

Abstract
The Small Heterodimer Partner (SHP) is an orphan nuclear receptor and an atypical member of the nuclear receptor superfamily Since its discovery, a growing body of evidences have pointed out a pivotal role for SHP in the transcriptional regulation of a variety of target genes involved in diverse metabolic pathways. While we have previously developed a homology model of the structure of SHP that was instrumental to identify a putative ligand binding pocket and suggest the possibility of the development of synthetic modulators, others reported that some atypical retinoids may represent the first synthetic ligands for this receptor. In this work, we report a combined computational approach aimed at shedding further lights on the binding mode and mechanism of action of some atypical retinoids as ligands of SHP. The results have been instrumental to design mutagenesis experiments whose preliminary data suggest the presence of a functional site in SHP as defined by residues Phe96, Arg138 and Arg238. While further experimental studies are ongoing, these findings constitute the basis for the design and identification of novel synthetic modulators of SHP functions.
Keywords: Nuclear receptors, Small hetedimer partner, Homology modeling, Molecular docking, Molecular dynamic simulation, Retinoids
Introduction
The Small Heterodimer Partner (SHP) is a member of the NR0B group of the nuclear receptor superfamily. Unlike common nuclear receptors (NRs), this group of receptors, that contains also DAX-1 (dosage-sensitive sex-reversal adrenal hypoplasia congenital critical region on the X chromosome gene 1), lacks the central DNA-binding domain (DBD) and the N-terminal ligand-independent activation domain (AF-1), while its members keep the C-terminal ligand binding domain (LBD) with the transcriptional activation function-2 (AF-2) [1, 2].
SHP is a pleiotropic orphan nuclear receptor that plays a pivotal role in regulating lipid, cholesterol and bile acid metabolism, drug metabolism, glucose homeostasis and steroidogenesis [3–5] More recently, it has been reported that SHP regulates cell survival by controlling mitochondrial function [6]. In line with these functions, diverse single nucleotide polymorphisms of SHP gene have been identified and associated to metabolic disorders such as obesity and diabetes [7–11].
While a thorough discussion of SHP regulation and function has been reported in recent in-depth reviews [3–5] below we summarize some key aspects of its mechanisms of action.
Beside very few reported exceptions [12], SHP is generally a transcriptional repressor that fulfills its endocrine functions through direct interactions with numerous nuclear receptors [3]. The repression mechanism of SHP, in particular, involves the following actions: (a) competition with the coactivators binding on the AF-2 of nuclear receptors; (b) recruitment of corepressors and chromatin remodeling factors; (c) inhibition of binding to DNA through interaction with transcriptional factors such as ARNT and HNF-3 [13–17].
The competition with coactivators binding on the AF-2 surface of interaction partners constitutes the molecular basis of the interaction between SHP and nuclear receptors. Briefly, the binding competition occurs through the interaction of at least two LxxLL-like motifs of SHP, also termed nuclear receptor boxes (NR box 1–2), that closely resemble the conserved motifs found in a large number of nuclear receptor AF-2 coregulators [14]. Noteworthy, SHP was found to interact with estrogen receptor (ER) through both NR boxes 1 and 2 [14]. Conversely, the interactions of SHP with androgen receptor (AR), glucocorticoid receptor (GR) and liver receptor homolog-1 (LRH-1) are only mediated by NR box 2 [18–20].
While crystallographic studies of the complexes between AR, LRH-1 and the NR box 2 have been instrumental to unveil the structural basis of the binding competition between SHP and coactivators [20, 21], these studies suggest that the redundancy of NR boxes is crucial to achieve a selective interaction between SHP and diverse nuclear receptors.
The second action of SHP involves the recruitment of corepressors and chromatin remodeling factors that lead to the formation of hetero-multimeric complexes with nuclear receptors. Båvner and coworkers firstly reported that E1A-like inhibitor of differentiation-1 (EID-1) is recruited by SHP as corepressor [15]. Using homology modeling and docking studies, we proposed an interaction model between SHP and EID-1 that was instrumental to identify a corepressor binding cleft on SHP and to gain insight into the structural mechanism of the transcriptional repression of the receptor [22]. Other corepressors and chromatin remodeling factors that interact with SHP include the Sin3A-Swi/Snf complex [23], histone methyltransferase G9a and hystone deacetylases [24–26].
As far as it concerns the inhibition of DNA binding, this mechanism of action has been described to explain the SHP mediated repression of the transcriptional activity of HNF3 and of the aryl hydrocarbon receptor (AHR)/aryl hydrocarbon receptor nuclear translocator (ARNT) complex [16, 17].
According to the view of proteins sending signal dynamically [27], it is likely that the above actions of SHP are mediated by a shift in the equilibrium population of receptor conformations that settle a rugged energy landscape. Since a correlation has been proposed between the degree of conformational diversity and promiscuity [28], the high functional promiscuity of SHP suggests that many conformational states may populate the energy landscape of this receptor. Upon either interaction with other nuclear receptors or corepressor binding, the energy landscape would change by shifting the equilibrium population and favoring specific conformational states of SHP.
Even ligands may alter such energy landscape. Although it is debated whether or not SHP may have natural ligands [3], a number of recent evidences suggest that synthetic small molecules may bind to and modulate SHP functions. While we firstly provided clues for the existence of a putative ligand binding site in SHP structure on the basis of homology modeling studies [22], Fontana and coworkers elegantly reported that the apoptotic activity of some atypical retinoids (Fig. 1), namely 6-[3-(1-adamantyl)-4-hydroxyphenyl]-2-naphthalenecarboxylic acid (AHPN, 1) and 4-[3-(1-adamantyl)-4-hydroxyphenyl]-3-chlorocinnamic acid (3-Cl-AHPC, 2), might be ascribed to their interaction with SHP and the ensuing recruitment of a complex containing Sin3A, NCoR, HDAC4 and HSP90 [29]. Interestingly, a previous work by Dawson and coworkers identified another atypical retinoid, (E)-4-[3′-(1-adamantyl)-4′-hydroxyphenyl]-3-(3′-acetamidopropyloxy) cinnamic acid (3-AAHPC, 3), as antagonist of the AHPN-induced apoptosis [30]. Thus, the combination of these studies gives rise to an intriguing scenario where small molecules oppositely regulate SHP functions. In line with these observations, further studies by Dawson and coworkers reported the importance of the carboxylic moiety of 1 and 2 for the apoptotic activity and, more recently, an interaction model of these compounds at the proposed ligand binding site of SHP has been suggested, though pending for further experimental appraisals [31, 32].
Fig. 1.

Chemical structures of selected atypical retinoids: AHPN (1), 3-Cl-AHPC (2), 3-A-AHPC (3), ST1926 (4)
Although there is little information on the molecular mechanism that agonists and antagonists may adopt to regulate SHP functions, the recent disclosure of the crystal structure of the closest SHP receptor homolog, namely DAX1, provides an unprecedented opportunity to revisit the structural features of this receptor and the molecular basis of its regulation [33].
In an attempt to address these issues and in the frame of a research project devoted to the understanding of the molecular basis of ligand binding to SHP [22, 32], we report herein a computational study composed of three parts and supported by mutagenesis experiments. Firstly, a series of homology models of the LBD of SHP endowed with different conformational features, including those of DAX1, is constructed and used to sample the rugged energy landscape of the receptor. In the second part of the work, docking experiments of reported agonists and antagonists are carried out in all of the homology models of SHP. Thirdly, molecular dynamic (MD) simulations are performed to identify the most stable complexes and design mutagenesis experiments. The results are then used to discuss the molecular basis responsible for the agonism and antagonism of some atypical retinoids at SHP.
