

Abstract
Nonsyndromic clefts of the lip and palate [NSCLP] are complex genetic traits. Together, they are classified as one of the most common birth defects with a prevalence of 1/700 live births. Genome-wide association studies [GWAS] for non-syndromic cleft lip with or without cleft palate [NSCL[P]] revealed significant association for common single nucleotide polymorphisms near genes involved in craniofacial development i.e. MAFB, PAX7, VAX1, ARHGAP29 (ABCA4 locus), and IRF6. Sequencing of protein coding regions of the NSCL[P] GWAS candidate genes or adjacent genes suggest a role for rare functional variants. Replication studies in the African population did not observe any significant association with the GWAS candidate genes. On the other hand, the role of rare functional variants in GWAS candidate genes has not been evaluated in the African population. We obtained saliva samples from case triads in Nigeria and Ethiopia for Sanger sequencing of the GWAS candidate genes [MAFB, PAX7, VAX1, ARHGAP29, and IRF6] in order to identify rare functional variants. A total of 220 African samples [140 Nigerians and 80 Ethiopians] were sequenced and we found the following new rare variants— p.His165Asn in the MAFB gene, p.Asp428Asn in the PAX7, a splice-site variant that creates a new donor splice-site in PAX7. We also found three previously reported missense variants p.Gly466Ser in PAX7; p.Leu913Ser and Arg955His in ARHGAP29. No de novo mutations were found. Future genome-wide association and sequencing studies should be conducted using samples from Africa in order to identify new molecular genetic factors that contribute to the etiology of NSCLP.
Keywords: Africa, GWAS, rare variants
INTRODUCTION
Nonsyndromic clefts of the lip and palate [NSCLP] are complex genetic traits and together, they are classified as one of the most common birth defect with a prevalence of 1/700 live births [Mossey and Modell, 2012]. These defects are easily recognizable, responsive to surgical correction and benefit from substantial multi-disciplinary team care. This team care will help restore aesthetics, function and psychosocial stability. However, this comes at a significant life time cost to the healthcare system and families [Wehby and Cassel, 2010]. The etiology of these complex traits has been widely studied in order for us to understand the risk factors and to design strategies for prevention. Genetic factors, environmental factors and the interaction between these two factors have been extensively reported in the literature as prime contributors to cleft outcomes [Dixon et al., 2012].
Genome-wide association studies for nonsyndromic cleft lip with or without palate [NSCL(P)] reported significant association for common variants in intergenic regions close to genes that are involved in craniofacial development i.e. MAFB, VAX1, PAX7 and IRF6 [Birnbaum et al., 2009, Grant et al., 2009, Beaty et al., 2010; Mangold et al., 2010]. A significant association was also reported in a gene desert in the Chr 8q.24 locus [Birnbaum et al., 2009, Grant et al., 2009, Beaty et al., 2010; Mangold et al., 2010]. This gene desert is enriched for epigenetic markers [Huppi et al., 2012]. A recent meta-analysis by Ludwig et al., [2012] confirmed previous GWAS findings and identified six new additional loci that are genome-wide significant. Additional studies in independent populations replicated the GWAS findings [Rojas-Martinez et al., 2010; Nikopensius et al., 2010; Fontoura et al., 2012; Butali et al., 2013].
Resequencing studies conducted on the NSCL[P] GWAS candidate genes or adjacent genes suggest a role for rare variants [Beaty et al., 2010; Leslie et al., 2012; Nasser et al., 2012; Butali et al., 2013]. Data on European and Asian cleft triads from the Beaty et al., [2010] GWAS study suggest that some genes/loci are population specific (chr8q24.21 for Europeans and MAFB for Asians), while other genes play a role in multiple populations [IRF6 in Europeans and Asians]. This conforms to reports from recent population genetics studies showing rare variants to be population specific [Keinan and Clark, 2012; Tennessen et al., 2012].
