayed endochondral ossification. Others have reported that in concert with the Sox5 and Sox6 co-factors, Sox9 inhibits terminal differentiation of chondrocytic cell lines [Ikeda et al., 2004; Saito et al., 2007]. It is possible that the activation of the TRPV4 channel in metatropic dysplasia leads to increased SOX9 and subsequently SOX5 and SOX6 activity, negatively regulating the terminal differentiation of growth plate chondrocytes. This could explain the histologic findings in the metatropic dysplasia growth plates, including the markedly diminished number of hypertrophic chondrocytes and the loss of normal terminal differentiation to establish a sharp transition between hypertrophic cells and the primary spongiosa.
We conclude that dominant mutations in TRPV4 can account for a range of phenotypes that include autosomal-dominant brachyolmia, SMDK, and both nonlethal and lethal forms of metatropic dysplasia. While the mutation analysis did not reveal a clear relationship between the domain of the protein affected and a particular phenotype, it is likely that the phenotypic consequence of each mutation reflects the extent to which the TRPV4 channel is activated and the downstream consequences of increased calcium in chondrocytes.
Supplementary Material
Acknowledgments
We thank the families for their participation. This research was supported in part by NIH grants HD22657 and AR43510. We thank A. Janssens for generation of the both metatropic mutant constructs. J.V. is a postdoctoral fellow of the Research Foundation-Flanders. This work was supported by grants from the Belgian Ministry for Science Policy (Interuniversity Attraction Pole IUAP P6/28), the Research Foundation-Flanders (G.0172.03 and G.0565.07), and the Research Council of the KU Leuven (GOA 2004/07 and EF/95/010).
Footnotes
Additional supporting information may be found in the online version of this article.
References
- Akiyama H, Chaboissier MC, Martin JF, Schedl A, de Crombrugghe B. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 2002;16:2813–2828. doi: 10.1101/gad.1017802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama H, Lyons JP, Mori-Akiyama Y, Yang X, Zhang R, Zhang Z, Deng JM, Taketo MM, Nakamura T, Behringer RR, McCrea PD, de Crombrugghe B. Interactions between Sox9 and beta-catenin control chondrocyte differentiation. Genes Dev. 2004;18:1072–1087. doi: 10.1101/gad.1171104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck M, Roubicek M, Rogers JG, Naumoff P, Spranger J. Heterogeneity of metatropic dysplasia. Eur J Pediatr. 1983;140:231–237. doi: 10.1007/BF00443368. [DOI] [PubMed] [Google Scholar]
- Bridgewater LC, Lefebvre V, de Crombrugghe B. Chondrocyte-specific enhancer elements in the Col11a2 gene resemble the Col2a1 tissue-specific enhancer. J Biol Chem. 1998;273:14998–15006. doi: 10.1074/jbc.273.24.14998. [DOI] [PubMed] [Google Scholar]
- Chen X, Alessandri-Haber N, Levine JD. Marked attenuation of inflammatory mediator-induced C-fiber sensitization for mechanical and hypotonic stimuli in TRPV4−/− mice. Mol Pain. 2007;3:31. doi: 10.1186/1744-8069-3-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark K, Middelbeek J, van Leeuwen FN. Interplay between TRP channels and the cytoskeleton in health and disease. Eur J Cell Biol. 2008;87:631–640. doi: 10.1016/j.ejcb.2008.01.009. [DOI] [PubMed] [Google Scholar]
- Damann N, Voets T, Nilius B. TRPs in our senses. Curr Biol. 2008;18:R880–R889. doi: 10.1016/j.cub.2008.07.063. [DOI] [PubMed] [Google Scholar]
- Geneviève D, Le Merrer M, Feingold J, Munnich A, Maroteaux P, Cormier-Daire V. Revisiting metatropic dysplasia: Presentation of a series of 19 novel patients and review of the literature. Am J Med Genet Part A. 2008;146A:992–996. doi: 10.1002/ajmg.a.32191. [DOI] [PubMed] [Google Scholar]
- Ikeda T, Kamekura S, Mabuchi A, Kou I, Seki S, Takato T, Nakamura K, Kawaguchi H, Ikegawa S, Chung UI. The combination of SOX5, SOX6, and SOX9 (the SOX trio) provides signals sufficient for induction of permanent cartilage. Arthritis Rheum. 2004;11:3561–3573. doi: 10.1002/art.20611. [DOI] [PubMed] [Google Scholar]
- Kannu P, Aftimos S, Mayne V, Donan L, Savarirayan R. Metatropic dysplasia: Clinical and radiographic findings in 11 patients demonstrating long-term natural history. Am J Med Gene Part A. 2007;143A:2512–2522. doi: 10.1002/ajmg.a.31941. [DOI] [PubMed] [Google Scholar]