Results
Homology modelling
Sequence similarity searches were used to identify a pool of nuclear receptors sharing homology relationships with the ligand binding domain of SHP and endowed with different conformational features. The selected nuclear receptors are reported in Table 1. Not surprisingly, the best hit of the similarity search resulted in the recently disclosed ligand binding domain (LBD) of DAX-1 (pdb code: 3F5C, E-value = 2.8e-23). Alike SHP, DAX-1 belongs to the NR0B group of the nuclear receptor superfamily. During embryogenesis, DAX-1 is involved in cell differentiation in testes and adrenal tissues and plays a key role in maintaining the pluripotent state of embryonic stem cells [34–36] The conformation of helix 12 (H12) of DAX-1 is in an antagonistic state, according to the receptor that acts as global repressor of many nuclear receptors [33]. Noteworthy, helix 10 (H10) is collapsed into the canonical binding site slightly lowering the druggability of this binding pocket as evidenced by the site score (site score = 0.98). This feature should apparently prevent the binding of ligands to DAX-1.
Table 1.
Results from sequence similarity searches using the primary sequence of the ligand binding domain of SHP versus the RCSB protein (PDB) database [34]
| Entry | PDB code | Name | E-value | Site score | Volume (Å3) | Type of site | Ligand | H12 conformation |
|---|---|---|---|---|---|---|---|---|
| a | 3F5C | DAX-1 | 2.8e-23 | 0.98 | 268 | Canonical | – | Antagonist |
| b | 3CJW | COUP-TF2 | 1.0e-16 | 0.88 | 154 | Canonical | – | Antagonist |
| c | 1Z5X | USP | 5.3e-08 | 0.82 | 142 | Canonical | – | Antagonist |
| d | 1G2N | USP | 3.1e-08 | 1.23 | 595 | Allosteric | EPH | Antagonist |
| e | 1MV9 | RXRα | 7.0e-07 | 1.14 | 374 | Canonical | HXA | Agonist |
| f | 1DKF | RXRα | 6.2e-07 | 1.32 | 277 | Canonical | OLA | Antagonist |
The chicken ovalbumin upstream promoter transcription factor-2 (COUP-TF2, pdb code: 3CJW, E-value = 1.0e-16) is the second receptor in the rank of E-values. It is one of the three functionally non redundant receptors belonging to the CUP family of orphan nuclear receptors. These nuclear receptors are differently involved in fetal development and in metabolic homeostasis of adult organisms [37]. From a structural point of view, the ligand binding domain of COUP-TF2 shares similar features with DAX-1, namely the antagonistic conformation of H12 and the collapse of H10 into the canonical binding site [38].
The next receptor in the rank is the Ultraspiracle protein (USP, pdb code: 1Z5X; E-value = 5.3e-8; pdb code: 1G2N; E-value = 3.1e-8). Ultraspiracle protein (USP) is the ortholog of the vertebrate retinoid X receptor (RXR) in insects and mediates the heterodimerization of several nuclear receptors like RXR does in vertebrates [39]. It is worth pointing out that two different conformational states of this receptor have been solved by X-ray crystallography [39, 40].
Although in either USP structures H12 adopts the antagonistic conformation and H10 collapses into the canonical ligand binding site (similarly to DAX-1 and COUP-TF2), there are two additional features in the structure of 1G2N. Firstly, the loop connecting helix H1 to helix H3 (L1-3) folds around helices H3, H11 and H12 [41]. This particular fold locks the H12 in a resting state and prevents any of its conformational movements. Remarkably, the loop L1-3 is partly or not at all solved in the structures of DAX-1, COUP-II and USP-1Z5X, thus evidencing its high flexibility. Secondly, the crystal structure of USP-1G2N shows a druggable allosteric binding site filled by a phospholipids molecule (site score = 1.23). Very interestingly, this seems to be not an artifact of the crystallization procedure since the same allosteric binding site has been found as functional in other nuclear receptors such as the steroidogenic factor-1 (SF-1) and LHR-1 [42, 43]. In previous works, we already used the 1G2N structure of USP as template to model the structural features of SHP and its interaction with compounds 1 and 2 [22, 32].
Thus, while entries a–c may resemble an antagonistic conformation of SHP with a collapsed canonical binding site, entry d may be a suitable template to construct an antagonistic conformational model of SHP endowed with an allosteric binding site.
In the bottom part of the E-value ranked proteins, we selected the canonical agonist and antagonist conformations of the retinoid X receptor-α (RXRα, pdb code: 1MV9; E-value = 7.0e-7; pdb code: 1DKF; E-value = 6.2e-7). This selection was motivated by the fact that the known small molecule modulators of SHP belong to the chemical class of synthetic retinoids and some members of this class may bind to the canonical binding site of nuclear retinoic acid receptors (RARs) or RXRs as well. Accordingly, we deemed advisable to not exclude at this stage the hypothesis that the molecular mechanism of SHP agonists and antagonists could comply with the classic principles for modulation of nuclear receptors [44]. Thus, entries e and f were instrumental to build two conformational models of SHP with a druggable canonical binding site and H12 in the agonistic and antagonistic states, respectively.
A multiple alignment between the sequences of SHP and the above receptors was generated (Fig. 2) and used to construct four 3D conformational models of the receptor: (a) SHPDAX-1 model (H12-antagonist, H10-collapsed) as representative of the conformational features observed in 3F5C (DAX-1), 3CJW (COUP-TF2) and 1Z5X (USP); (b) SHPUSP (H12-antagonist, allosteric site) as example of the conformational features observed in 1G2N (USP); (c) SHPRXRα (H12-agonist, canonical site) as representative of the conformational features observed in 1MV9 (RXRα); (d) SHPRXRα* (H12-antagonist, canonical site) as example of the conformational features observed in 1DKF (RXRα). The resulting 3D models of SHP were geometrically validated and their folding was checked using the Verify3D server (see supporting information).
Fig. 2.

Multiple alignment between the sequences of SHP and selected template receptors. Atomic coordinates of residues marked with a star were modeled according to the methodology previously reported (see Ref. 21). Mutated residues are shaded with black boxes
Docking experiments
In this part of the study, we used the homology models of SHP (models a–d, Table 1) to perform docking experiments of agonist (3Cl-AHPC, 2), competitive antagonist (3A-AHPC, 3) and an additional atypical retinoid molecule (ST1926, 4) whose structure is closely related to 3Cl-AHPC (2), albeit no data is available about its binding to SHP [45, 46]. Concerning ST1926 (4), in particular, the idea was to assess whether it could also bind and modulate the receptor by adopting a molecular mechanism similar to 3Cl-AHPC (2). Docking experiments were carried out into both the canonical and allosteric binding sites of SHP. Briefly, the canonical binding site refers to the hydrophobic cleft that is almost conserved in the ligand binding domain (LBD) of all nuclear receptors and is linked to the stabilization of the activation function on helix 12 (H12). The allosteric pocket refers to the phospholipids binding site that has been unexpectedly discovered in some crystal structures of nuclear receptors (e.g. USP, LRH-1, SF-1). Noteworthy, a growing number of experimental evidences suggest a modulatory role of this site upon phospholipids binding at least in LRH-1 and SF-1 [42, 43, 47, 48]. The canonical and allosteric sites share a common volume, forming together an “L” shaped cavity (Fig. 3a). The “L” shaped cavity is wedged between the Loop108-138 and the helix H12, with positively charged residues being located at the bottom of each arm.
Fig. 3.