GWAS replication studies using samples in the African population did not observe any significant association with the GWAS candidate genes [Butali et al., 2011; Weatherly et al., 2011]. Nonetheless, the role of rare functional variants in GWAS candidate genes has not been evaluated in the African population. We obtained saliva samples from case triads in Nigeria and Ethiopia for Sanger sequencing of the GWAS candidate genes in order to potential etiologic rare variants.
MATERIALS AND METHODS
Samples
The samples used for this genetic study were obtained in Nigeria and Ethiopia following ethical approval by the Institutional Review Boards at the Lagos University Teaching Hospital Idi-Araba, Lagos [IRB approval number: ADM/DCST/HREC/VOL.XV/321], Obafemi Awolowo University Teaching Hospital Ile-Ife [IRB approval number:ERC/2011/12/01], and the Addis Ababa University [IRB approval number: 003/10/surg]. A signed informed consent was obtained from every family recruited after they had read the study information and personal discussions with a member of the research team at the cleft clinic. All patients were examined by maxillofacial surgeons in Nigeria and plastic surgeons in Ethiopia for the presence of other congenital anomalies, features of syndromic forms of clefts and other major structural anomalies and were excluded if these were present [described in Butali et al., 2011]. We collected samples from 220 affected probands [191 non-syndromic cleft lip with or without cleft palate [NSCL(P)] and 29 non-syndromic cleft palate [NSCP]]and both parents [where possible]. In total, samples were collected from 50 complete triads and 170 dyads (these include 47 triads and 93 dyads from Nigeria and 3 triads and 77 dyads from Ethiopia).
The Oragene collection kit [www.dnagenotek.com] was used to collect saliva samples from adults and children who were able to spit into the tube. For the newborn and children who were unable to spit, we used saliva sponges to soak saliva sublingually or in the buccal sulcus and these sponges were cut into the Oragene collection tubes. Blood samples were also collected during surgery for all affected probands.
Samples were shipped to the US for downstream applications that include DNA processing and sequencing. The protocol for DNA processing can be accessed via the Murray Lab website [ genetics@uiowa.edu]. We measured the DNA concentration for all the samples using Qubit [http://www.invitrogen.com/site/us/en/home/brands/Product-Brand/Qubit.html] and a 4ng/ul DNA concentration was used for Sanger sequencing.
Sequencing
We selected the genes identified from the GWAS reported by Beaty et al [2010]. These include MAFB, PAX7, VAX1, ARHGAP29 (ABCA4 locus), and IRF6. We included ARHGAP29 reported by Leslie et al., [2012] which was identified as the candidate gene following resequencing of the GWAS signal around the ABCA4. The method used for sequencing has been previously reported [Leslie et al., 2012; Butali et al., 2013]. In summary, primers for the coding regions of all the candidate genes (MAFB NM_00546; PAX7 NM_001135254.1; VAX1 NM_001112704.1; ARHGAP29 NM_004815.3; and IRF6 NM_006147.2) were designed using Primer 3 (http://bioinfo.ut.ee/primer3-0.4.0/) and optimized in the Murray lab using a gradient Polymerase Chain Reaction (PCR) to determine the annealing temperature for each primer set. A master mix containing 10xNH4 buffer, 5% DMSO, 200 μM DNTPs, 50 μM MgCl, water, 20 μM of forward and reverse primers and the 5u/μl Taq polymerase enzyme was prepared. We added 9 μl of the master mix to 1 μl of DNA in a 96 well plate. Two Centre d’Etude du Polymorphisme Humain (CEPH) samples and two water samples were added as controls. The primers used and annealing temperatures are available on request. Amplified DNA products were shipped to Functional Biosciences (http://order.functionalbio.com/seq/index) in Wisconsin for sequencing using an ABI 3730XL. Chromatograms were transferred to a Unix workstation, base-called with PHRED (v.0.961028), assembled with PHRAP (v. 0.960731), scanned by POLYPHRED (v. 0.970312), and viewed with the CONSED program (v. 4). Variants found were compared to the variants that are present in the 1000 genome (1 KG) database (http://www.1000genomes.org/) and the Exome Variant Server (EVS) database (http://snp.gs.washington.edu/EVS/). We predicted the functional effects of these variants on the protein by using bioinformatics tools such as polyphen (http://genetics.bwh.harvard.edu/pph2/) [Adzhubei et al., 2010], SIFT (http://sift.jcvi.org/) [Kumar et a., 2009] and HOPE (http://www.cmbi.ru.nl/hope) [Venselaar et al., 2010], and the Human Splice Finder (HSF) [Desmet et al., 2009]. We first sequenced only the probands in order to identify rare functional variants. In probands with new rare functional variants, we sequenced samples from their parents to tease out variants that are de novo or segregating in the family.