- Karsenty G, Kronenberg HM, Settembre C. Genetic control of bone formation. Ann Rev Cell Dev Biol. 2009;25:629–648. doi: 10.1146/annurev.cellbio.042308.113308. [DOI] [PubMed] [Google Scholar]
- Kozlowski K, Maroteaux P, Spranger JW. La dysostose spondylometaphisaire. Presse Med. 1967;75:2769–2774. [Google Scholar]
- Krakow D, Vriens J, Camacho N, Luong P, Deixler H, Funari TL, Bacino CA, Irons MB, Holm IA, Sadler L, Okenfuss EB, Janssens A, Voets T, Rimoin DL, Lachman RS, Nilius B, Cohn DH. Mutations in the gene encoding the calcium-permeable ion channel TRPV4 produce spondylometaphyseal dysplasia, Kozlowski type and metatropic dysplasia. Am J Hum Genet. 2009;84:307–315. doi: 10.1016/j.ajhg.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leet AI, Sampath JS, Scott CI, Jr, MacKenzie WG. Cervical spinal stenosis in metatropic dysplasia. J Pediatr Orthop. 2006;26:347–352. doi: 10.1097/01.bpo.0000214923.95314.ad. [DOI] [PubMed] [Google Scholar]
- Lefebvre V, Huang W, Harley VR, Goodfellow PN, de Crombrugghe B. SOX9 is a potent activator of the chondrocyte-specific enhancer of the pro alpha1(II) collagen gene. Mol Cell Biol. 1997;17:2336–2346. doi: 10.1128/mcb.17.4.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loukin SH, Su Z, Kung C. Hypotonic shocks activate rat TRPV4 in yeast in the absence of polyunsaturated fatty acids. FEBS Lett. 2009;583:754–758. doi: 10.1016/j.febslet.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroteaux P, Spranger JW, Wiedemann H-R. Der metatropische Zwergwuchs (metatrophic dwarfism) Arch Kinderheilk. 1966;173:211–226. [PubMed] [Google Scholar]
- Masuyama R, Vriens J, Voets T, Karashima Y, Owsianik G, Vennekens R, Lieben L, Torrekens S, Moermans K, Vanden Bosch A, Bouillon R, Nilius B, Carmeliet G. TRPV4-mediated calcium influx regulates terminal differentiation of osteoclasts. Cell Metab. 2008;8:257–265. doi: 10.1016/j.cmet.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Mizoguchi F, Mizuno A, Hayata T, Nakashima K, Heller S, Ushida T, Sokabe M, Miyasaka N, Suzuki M, Ezura Y, Noda M. Transient receptor potential vanilloid 4 deficiency suppresses unloading-induced bone loss. J Cell Physiol. 2008;216:47–53. doi: 10.1002/jcp.21374. [DOI] [PubMed] [Google Scholar]
- Muramatsu S, Wakabayashi M, Ohno T, Amano K, Ooishi R, Sugahara T, Shiojiri S, Tashiro K, Suzuki Y, Nishimura R, Kuhara S, Sugano S, Yoneda T, Matsuda A. Functional gene screening system identified TRPV4 as a regulator of chondrogenic differentiation. J Biol Chem. 2007;282:32158–32167. doi: 10.1074/jbc.M706158200. [DOI] [PubMed] [Google Scholar]
- Rock MJ, Prenen J, Funari VA, Funari TL, Merriman B, Nelson SF, Lachman RS, Wilcox WR, Reyno S, Quadrelli R, Vaglio A, Owsianik G, Janssens A, Voets T, Ikegawa S, Nagain T, Rimoin DL, Nilius B, Cohn DH. Gain-of-function mutations in TRPV4 cause autosomal dominant brachyolmia. Nat Genet. 2008;40:999–1003. doi: 10.1038/ng.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Ikeda T, Nakamura K, Chung UI, Kawaguchi H. S100A1 and S100B, transcriptional targets of SOX trio, inhibit terminal differentiation of chondrocytes. EMBO Rep. 2007;8:504–509. doi: 10.1038/sj.embor.7400934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vriens J, Owsianik G, Fisslthaler B, Suzuki M, Janssens A, Voets T, Morisseau C, Hammock BD, Fleming I, Busse R, Nilius B. Modulation of the Ca2 permeable cation channel TRPV4 by cytochrome P450 epoxygenases in vascular endothelium. Circ Res. 2005;97:908–915. doi: 10.1161/01.RES.0000187474.47805.30. [DOI] [PubMed] [Google Scholar]
- Vriens J, Owsianik G, Janssens A, Voets T, Nilius B. Determinants of 4 alpha-phorbol sensitivity in transmembrane domains 3 and 4 of the cation channel TRPV4. J Biol Chem. 2007;282:12796–12803. doi: 10.1074/jbc.M610485200. [DOI] [PubMed] [Google Scholar]
- Xu H, Zhao H, Tian W, Yoshida K, Roullet JB, Cohen DM. Regulation of a transient receptor potential (TRP) channel by tyrosine phosphorylation. SRC family kinase-dependent tyrosine phosphorylation of TRPV4 on TYR-253 mediates its response to hypotonic stress. J Biol Chem. 2003;278:11520–11527. doi: 10.1074/jbc.M211061200. [DOI] [PubMed] [Google Scholar]
- Zhang P, Jimenez SA, Stokes DG. Regulation of human COL9A1 gene expression. Activation of the proximal promoter region by SOX9. J Biol Chem. 2003;278:117–123. doi: 10.1074/jbc.M208049200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.