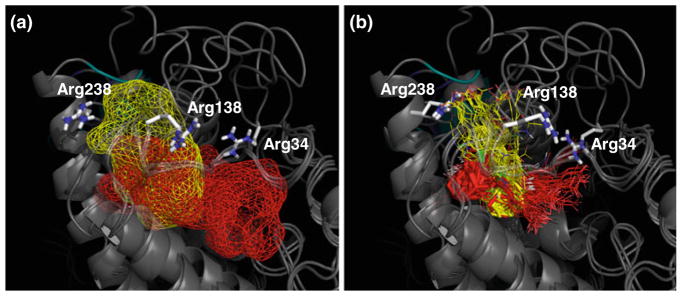
a Canonical (red) and allosteric (yellow) sites as mapped into superimposed SHP models. Positively charged residues are shown in sticks. b Superimposition of the selected binding poses of compounds 2–4 as resulting from docking experiments. Solutions occupying the allosteric site are yellow colored, solutions occupying the canonical site are red colored. The adamantane moiety is highlighted in sticks. The conformation of the side chain of arginines changes in response to ligand induced fit
From docking experiments, two alternative binding poses for each compound in either the canonical or allosteric binding sites of models a–d of SHP were selected for further studies (Table 2). In all of these complexes, the adamantane moiety of compounds 2–4 occupies the wedge of the “L” shaped cavity, whereas the carboxylic group alternatively points to the positively charged residues at the bottom of each arm of the cavity (Fig. 3b).
Table 2.
Docking results of compounds into the canonical and allosteric binding sites of SHP models
| No | SHPDAX-1
|
SHPUSP
|
SHPRXRα
|
SHPRXRα*
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IFD | RMSD (Å) Rel. | RMSD (Å) Abs. | IFD | RMSD (Å) Rel. | RMSD (Å) Abs. | IFD | RMSD (Å) Rel. | RMSD (Å) Abs. | IFD | RMSD (Å) Rel. | RMSD (Å) Abs. | |
| 2 | −396.309 | 0.000 | 2.418 | −392.812 | 0.000 | 0.000 | −391.782 | 0.000 | 10.363 | −394.816 | 0.000 | 1.893 |
| −394.044 | 2.007 | 2.538 | −389.583 | 5.614 | 5.700 | −391.543 | 10.772 | 2.561 | −390.060 | 9.621 | 9.613 | |
| 3 | −405.863 | 0.000 | 0.000 | −391.955 | 0.000 | 5.060 | −401.278 | 0.000 | 0.000 | −398.136 | 0.000 | 0.000 |
| −404.027 | 1.579 | 1.623 | −387.746 | 6.206 | 5.733 | −397.750 | 4.181 | 4.711 | −392.743 | 9.185 | 9.701 | |
| 4 | −395.084 | 0.000 | 1.948 | −389.959 | 0.000 | 5.195 | −393.433 | 0.000 | 7.418 | −391.616 | 0.000 | 2.589 |
| −394.230 | 1.571 | 2.185 | −387.678 | 5.706 | 6.210 | −388.145 | 7.939 | 2.542 | −388.334 | 1.069 | 2.088 | |
IFD Induced fit docking score, RMSD Rel is calculated taking the top docked solution of the same compound as reference pose, RMSD Abs is calculated taking the best solution among all of the compounds as reference pose
Molecular dynamic simulations
Since it was not possible to uniquely identify the most energetic favored solutions on the basis of the scoring function of docking experiments (IFD score, Table 2), we performed 10 ns MD simulations of the complexes in full water solvated box. The following tasks, in particular, were the goals of this last part of the work: (a) to optimize the binding energies of compounds 2–4 taking into account any possible ligand induced fit effect of the receptor and, thereby, aiding the identification of their most energetic favored solutions into a specific binding cleft and conformation of SHP; (b) to design mutagenesis experiments; (c) to understand the molecular mechanisms responsible of the agonistic and antagonistic modulation of the receptor by the atypical retinoid molecules.
Concerning the first task, from the inspection of the average and standard deviations of the interaction energies of compounds 2–4 with the receptor (Table 3), it is found that they show a stable and energetic favored solution when docked into the allosteric site of the conformational model of SHPUSP. In addition, the agonist (3Cl-AHPC, 2) displays an alternative favored binding pose into the canonical cleft of the conformational model SHPDAX-1, as evidenced by the slight difference of interaction energy from the selected solution of SHPUSP (ΔE = 5.6 kcal/mol). Figures 4 and 5 show the interactions of compounds 2–4 into the conformational models of SHP according to the solutions selected from MD simulations.
Table 3.
Averages and standard deviations of the interaction energy (kcal/mol) of the complex
| No | SHPDAX-1
|
SHPUSP
|
SHPRXRα
|
SHPRXRα*
|
||||
|---|---|---|---|---|---|---|---|---|
| Energy | RMSD | Energy | RMSD | Energy | RMSD | Energy | RMSD | |
| Average | Average | Average | Average | Average | Average | Average | Average | |
| SD | SD | SD | SD | SD | SD | SD | SD | |
| 2 | −145.962 | 4.734 | −151.591 | 3.998 | −117.696 | 2.820 | −118.340 | 2.619 |
| ±28.394 | ±0.431 | ±21.721 | ±0.483 | ±28.197 | ±0.430 | ±13.021 | ±0.286 | |
| / | / | −108.364 | 3.902 | −111.411 | 4.129 | −43.405 | 4.014 | |
| ±11.195 | ±0.465 | ±33.811 | ±0.607 | ±13.254 | ±0.846 | |||
| 3 | −138.822 | 2.252 | −135.318 | 4.182 | −65.691 | 4.029 | −50.503 | 3.839 |
| ±14.885 | ±0.425 | ±19.109 | ±0.369 | ±20.397 | ±0.542 | ±12.881 | ±0.418 | |
| / | / | −194.395 | 3.151 | −39.512 | 3.986 | −69.928 | 2.674 | |
| ±25.340 | ±0.499 | ±11.607 | ±0.460 | ±9.655 | ±0.463 | |||
| 4 | −141.538 | 4.362 | −157.385 | 3.677 | −82.558 | 3.760 | −10.556 | 3.716 |
| ±24.897 | ±0.403 | ±22.642 | ±0.378 | ±27.442 | ±0.241 | ±14.510 | ±1.143 | |
| / | / | −127.603 | 4.672 | −10.448 | 4.719 | / | / | |
| ±28.081 | ±0.595 | ±11.739 | ±0.552 | |||||
Averages and standard deviations of the RMSD (Å) calculated using the heavy atoms of the ligand and the binding pose obtained from docking experiments as reference structure. All the values are calculated along the last 8 ns of the MD simulations. Selected solutions are highlighted in italic
Fig. 4.

a Interaction of 3-Cl-AHPC (2) into the canonical binding site of SHPDAX-1 model after 10 ns of MD simulation. b Interaction of 3-Cl-AHPC (2) into the allosteric binding site of SHPUSP model after 10 ns of MD simulation. c Interaction of 3-A-AHPC (3) into the allosteric binding site of SHPUSP model after 10 ns of MD simulation. d Interaction of ST1926 (4) into the allosteric binding site of SHPUSP model after 10 ns of MD simulation
Fig. 5.

a Schematic interactions of 3-Cl-AHPC (2) into the canonical binding site of SHPDAX-1 model after 10 ns of MD simulation. b Schematic interactions of 3-Cl-AHPC (2) into the allosteric binding site of SHPUSP model after 10 ns of MD simulation. c Schematic interaction of 3-A-AHPC (3) into the allosteric binding site of SHPUSP model after 10 ns of MD simulation. d Schematic interactions of ST1926 (4) into the allosteric binding site of SHPUSP model after 10 ns of MD simulation
On the basis of these results, a number of residues was selected for mutagenesis experiments including Arg34, Trp92, Phe96, Gln134, Arg138, Trp148 and Arg238 (Fig. 2). These experiments are detailed in the next section.