RESULTS
Sequencing Results
We found three new variants; a missense variant c.493C>G in MAFB, c.1282G>A in PAX7 and a splice-site variant c.952+2T>A that changes the donor site in exon 5 of PAX7. Two new variants were also found: c.493C>G in one unaffected parent and c.952+2T>A in another unaffected parent. For the proband with the c.1282G>A variant, we only have a sample from the father [who did not have the variant] and cannot confirm if the variant segregates or not. The other variants c.2864G>A and c.2738C>A in ARHGAP29 and c.1396G>A in PAX7 have been previously identified. We did not identify any rare variant in VAX1.
The mutant residue (Serine) is bigger than the wild-type residue (Glycine) at position 466 of the PAX7 protein (Figure 1) and will probably not the fit core of the protein occupied by the wild type. Although the glycine residue is conserved at this position, a few other residue types have been observed at this position too. This includes the mutant Serine residue. This mutation is possibly not damaging to the protein based on conservation since homologous proteins exist with the same residue type as Serine mutant at this position.
Figure 1.
Shows the structures of Glycine [wild type] and Serine [mutant] at position 466 of the PAX7 protein.
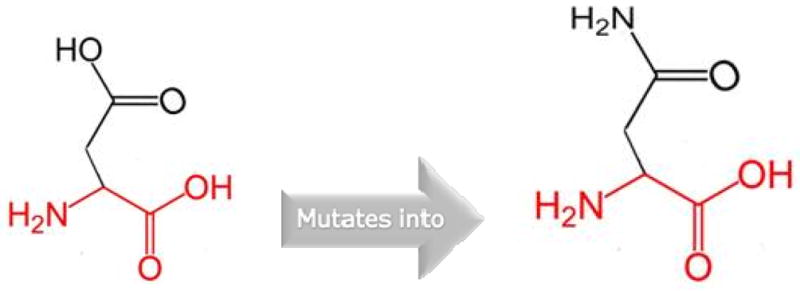
The wild-type residue (Aspartic acid) is negatively charged and the mutant residue (Asparagine) is neutral at position 428 of the PAX7 protein (Figure 2). The negative charge of the buried wild-type residue in the core domain is lost when replaced by neutral mutation. The loss of negative charge might disturb the core structure of this domain.
Figure 2.

Structures of Aspartic acid [wild type] and Asparagine [mutant] at position 428 of the PAX7 protein.
The wild-type residue (Histidine) is not conserved at position 165 of the MAFB protein (Figure 3). The mutant residue (Asparagine) is among the observed residue types at this position in other homologous sequences. This suggests that this variant is not damaging to the protein’s structure and function.
Figure 3.
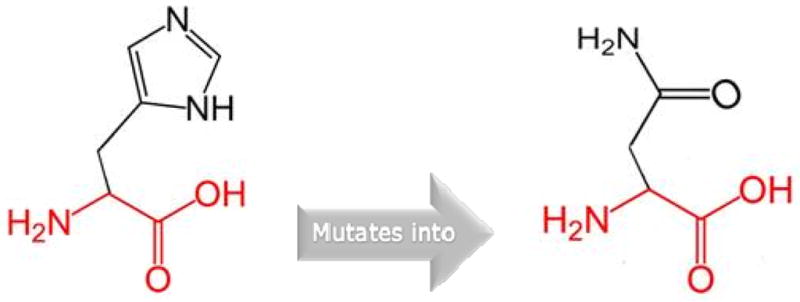
The schematic structures of Histidine [wild type] and Asparagine [mutant] position 165 of the MAFB protein.