The final task of molecular dynamic simulations was to shed light on the molecular mechanisms responsible of the agonistic and antagonistic modulation of SHP. To this end, we investigated the effect of the agonist (2) and antagonist (3) on the stability of the three nuclear receptor boxes and helix H12 in the conformational model SHPUSP. Indeed, being in agreement with the competitive profile of the agonist (2) and antagonist (3) as well as mutagenesis experiments (see below), it is only the conformational model SHPUSP that is able to host compounds 2–3 in a common binding site, namely the allosteric site.
Table 4 reports the averages and the standard deviations of RMSD values calculated on the backbone atoms of the receptor and, more specifically, on the backbone atoms of the selected structural motifs. From the inspection of the table, it is found that 3Cl-AHPC (2) and (3A-AHPC, 3) induce a similar pattern of stabilization (RMSD<3.0) and destabilization (RMSD>3.0) of the NR boxes, with the NR boxes 1, 3 being stabilized and NR box-2 being destabilized. Remarkably, compounds 2 and 3 show a different stabilization degree of helix H12 that makes sense in view of their pharmacological profile. Thus, while the agonist (2) destabilizes the antagonistic conformation of H12, the antagonist (3) stabilizes it in SHP.
Table 4.
Averages and standard deviations of the RMSD (Å) calculated on the backbone atoms of SHPUSP and its functional motifs
| No | SHPUSP
|
||||
|---|---|---|---|---|---|
| NRbox 1 | NRbox 2 | NRbox 3 | H12 | Backbone | |
| 2 | 2.659 | 9.436 | 2.476 | 3.895 | 4.506 |
| ±0.557 | ±0.889 | ±0.651 | ±0.399 | ±0.197 | |
| 3 | 2.150 | 8.098 | 3.036 | 1.875 | 3.858 |
| ±0.450 | ±2.024 | ±0.505 | ±0.295 | ±0.386 | |
| 4 | 1.606 | 3.902 | 1.970 | 4.868 | 3.588 |
| ±0.471 | ±1.227 | ±0.728 | ±0.817 | ±0.386 | |
The values are calculated along the last 8 ns of the MD simulations
ST1926 (4), a compound not yet tested in SHP functional assays, shows a behavior akin to the agonist 3Cl-AHPC (2), with the destabilization of the antagonistic conformation of H12. However, conversely to compounds 2 and 3, it shows a higher stabilization of the NR boxes 1, 3 and a lower destabilization of the NR box 2.
Mutational studies
To verify the presence of a functional binding site in SHP, both the allosteric and canonical binding sites hypothesized were studied using preliminary mutagenesis experiments. One or more amino acids which were thought to be significantly interacting with the ligand in these hypothesized binding sites were mutated. All the mutants were studied for their functional changes using a reporter assay in which SHP was used to repress the HNF4α induced luciferase expression driven by ApoCIII promoter (Table 5; Fig. 6).
Table 5.
Averages and standard deviations of the repression activity of wild type and SHP mutants on HNF4α mediated transactivation of the ApoCIII promoter luciferase reporter
| pApoCIII-Luc. | Average (Luc/β-gal) | SD |
|---|---|---|
| HNF4α | 0.540 | 0.034 |
| HNF4α + SHPwt | 0.304 | 0.014 |
| HNF4α + Trp148Ala | 0.259 | 0.043 |
| HNF4α + Phe96Ala | 0.633 | 0.047 |
| HNF4α + Arg238Ala | 0.244 | 0.011 |
| HNF4α + Arg138Ala | 0.325 | 0.033 |
| HNF4α + Trp92Ala | 0.349 | 0.010 |
| HNF4α + Arg138Asp − Arg238Asp | 0.592 | 0.003 |
| HNF4α + Arg34Ala | 0.232 | 0.004 |
| HNF4α + Arg34Gln − Gln134Arg | 0.279 | 0.011 |
Fig. 6.

Transient transfection assays to determine the activity of SHP mutants in inhibiting HNF4a transactivation of the ApoCIII promoter luciferase reporter. Luc activities were determined and normalized to β-gal activities. Data are expressed as means ± SD (*p<0.01 vs. gray bar-HNF4a; ¶p<0.01 vs. white bar-SHPwt). Each SHP mutant construct was generated using site directed mutagenesis, which was confirmed by sequencing
In case of the allosteric binding site (hypothesis A), a double SHP mutant with Arg238 and Arg138 replaced by acidic Asp residue was constructed in order to invert the electrostatic profile of the receptor at the entrance of the allosteric site, thereby making repulsive the interaction with the carboxylic group of a putative ligand. Two additional single mutants of SHP, with Arg238 and Arg138 replaced by alanine residue, were also made to understand the specific contribution of the single residue to SHP function. Finally, SHP mutants Trp92Ala, Phe96Ala and Trp148Ala were studied to test the involvement of these aromatic amino acids in the binding of a putative SHP ligand. Two mutants, SHP(Arg238Asp + Arg138Asp) and SHP(Phe96Ala) completely lost their function in repressing the HNF4a induced luciferase activity on pApoCIII-Luc while the other mutants did not show any significant changes in their function in comparison to the wtSHP protein.
On the other hand, the canonical binding site (hypothesis B) was verified by constructing two other SHP mutants: SHP(Arg34Ala) and a swap double mutant SHP(Arg34Gln + Gln134Arg). None of these two mutants showed any difference in their repression function as compared to the wild type SHP protein.
Discussion
SHP is a pleiotropic protein that is involved in many cellular functions. Recently, some small molecules (2, 3) belonging to the chemical class of atypical retinoids were found to bind and modulate this nuclear receptor. In this study, as continuation of our previous works in the field [22, 32], we have been committed to get insights into the molecular basis of ligand binding to SHP.
In the first part of the study, the construction of four homology models was instrumental to generate four diverse conformations of the receptor. Indeed, it is becoming widely accepted that pleiotropic proteins are endowed with a particularly rugged energy landscape where diverse conformational states mediate different functions. Pursuing this paradigm, the intent of the four models was to sample distinct regions of the conformational energy landscape of SHP where the receptor is found in conformational states that resemble those observed in the template structures (Table 1), respectively.
In the second part of the work, docking experiments of compounds 2–4 were carried out into the four conformational models of SHP. The aim of these experiments was to identify the conformation and the putative binding site of SHP involved in the interaction with ligands 2–4.
ST1926 (4) [45, 46], in particular, is an orally active adamantyl retinoid derivative which is currently in clinical studies for ovarian cancer disease [49–51] Because the mechanism of action leading to its apoptotic activity is still not fully understood [52–55] we wondered whether compound 4 could also bind to SHP in docking experiments, putting forward this receptor as one of the potential biological targets of ST1926 (4).
Unfortunately, docking results were not able to provide a unique solution for compounds 2–4 on the basis of the scoring function (IFD score, Table 2), though the adamantane moiety was always placed in the wedge of the “L” shaped cavity formed by the allosteric and canonical binding sites (Fig. 3). Accordingly, 10 ns MD simulations were performed for each complex in full water solvated box with a double intent: firstly, to optimize the binding energies of compounds 2–4, thereby favoring the identification of the most energetic favored solutions of the compounds; in the second instance, to get insights into the molecular mechanisms responsible for the modulation of the receptor by the agonist and antagonist.
As a first result, it was found that compounds 2–4 bind the allosteric site of the conformational model SHPUSP in a stable and energetic favored solution, with compound 2 showing, however, an alternative and stable binding mode into the canonical cleft of the conformational model SHPDAX-1 (Figs. 4, 5).
This observation leads to envisage two hypotheses of ligand binding: the hypothesis A where agonist and antagonist bind to SHP occupying the allosteric site; the hypothesis B where the agonist 2 may alternatively bind the canonical cleft.