DISCUSSION
We found three new variants (p.Asp428Asn in PAX7; p.His165Asn in MAFB, and a donor splice site mutation in PAX7) following resequencing of GWAS candidate genes in the African population. The wild-type residue (Aspartic acid) in the variant p.Asp428Asn in PAX7 is highly conserved. Nonetheless, other residue types have been identified at this position too in homologous sequences. Neither the Asparagine residue nor another residue with similar properties was observed at this position in other homologous sequences. Therefore, based on conservation scores and amino acid properties, the variant in PAX7 was predicted to be probably damaging and deleterious. Glycine, the wild-type residue in the variant p.Gly466Ser in PAX7, is the most flexible of all residues and this unique property might be necessary for the protein’s function. This flexibility may be required at this position to make a greater supporting structure or to ease movement of the protein. Therefore, mutation of glycine can eliminate these molecular properties by affecting the structure and movement of the protein leading to a less flexible residue. This mutation was previously reported by us in a Filipino case with CLP [Butali et al., 2013].
The variant p.His165Asn in MAFB was predicted to be benign by polyphen and tolerated by SIFT. HOPE shows that the mutant residue is smaller than the wild-type and this change in size can be detrimental to the molecular interactions and deleterious to the protein. A previous study reported a rare damaging variant at position p.His131Glu in a Filipino case [Beaty et al., 2010]. The variants in ARHGAP29 are known variants and previously reported in NSCL[P] by Leslie et al. [2012]. The ARHGAP29 variants were also found in the 1000 genome database and Exome Variant Server [EVS]. The individuals in the 1000 genome database have limited clinical information and family history; and the individuals in the Exome Variant Server were selected for specific heart, lung and blood diseases. Therefore, it is possible that these variants may be rare normal variants, but because the cleft status of individuals in the EVS database is unknown we cannot be sure. There are Nigerians and Kenyans [from East Africa – sharing the same border and similar geographical location as Ethiopia] in the 1KG database. In the EVS, there are African Americans who are historically from West Africa [the geographical region where Nigeria is]. Individuals in both databases serve as population controls for our cases. We observed that two of the new mutations [c.493C>G and c.952+2T>A] are in unaffected parents suggesting that there is incomplete penetrance of the phenotype in these individuals.
Previous studies have argued that it is important to investigate diverse populations in the study of complex diseases [Ramos et al., 2012]. A recent GWAS clearly showed the existence of population heterogeneity for some candidate genes [Beaty et al., 2010]. In addition, rare coding variants have been reported to play a role in the etiology of NSCLP [Leslie and Murray, 2012]. The data on the new rare variants in PAX7 and MAFB [conservation score, amino acid properties and disruption of core domain] in the cases and not in controls suggest they are etiologic and specific to the African population. Our study thus provides additional evidence that studies in diverse populations may identify rare variants that are population specific.
The etiology of NSCL[P] is complex and it is possible that there are new candidate genes that may be specific to the African population – but this requires further verification. The findings also support the notion that rare variants may be population-specific or individual family specific. Therefore, future genome-wide studies in an independent African population should be conducted in order to unravel these additional molecular genetic etiologies in addition to considering the role of common and unique environmental etiologies as co-variate in NSCLP risk.
Table 1.
Rare functional Variants in GWAS candidate Genes
| Genes | HGVSa | Amino acid change | Polyphen/SIFT predictions | Cleft details | Nigerian Cases [N=140] | Ethiopian Cases [N=80] | 1KG | EVS |
|---|---|---|---|---|---|---|---|---|
| ARHGAP29 | c.2864G>A | p.Arg955His | Benign/tolerated | CLP | 1 | 0 | 1 | 0 |
| c.2738C>A | p.Ser913Leu | Stop retained | CL | 1 | 0 | 3 | 11 | |
| PAX7 | c.1396G>A | p.Gly466Ser | Benign/tolerated | CL | 0 | 1 | 0 | 0 |
| c.952+2T>A** | Donor splice site variant | CL | 0 | 1 | 0 | 0 | ||
| c.1282G>A** | p.Asp428Asn | Possibly damaging/deleterious | CL | 1 | 0 | 0 | 0 | |
| MAFB | c.493C>G** | p.His165Asn | Benign/Tolerated | CP | 1 | 0 | 0 | 0 |
1KG = 1000 genomes, EVS= Exome variant server
New variants
Ref Seq [ARHGAP29 NM_004815.3; PAX7 NM_001135254.1; MAFB NM_00546
CL = Cleft lip CP= Cleft palate CLP = Cleft lip and palate
Acknowledgments
We owe our gratitude to the families who voluntarily participated in Nigeria and Ethiopia. We are also very grateful to Erin Brothers-Smith for her administrative assistance, to all the nurses and research assistants in Nigeria and Ethiopia for patient recruitment, consenting, sample and data entry and collection. Our gratitude goes to the Smile Train for funding free cleft surgeries in Nigeria and Ethiopia and to Transforming Faces Worldwide for supporting holistic cleft care in Ethiopia. This project was supported by grants from the NIDCR K99/R00 DE022378 [AB], NIDCR R37 Grants DE-08559 and DE-016148 [JCM].
Footnotes
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaty TH, Murray JC, Marazita ML, Munger RG, Ruczinski I, Hetmanski JB, Liang KY, Wu T, Murray T, Fallin MD, Redett RA, Raymond G, Schwender H, Jin SC, Cooper ME, Dunnwald M, Mansilla MA, Leslie E, Bullard S, Lidral A, Moreno LM, Menezes R, Vieira AR, Petrin A, Wilcox A, Lie RT, Jabs EW, Wu-Chou YH, Chen PK, Wang H, Ye X, Huang S, Yeow V, Chong SS, Jee SH, Shi B, Christensen K, Doheny K, Pugh EW, Ling H, Castilla EE, Czeizel AE, Ma L, Field LL, Brody L, Pangilinan F, Mills JL, Molloy AM, Kirke PN, Scott JM, Arcos Burgos M, Scott AF. A genome wide association study of cleft lip with/without cleft palate using case-parent trios of European and Asian ancestry identifies MAFB and ABCA4 as new candidate genes. Nat Genet. 2010;42:525–529. doi: 10.1038/ng.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum S, Ludwig KU, Reutter H, Herms S, Steffens M, Rubini M, Baluardo C, Ferrian M, Almeida de Assis N, Alblas MA, Barth S, Freudenberg J, Lauster C, Schmidt G, Scheer M, Braumann B, Bergé SJ, Reich RH, Schiefke F, Hemprich A, Pötzsch S, Steegers-Theunissen RP, Pötzsch B, Moebus S, Horsthemke B, Kramer FJ, Wienker TF, Mossey PA, Propping P, Cichon S, Hoffmann P, Knapp M, Nöthen MM, Mangold E. Key susceptibility locus for nonsyndromic cleft lip with or withgout cleft palate on chromosome 8q24. Nature Genet. 2009;41:473–477. doi: 10.1038/ng.333. [DOI] [PubMed] [Google Scholar]