According to the hypothesis A (Figs. 4b and 5b), the agonist (3Cl-AHPC, 2) forms stable hydrogen bonds along the simulation with Arg238 (80% occupancy), and less stable interactions with Gln140 (18% occupancy) and Arg138 (17% occupancy). The adamantane moiety of 2 is buried into the bottom part of the allosteric site where it is caged by the aromatic side chains of Phe96 and Trp148 as well as the hydrophobic side chains of Leu97, Leu100 and Leu231.
Alike the agonist, the antagonist (3A-AHPC, 3) is involved in extended hydrogen bond interactions with Arg238 (43% occupancy), Arg54 (36% occupancy) and Arg138 (33% occupancy). Additionally, while its para hydroxyl group forms hydrogen bonds with the side chain of Asp234 (60% occupancy), the acetamidopropanol arm makes hydrophobic contacts with Pro136.
Again, the adamantane moiety of 3A-AHPC (3) is packed with the side chains of Phe96 and Trp148. It is worth noting that the antagonist (3) is endowed with a higher number of interactions then the agonist (2), reflecting a better fit of the former into the antagonistic conformation of SHP (Figs. 4c and 5c). As far as hypothesis B is concerned (Figs. 4a and 5a), from the inspection of the binding pose of 3Cl-AHPC (2) into the canonical cleft of SHP, it is found that the carboxylic group establishes strong hydrogen bonds with the side chain of Arg34 (94% occupancy) and the backbones of Ser132 (68% occupancy) and Gln134 (44% occupancy). Conversely, the aromatic scaffold and the adamantane moiety of 2 are wrapped with hydrophobic side chains from Phe96, Leu231, Leu235 and Phe236.
Remarkably, there are data in literature that support the plausibility of both hypothesis A and B. Indeed, it has been previously reported that the antagonist (3) is competitive with respect to the agonist (2) [30], thereby suggesting a common binding site at which these compounds may interact. From the analysis of MD simulations, only the allosteric binding may explain the competitive profile of the compounds, being able to host both the agonist (2) and the antagonist (3). Furthermore, mutagenesis experiments have been reported that identify Lys474 in LRH-1 and Lys440 in SF-1 as affecting phospholipids binding and the activity to these nuclear receptors [42, 43]. Such residues align with Arg238 in the sequence of SHP, thereby sustaining a key role for Arg238 in ligand binding to the allosteric site according to the hypothesis A (Fig. 7).
Fig. 7.

Sequence and structure alignments of helix H11 between LRH-1, SF-1 and SHP. The conserved Lys474 affecting phospholipids binding in LRH-1 and SF-1 is highlighted
On the other hand, it is worth noting that Arg34 has been identified as belonging to a set of missense mutations that affect the activity of SHP [7, 8]. While this observation may support the binding of the agonist (2) to the canonical cleft in accordance with the hypothesis B, it cannot be ruled out that Arg34 may be alternatively involved in corepressor recruitment [22].
Thus, in order to shed lights on which of the two hypotheses is more likely to occur, preliminary mutagenesis experiments were designed including a number of residues from both the allosteric site and the canonical binding site of SHP (Fig. 2). As an interesting finding, the results have shown that either the replacement of Arg138 and Arg238 with Asp or Phe96 with Ala reduce the repression functions of the receptor (Table 5; Fig. 6), thereby supporting the presence of a functional site in SHP as defined by the allosteric cleft.
In the first case, the double replacement Arg138Asp and Arg238Asp introduces negative net charges at the entrance of the putative allosteric site of the receptor. Likewise to LHR-1 and SF-1, [42, 43] we may speculate that the inverted electrostatic profile of SHP promotes repulsive interactions with the acidic head of potential endogenous ligands, such as phospholipids, hampering their binding and reducing the repressive activity of the receptor.
In this context, the marginal effects shown by the single Arg138Ala and Arg238Ala mutants suggest that the presence of only one basic residue at the allosteric cleft is sufficient to compensate for the absence of the other in keeping the activity of SHP.
As far as it concerns Trp92Ala, Phe96Ala and Trp148Ala replacements, only Phe96Ala mutant shows a significant reduction of the repressive activity of SHP. This observation pinpoints a key role for the hydrophobic interactions, rather than the aromatic interactions, in stabilizing the active conformation of the receptor. Indeed, while both phenylalanine and tryptophane residue can make aromatic interactions, it is the lower polarity of the former that mostly favours the hydrophobic packing.
Although further experimental work is ongoing to provide conclusive evidences on ligand binding to SHP, on the basis of these preliminary findings as well as the aforementioned bibliographic data, we deemed advisable to pursue the hypothesis A of ligand binding. Accordingly, we analyzed the stability of the three nuclear receptor boxes and helix H12 in the conformational model SHPUSP along the trajectories of MD simulations in order to gain insight into the molecular mechanism adopted by agonist (2) and antagonist (3) to modulate the receptor.
As a result, it was found that the molecular mechanism adopted by the agonist (2) and antagonist (3) to modulate the pleiotropic functions of SHP relies on a different pattern of stabilization and destabilization of the H12 and NR boxes (Table 4). This makes sense in the light of the pharmacological profile of the compounds as well as SHP exploiting NR boxes as part of its repression mechanism to compete with coactivators binding on the AF-2 surface of nuclear receptors [14, 18–21].
Accordingly, diverse ligand induced patterns of stabilization of these structural motifs may affect the potency and selectivity of binding between SHP and the nuclear interaction partners as well as corepressors, thereby providing a distinct functional outcome. Furthermore, since ST1926 (4) binds SHP in silico and shows a similar behavior to that of the agonist 3Cl-AHPC (2), it is likely that other atypical retinoids may exploit SHP modulation as part of the mechanism of action for their antiproliferative or pro-apoptotic activities.
Conclusions
Although our results put forward the combination of different conformational homology models with docking experiments and MD simulations as suitable approach to investigate the structural aspects of SHP and the behaviour of the receptor in response to agonist or antagonist binding, some issues still require further clarifications. In particular, our results indicate two possible hypotheses of ligand binding to SHP where either the agonist (2) and antagonist (3) bind to the allosteric site of the receptor, or the agonist (2) binds to the canonical cleft of the receptor, respectively. It is apparent from bibliographic evidences that both hypotheses are plausible, with a conserved residue of the allosteric site (Arg238) playing a functional role in other nuclear receptors such as LRH-1 and SF-1 and a residue of the canonical site (Arg34) being identified in a group of missense mutations of SHP. While these hypotheses are the object of ongoing thorough experimental studies whose results will be reported in due course, results of preliminary mutagenesis experiments suggest that some residues of the allosteric site such as Phe96, Arg138 and Arg238, are indeed involved in the repression mechanism of SHP. Accordingly, also in view of the competitive profile of ligands 2 and 3, we have pursued the hypothesis that the atypical retinoids 2–4 may bind to the allosteric site of the receptor. In this scenario, MD simulations indicate that agonist (2) and antagonist (3) induce different patterns of stabilization of the helix H12 and NR boxes that, in turn, affect the repression mechanism of SHP by regulating the corepressor recruitment and the binding of this receptor to the AF-2 surface of nuclear receptors, respectively. Furthermore, our studies show that additional atypical retinoids, such as ST1926 (4), may interact with SHP as part of their mechanism of pro-apoptotic activity.
Methods
Homology modelling
The sequence of human SHP (Entrez code: NP_068804) was submitted to a BLAST [56] search versus the RCSB protein (PDB) database [57] using the WU-BLAST server [58]. The resulting top ranked proteins are shown in Table 1. SiteMap algorithm, as implemented in the Schrodinger software package [59], was used to locate binding sites into each of the resulting structures, assigning a site score (S) [60]. This score is calibrated so that a satisfactory binding site is endowed with S ≥ 1.0.