- Butali A, Mossey PA, Adeyemo WL, Jezewski PA, Onwuamah CK, Ogunlewe MO, Ugboko VI, Adejuyigbe O, Adigun AI, Abdur-Rahman LO, Onah, Audu RA, Idigbe EO, Mansilla MA, Dragan EA, Petrin AL, Bullard SA, Uduezue AO, Akpata O, Osaguona AO, Olasoji HO, Ligali TO, Kejeh BM, Iseh KR, Olaitan PB, Adebola AR, Efunkoya E, Adesina OA, Oluwatosin OM, Murray JC NigeriaCRAN Collaboration. Genetic studies in the Nigerian population implicate an MSX1 mutation in complex oral facial clefting disorders. Cleft Palate Craniofac J. 2011;48:646–653. doi: 10.1597/10-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butali A, Suzuki S, Cooper ME, Mansilla AM, Cuenco K, Leslie EJ, Suzuki Y, Niimi T, Yamamoto M, Ayanga G, Erkhembaatar T, Furukawa H, Fujiwawa K, Imura H, Petrin AL, Natsume N, Beaty TH, Marazita ML, Murray JC. Replication of genome wide association identified candidate genes confirm the role of common and rare variants in PAX7 and VAX1 in the etiology of nonsyndromic CL[P] Am J Med Genet A. 2013;161A:965–972. doi: 10.1002/ajmg.a.35749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acid Research. 2009;37:e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon MJ, Marazita ML, Beaty TH, Murray JC. Cleft lip and palate: understanding genetic and environmental influences. Nat Rev Genet. 2011;12:167–178. doi: 10.1038/nrg2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontoura C, Silva RM, Granjeiro JM, Letra A. Further evidence of association of the ABCA4 gene with cleft lip/palate. Eur J Oral Sci. 2012;120:553–557. doi: 10.1111/eos.12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant SFA, Wang K, Zhang H, Glaberson W, Annaiah K, Kim CE, Bradfield PJ, Glessner JT, Thomas KA, Garris M, Frackelton EC, Otieno FG, Chiavacci RM, Nah HD, Kirschner RE, Hakonarson H. A genome-wide association study identifies a locus for non-syndromic cleft lip with or without cleft palate on 8q.24. Journal of Pediatr. 2009;155:909–913. doi: 10.1016/j.jpeds.2009.06.020. [DOI] [PubMed] [Google Scholar]
- Huppi K, Pitt JJ, Wahlberg BM, Caplen NJ. The 8q24 gene desert: an oasis of non-coding transcriptional activity. Front Genet. 2012;3:69. doi: 10.3389/fgene.2012.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keinan A, Clark AG. Recent explosive human population growth has resulted in an excess of rare genetic variants. Science. 2012;336:740–743. doi: 10.1126/science.1217283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Leslie EJ, Mansilla MA, Biggs LC, Schuette K, Bullard S, Cooper M, Dunnwald M, Lidral AC, Marazita ML, Beaty TH, Murray JC. Expression and mutation analyses implicate ARHGAP29 as the etiologic gene for the cleft lip with or without cleft palate locus identified by genome-wide association on chromosome 1p22. Birth Defects Res A Clin Mol Teratol. 2012 Nov;94:934–942. doi: 10.1002/bdra.23076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie E, Murray J. Evaluating rare coding variants as contributing causes to non-syndromic cleft lip and palate. Clin Genet. 2012 doi: 10.1111/cge.12018. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig KU, Mangold E, Herms S, Nowak S, Reutter H, Paul A, Becker J, Herberz R, Alchawa T, Nasser E, Böhmer AC, Mattheisen M, Alblas MA, Barth S, Kluck N, Lauster C, Braumann B, Reich RH, Hemprich A, Pötzsch S, Blaumeiser B, Daratsianos N, Kreusch T, Murray JC, Marazita ML, Ruczinski I, Scott AF, Beaty TH, Kramer FJ, Wienker TF, Steegers-Theunissen RP, Rubini M, Mossey PA, Hoffmann P, Lange C, Cichon S, Propping P, Knapp M, Nöthen MM. Genome-wide meta-analyses of nonsyndromic cleft lip with or without cleft palate identify six new risk loci. Nat Genet. 