After this analysis, four structures (pdb codes: 3F5C; 1G2N; 1MV9; 1DKF) were identified as suitable templates to build different homology models of SHP on the basis of conformational criteria and the site score. Despite its slightly low site score (=0.98), DAX-1 (pdb code: 3F5C) was included in the template structures because of its closest sequence similarity relationship to SHP. Multiple sequence alignments and the construction of SHP homology models were carried out using Prime v. 2.1. The multiple alignments were carefully checked to avoid gap insertion where conserved secondary structure motifs were present. At this regard, it should be mentioned that the large loop insertion of SHP (residues 110–138) was modeled as previously reported [22]. For each of the generated model, the side chains of residues were refined identifying the low energy conformations using a rotamer library as implemented in Prime v. 2.1. Finally, the homology models were submitted to the protein preparation refinement protocol as implemented in the Schrodinger package, consisting in the optimization of the hydrogen bond network and a restrained minimization (maximum RMSD allowed 0.30 Å) using the OPLS-2001 force field. The statistical and geometrical validations of the final models were carried using Molprobity and Verify 3D servers [61–63].
Docking studies
Ligands 2–4 were prepared with the ligand preparation tool implemented in the Schrödinger Suite 2009. In particular, we considered for each compound different tautomers and ionization states at the physiological pH of 7.0. Docking experiments into the four conformational models of SHP were carried out using Glide 5.5 and the extra precision (XP) protocol. During these experiments, the induced fit protocol of Glide was employed to take into account any possible ligand induced conformational rearrangement of the binding site. Two solutions for each ligand, namely the top scored one and its most diverse solution among the first ten ranked solutions, were stored for the analysis of the binding modes. In particular, the calculation of the RMSD of heavy atoms was used to identify the second solution of docking experiments.
Molecular dynamics
The complexes resulting from docking experiments (Table 2) were solvated in an orthorhombic box using TIP3P water molecules [64]. Sodium and chlorine atoms were also placed to counter-balance the net charges of the receptor. All the molecular dynamic simulations (MDs) were performed using Desmond 2.2 software [65, 66] and the force field OPLS-2005 as implemented in the Schrödinger Suite 2009. In particular, the simulation protocol was constituted of the Desmond default relaxation steps, 2 ns equilibration phase and a final production phase of 8 ns. In particular, Desmond default relaxation steps comprised: (a) the energetic minimization of the system over a maximum of 2000 steps, with the first 10 steps using the steepest descendent and the convergence criterion being set to 50 kcal/mol Å−1, harmonic restrains placed on the solute atoms (force constant = 50.0 kcal/mol); (b) an analogous energetic refinement of the system without restrains; (c) MDs of 12 ps at temperature 10°K with harmonic restrains on the solute heavy atoms (force constant = 50.0 kcal/mol), NVT Berendesen ensemble; (d) MDs of 12 ps at temperature 10°K retaining the harmonic restrains, NPT Berendesen ensemble; (e) MDs of 24 ps heating phase to temperature 300°K retaining the harmonic restrains, NPT Berendesen ensemble; (f) MDs of 24 ps at temperature 300°K without harmonic restrains, NPT Berendesen ensemble. Both equilibration and productions phases were run using the NPT Berendesen ensemble at temperature 300°K. During all the simulations, a time step of 2 fs was used. Other not specified parameters were set to the default values. The atomic coordinates of each system were saved every 4.8 ps. The occupancy of ligand-receptor hydrogen bond interactions was calculated in the production phase of the simulations (last 8 ns). Schematic interactions shown in Fig. 5 were drawn using MOE [67].
Mutagenesis experiments
Mutations were introduced in Human SHP gene by PCR based site directed mutagenesis using quick change site directed mutagenesis kit (Stratagene, CA, USA). Two complementary oligonucleotide sets were designed as PCR primers to introduce each mutation whose sequences were listed in the Table s2. Reaction mixtures were set up according to the manufacturer’s instructions using 50 ng of template DNA (hSHP cloned in pcDNA3) and 125 ng of primers. Cyclic parameters: denaturing at 95 °C for 30 s, followed by 18 cycles at 95 °C for 30 s, 55 °C for 1 min and 68 °C for 7 min. The reaction mixture was digested with Dpn1 for 1 h at 37 °C and finally transformed into XL1 blue super competent cells. Sequences of all constructs were confirmed using DNA sequencing.
Luciferase reporter assay
Cos7 cells were maintained in Dulbecco’s minimal essential medium in the presence of 10% FBS. pApoCIII-Luc and pcDNA3-HNF4α were kindly provided by Dr. Frances Sladek while human SHP was cloned into pcDNA3. Different hSHP mutants were constructed as described earlier. For luciferase assays, cells were plated in 24 well plates 1 day before transfection and transfection was carried out using lipofectamine 2000 (Promega, Madison, USA). Total DNA in each transfection was adjusted by adding appropriate amounts of pcDNA3 empty vector. Approximately 48 h after transfection, cells were harvested and luciferase activities were measured using luciferase assay systems (Promega) according to manufacturer’s instructions. The luciferase readings were normalized against β-galactosidase activity as an internal control. The transfection experiments were repeated thrice, each time carried out in triplicates.
Supplementary Material
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10822-010-9386-9) contains supplementary material, which is available to authorized users.
Contributor Information
Marco Cellanetti, Dipartimento di Chimica e Tecnologia del Farmaco, Università di Perugia, via del Liceo 1, 06123 Perugia, Italy.
Viswanath Gunda, Departments of Medicine, Oncological Sciences and Huntsman Cancer Institute, University of Utah School of Medicine, Salt Lake City, UT 84132, USA.
Li Wang, Departments of Medicine, Oncological Sciences and Huntsman Cancer Institute, University of Utah School of Medicine, Salt Lake City, UT 84132, USA.
Antonio Macchiarulo, Email: antonio@chimfarm.unipg.it, Dipartimento di Chimica e Tecnologia del Farmaco, Università di Perugia, via del Liceo 1, 06123 Perugia, Italy.
Roberto Pellicciari, Dipartimento di Chimica e Tecnologia del Farmaco, Università di Perugia, via del Liceo 1, 06123 Perugia, Italy.