2012;44:968–971. doi: 10.1038/ng.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangold E, Ludwig KU, Birnbaum S, Baluardo C, Ferrian M, Herms S, Reutter H, Almeida de Assis N, Al Chawa T, Mattheisen M. Genome-wide association study identifies two susceptibility loci for nonsyndromic cleft lip with or without cleft palate. Nat Genet. 2010;42:24–26. doi: 10.1038/ng.506. [DOI] [PubMed] [Google Scholar]
- Mossey PA, Modell B. Epidemiology of oral clefts 2012: an international perspective. Front Oral Biol. 2012;16:1–18. doi: 10.1159/000337464. [DOI] [PubMed] [Google Scholar]
- Nasser E, Mangold E, Tradowsky DC, Fier H, Becker J, Boehmer AC, Herberz R, Fricker N, Barth S, Wahle P, Nowak S, Reutter H, Reich RH, Lauster C, Braumann B, Kreusch T, Hemprich A, Pötzsch B, Hoffmann P, Kramer FJ, Knapp M, Lange C, Nöthen MM, Ludwig KU. Resequencing of VAX1 in patients with nonsyndromic cleft lip with or without cleft palate. Birth Defects Res A Clin Mol Teratol. 2012 Nov;94:925–933. doi: 10.1002/bdra.23078. [DOI] [PubMed] [Google Scholar]
- Nikopensius T, Birnbaum S, Ludwig KU, Jagomägi T, Saag M, Herms S, Knapp M, Hoffmann P, Nöthen MM, Metspalu A, Mangold E. Susceptibility locus for non-syndromic cleft lip with or without cleft palate on chromosome 10q25 confers risk in Estonian patients. Eur J Oral Sci. 2010;18:317–319. doi: 10.1111/j.1600-0722.2010.00741.x. [DOI] [PubMed] [Google Scholar]
- Rojas-Martinez A, Reutter H, Chacon-Camacho O, Leon-Cachon RBR, Munoz-Jimenez SG, Nowak S, Becker J, Herberz R, Ludwig KU, Paredes-Zenteno M, Arizpe-Cantú A, Raeder S, Herms S, Ortiz-Lopez R, Knapp M, Hoffmann P, Nöthen MM, Mangold E. Genetic Risk Factors for Non-syndromic Cleft Lip with or without Cleft Palate in a Mesoamerican Population: Evidence for IRF6 and Variants at 8q24 and 10q25. Birth Defects Research Part A. 2010;88:535–537. doi: 10.1002/bdra.20689. [DOI] [PubMed] [Google Scholar]
- Ramos E, Callier SL, Rotimi CN. Why personalized medicine will fail if we stay the course. Per Med. 2012;9:839–847. doi: 10.2217/PME.12.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherley-White RC, Ben S, Jin Y, Riccardi S, Arnold TD, Spritz RA. Analysis of genome-wide association signals for nonsyndromic cleft lip/palate in a Kenya African Cohort. Am J Med Genet A. 2011;155A:2422–2425. doi: 10.1002/ajmg.a.34191. [DOI] [PubMed] [Google Scholar]
- Wehby G, Cassell C. The impact of orofacial clefts on quality of life and healthcare use and costs. Oral Dis. 2010;16:3–10. doi: 10.1111/j.1601-0825.2009.01588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennessen JA, Bigham AW, O’Connor TD, Fu W, Kenny EE, Gravel S, McGee S, Do R, Liu X, Jun G, Kang HM, Jordan D, Leal SM, Gabriel S, Rieder MJ, Abecasis G, Altshuler D, Nickerson DA, Boerwinkle E, Sunyaev S, Bustamante CD, Bamshad MJ, Akey JM, Broad GO, Seattle GO. NHLBI Exome Sequencing Project. 2012. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venselaar H, Te Beek TA, Kuipers RK, Hekkelman ML, Vriend G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics. 2010;11:548. doi: 10.1186/1471-2105-11-548. [DOI] [PMC free article] [PubMed] [Google Scholar]