References
- 1.Seol W, Choi HS, Moore DD. An orphan nuclear hormone receptor that lacks a DNA binding domain and heterodimerizes with other receptors. Science. 1996;272:1336–1339. doi: 10.1126/science.272.5266.1336. [DOI] [PubMed] [Google Scholar]
- 2.Lee HK, Lee YK, Park SH, Kim YS, Lee JW, et al. Structure and expression of the orphan nuclear receptor SHP gene. J Biol Chem. 1998;273:14398–14402. doi: 10.1074/jbc.273.23.14398. [DOI] [PubMed] [Google Scholar]
- 3.Chanda D, Park JH, Choi HS. Molecular basis of endocrine regulation by orphan nuclear receptor Small Heterodimer Partner. Endocr J. 2008;55:253–268. doi: 10.1507/endocrj.k07e-103. [DOI] [PubMed] [Google Scholar]
- 4.Lee YS, Chanda D, Sim J, Park YY, Choi HS. Structure and function of the atypical orphan nuclear receptor small heterodimer partner. Int Rev Cytol. 2007;261:117–158. doi: 10.1016/S0074-7696(07)61003-1. [DOI] [PubMed] [Google Scholar]
- 5.Kim MK, Chanda D, Lee IK, Choi HS, Park KG. Targeting orphan nuclear receptor SHP in the treatment of metabolic diseases. Expert Opin Ther Targets. 2010;14:453–466. doi: 10.1517/14728221003652463. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Y, Soto J, Park K, Viswanath G, Kuwada S, et al. Nuclear receptor SHP, a death receptor that targets mitochondria, induces apoptosis and inhibits tumor growth. Mol Cell Biol. 2010;30:1341–1356. doi: 10.1128/MCB.01076-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hung CC, Farooqi IS, Ong K, Luan J, Keogh JM, et al. Contribution of variants in the small heterodimer partner gene to birthweight, adiposity, and insulin levels: mutational analysis and association studies in multiple populations. Diabetes. 2003;52:1288–1291. doi: 10.2337/diabetes.52.5.1288. [DOI] [PubMed] [Google Scholar]
- 8.Nishigori H, Tomura H, Tonooka N, Kanamori M, Yamada S, et al. Mutations in the small heterodimer partner gene are associated with mild obesity in Japanese subjects. Proc Natl Acad Sci U S A. 2001;98:575–580. doi: 10.1073/pnas.021544398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cao H, Hegele RA. Identification of polymorphisms in the human SHP1 gene. J Hum Genet. 2002;47:445–447. doi: 10.1007/s100380200062. [DOI] [PubMed] [Google Scholar]
- 10.Echwald SM, Andersen KL, Sorensen TI, Larsen LH, Andersen T, et al. Mutation analysis of NR0B2 among 1545 Danish men identifies a novel c.278G>A (p.G93D) variant with reduced functional activity. Hum Mutat. 2004;24:381–387. doi: 10.1002/humu.20090. [DOI] [PubMed] [Google Scholar]
- 11.Mitchell SM, Weedon MN, Owen KR, Shields B, Wilkins-Wall B, et al. Genetic variation in the small heterodimer partner gene and young-onset type 2 diabetes, obesity, and birth weight in U.K. subjects. Diabetes. 2003;52:1276–1279. doi: 10.2337/diabetes.52.5.1276. [DOI] [PubMed] [Google Scholar]
- 12.Nishizawa H, Yamagata K, Shimomura I, Takahashi M, Kuriyama H, et al. Small heterodimer partner, an orphan nuclear receptor, augments peroxisome proliferator-activated receptor gamma transactivation. J Biol Chem. 2002;277:1586–1592. doi: 10.1074/jbc.M104301200. [DOI] [PubMed] [Google Scholar]
- 13.Lee YK, Dell H, Dowhan DH, Hadzopoulou-Cladaras M, Moore DD. The orphan nuclear receptor SHP inhibits hepatocyte nuclear factor 4 and retinoid X receptor transactivation: two mechanisms for repression. Mol Cell Biol. 2000;20:187–195. doi: 10.1128/mcb.20.1.187-195.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johansson L, Bavner A, Thomsen JS, Farnegardh M, Gustafsson JA, et al. The orphan nuclear receptor SHP utilizes conserved LXXLL-related motifs for interactions with ligand-activated estrogen receptors. Mol Cell Biol. 2000;20:1124–1133. doi: 10.1128/mcb.20.4.1124-1133.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bavner A, Johansson L, Toresson G, Gustafsson JA, Treuter E. A transcriptional inhibitor targeted by the atypical orphan nuclear receptor SHP. EMBO Rep. 2002;3:478–484. doi: 10.1093/embo-reports/kvf087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klinge CM, Jernigan SC, Risinger KE, Lee JE, Tyulmenkov VV, et al. Short heterodimer partner (SHP) orphan nuclear receptor inhibits the transcriptional activity of aryl hydrocarbon receptor (AHR)/AHR nuclear translocator (ARNT) Arch Biochem Biophys. 2001;390:64–70. doi: 10.1006/abbi.2001.2366. [DOI] [PubMed] [Google Scholar]
- 17.Kim JY, Kim HJ, Kim KT, Park YY, Seong HA, et al. Orphan nuclear receptor small heterodimer partner represses hepatocyte nuclear factor 3/Foxa transactivation via inhibition of its DNA binding. Mol Endocrinol. 2004;18:2880–2894. doi: 10.1210/me.2004-0211. [DOI] [PubMed] [Google Scholar]
- 18.Gobinet J, Auzou G, Nicolas JC, Sultan C, Jalaguier S. Characterization of the interaction between androgen receptor, a new transcriptional inhibitor, SHP. Biochemistry. 2001;40:15369–15377. doi: 10.1021/bi011384o. [DOI] [PubMed] [Google Scholar]
- 19.Borgius LJ, Steffensen KR, Gustafsson JA, Treuter E. Glucocorticoid signaling is perturbed by the atypical orphan receptor and corepressor SHP. J Biol Chem. 2002;277:49761–49766. doi: 10.1074/jbc.M205641200. [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Choi M, Suino K, Kovach A, Daugherty J, et al. Structural and biochemical basis for selective repression of the orphan nuclear receptor liver receptor homolog 1 by small heterodimer partner. Proc Natl Acad Sci U S A. 2005;102:9505–9510. doi: 10.1073/pnas.0501204102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jouravel N, Sablin E, Arnold LA, Guy RK, Fletterick RJ. Interaction between the androgen receptor and a segment of its corepressor SHP. Acta Crystallogr D Biol Crystallogr. 2007;63:1198–1200. doi: 10.1107/S0907444907045702. [DOI] [PubMed] [Google Scholar]
- 22.Macchiarulo A, Rizzo G, Costantino G, Fiorucci S, Pellicciari R. Unveiling hidden features of orphan nuclear receptors: the case of the small heterodimer partner (SHP) J Mol Graph Model. 2006;24:362–372. doi: 10.1016/j.jmgm.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 23.Kemper JK, Kim H, Miao J, Bhalla S, Bae Y. Role of an mSin3A-Swi/Snf chromatin remodeling complex in the feedback repression of bile acid biosynthesis by SHP. Mol Cell Biol. 2004;24:7707–7719. doi: 10.1128/MCB.24.17.7707-7719.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boulias K, Talianidis I. Functional role of G9a-induced histone methylation in small heterodimer partner-mediated transcriptional repression. Nucleic Acids Res. 2004;32:6096–6103. doi: 10.1093/nar/gkh947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fang S, Miao J, Xiang L, Ponugoti B, Treuter E, et al. Coordinated recruitment of histone methyltransferase G9a and other chromatin-modifying enzymes in SHP-mediated regulation of hepatic bile acid metabolism. Mol Cell Biol. 2007;27:1407–1424. doi: 10.1128/MCB.00944-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gobinet J, Carascossa S, Cavailles V, Vignon F, Nicolas JC, et al. SHP represses transcriptional activity via recruitment of histone deacetylases. Biochemistry. 2005;44:6312–6320. doi: 10.1021/bi047308d. [DOI] [PubMed] [Google Scholar]
- 27.Smock RG, Gierasch LM. Sending signals dynamically. Science. 2009;324:198–203. doi: 10.1126/science.1169377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tokuriki N, Tawfik DS. Protein dynamism and evolvability. Science. 2009;324:203–207. doi: 10.1126/science.1169375. [DOI] [PubMed] [Google Scholar]
- 29.Farhana L, Dawson MI, Leid M, Wang L, Moore DD, et al. Adamantyl-substituted retinoid-related molecules bind small heterodimer partner and modulate the Sin3A repressor. Cancer Res. 2007;67:318–325. doi: 10.1158/0008-5472.CAN-06-2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dawson MI, Harris DL, Liu G, Hobbs PD, Lange CW, et al. Antagonist analogue of 6-[3′-(1-adamantyl)-4′-hydroxyphenyl]-2-naphthalenecarboxylic acid (AHPN) family of apoptosis inducers that effectively blocks AHPN-induced apoptosis but not cell-cycle arrest. J Med Chem. 2004;47:3518–3536. doi: 10.1021/jm030524k. [DOI] [PubMed] [Google Scholar]
- 31.Dawson MI, Xia Z, Liu G, Ye M, Fontana JA, et al. An adamantyl-substituted retinoid-derived molecule that inhibits cancer cell growth and angiogenesis by inducing apoptosis and binds to small heterodimer partner nuclear receptor: effects of modifying its carboxylate group on apoptosis, proliferation, and protein-tyrosine phosphatase activity. J Med Chem. 2007;50:2622–2639. doi: 10.1021/jm0613323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dawson MI, Xia Z, Jiang T, Ye M, Fontana JA, et al. Adamantyl-substituted retinoid-derived molecules that interact with the orphan nuclear receptor small heterodimer partner: effects of replacing the 1-adamantyl or hydroxyl group on inhibition of cancer cell growth, induction of cancer cell apoptosis, and inhibition of SRC homology 2 domain-containing protein tyrosine phosphatase-2 activity. J Med Chem. 2008;51:5650–5662. doi: 10.1021/jm800456k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sablin EP, Woods A, Krylova IN, Hwang P, Ingraham HA, et al. The structure of corepressor Dax-1 bound to its target nuclear receptor LRH-1. Proc Natl Acad Sci U S A. 2008;105:18390–18395. doi: 10.1073/pnas.0808936105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lalli E, Sassone-Corsi P. DAX-1, an unusual orphan receptor at the crossroads of steroidogenic function and sexual differentiation. Mol Endocrinol. 2003;17:1445–1453. doi: 10.1210/me.2003-0159. [DOI] [PubMed] [Google Scholar]
- 35.Wang J, Rao S, Chu J, Shen X, Levasseur DN, et al. A protein interaction network for pluripotency of embryonic stem cells. Nature. 2006;444:364–368. doi: 10.1038/nature05284. [DOI] [PubMed] [Google Scholar]
- 36.Niakan KK, Davis EC, Clipsham RC, Jiang M, Dehart DB, et al. Novel role for the orphan nuclear receptor Dax1 in embryogenesis, different from steroidogenesis. Mol Genet Metab. 2006;88:261–271. doi: 10.1016/j.ymgme.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 37.Pereira FA, Tsai MJ, Tsai SY. COUP-TF orphan nuclear receptors in development and differentiation. Cell Mol Life Sci. 2000;57:1388–1398. doi: 10.1007/PL00000624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kruse SW, Suino-Powell K, Zhou XE, Kretschman JE, Reynolds R, et al. Identification of COUP-TFII orphan nuclear receptor as a retinoic acid-activated receptor. PLoS Biol. 2008;6:e227. doi: 10.1371/journal.pbio.0060227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Billas IM, Moulinier L, Rochel N, Moras D. Crystal structure of the ligand-binding domain of the ultraspiracle protein USP, the ortholog of retinoid X receptors in insects. J Biol Chem. 2001;276:7465–7474. doi: 10.1074/jbc.M008926200. [DOI] [PubMed] [Google Scholar]
- 40.Carmichael JA, Lawrence MC, Graham LD, Pilling PA, Epa VC, et al. The X-ray structure of a hemipteran ecdysone receptor ligand-binding domain: comparison with a lepidopteran ecdysone receptor ligand-binding domain and implications for insecticide design. J Biol Chem. 2005;280:22258–22269. doi: 10.1074/jbc.M500661200. [DOI] [PubMed] [Google Scholar]
- 41.Clayton GM, Peak-Chew SY, Evans RM, Schwabe JW. The structure of the ultraspiracle ligand-binding domain reveals a nuclear receptor locked in an inactive conformation. Proc Natl Acad Sci U S A. 2001;98:1549–1554. doi: 10.1073/pnas.041611298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sablin EP, Blind RD, Krylova IN, Ingraham JG, Cai F, et al. Structure of SF-1 bound by different phospholipids: evidence for regulatory ligands. Mol Endocrinol. 2009;23:25–34. doi: 10.1210/me.2007-0508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang W, Zhang C, Marimuthu A, Krupka HI, Tabrizizad M, et al. The crystal structures of human steroidogenic factor-1 and liver receptor homologue-1. Proc Natl Acad Sci U S A. 2005;102:7505–7510. doi: 10.1073/pnas.0409482102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gronemeyer H, Gustafsson JA, Laudet V. Principles for modulation of the nuclear receptor superfamily. Nat Rev Drug Discov. 2004;3:950–964. doi: 10.1038/nrd1551. [DOI] [PubMed] [Google Scholar]
- 45.Cincinelli R, Dallavalle S, Nannei R, Carella S, De Zani D, et al. Synthesis and structure-activity relationships of a new series of retinoid-related biphenyl-4-ylacrylic acids endowed with antiproliferative and proapoptotic activity. J Med Chem. 2005;48:4931–4946. doi: 10.1021/jm049440h. [DOI] [PubMed] [Google Scholar]
- 46.Cincinelli R, Dallavalle S, Merlini L, Penco S, Pisano C, et al. A novel atypical retinoid endowed with proapoptotic and antitumor activity. J Med Chem. 2003;46:909–912. doi: 10.1021/jm025593y. [DOI] [PubMed] [Google Scholar]
- 47.Krylova IN, Sablin EP, Moore J, Xu RX, Waitt GM, et al. Structural analyses reveal phosphatidyl inositols as ligands for the NR5 orphan receptors SF-1 and LRH-1. Cell. 2005;120:343–355. doi: 10.1016/j.cell.2005.01.024. [DOI] [PubMed] [Google Scholar]
- 48.Li Y, Choi M, Cavey G, Daugherty J, Suino K, et al. Crystallographic identification and functional characterization of phospholipids as ligands for the orphan nuclear receptor steroidogenic factor-1. Mol Cell. 2005;17:491–502. doi: 10.1016/j.molcel.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 49.Garattini E, Gianni M, Terao M. Retinoid related molecules an emerging class of apoptotic agents with promising therapeutic potential in oncology: pharmacological activity and mechanisms of action. Curr Pharm Des. 2004;10:433–448. doi: 10.2174/1381612043453351. [DOI] [PubMed] [Google Scholar]
- 50.Pisano C, Vesci L, Fodera R, Ferrara FF, Rossi C, et al. Antitumor activity of the combination of synthetic retinoid ST1926 and cisplatin in ovarian carcinoma models. Ann Oncol. 2007;18:1500–1505. doi: 10.1093/annonc/mdm195. [DOI] [PubMed] [Google Scholar]
- 51.Sala F, Zucchetti M, Bagnati R, D’Incalci M, Pace S, et al. Development and validation of a liquid chromatography-tandem mass spectrometry method for the determination of ST1926, a novel oral antitumor agent, adamantyl retinoid derivative, in plasma of patients in a Phase I study. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:3118–3126. doi: 10.1016/j.jchromb.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 52.Pisano C, Merlini L, Penco S, Carminati P, Zunino F. Cellular and pharmacological bases of the antitumor activity of a novel adamantyl retinoid ST1926. J Chemother. 2004;16(supply 4):74–76. doi: 10.1179/joc.2004.16.Supplement-1.74. [DOI] [PubMed] [Google Scholar]
- 53.Zuco V, Zanchi C, Lanzi C, Beretta GL, Supino R, et al. Development of resistance to the atypical retinoid, ST1926, in the lung carcinoma cell line H460 is associated with reduced formation of DNA strand breaks and a defective DNA damage response. Neoplasia. 2005;7:667–677. doi: 10.1593/neo.05127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zuco V, Zanchi C, Cassinelli G, Lanzi C, Supino R, et al. Induction of apoptosis and stress response in ovarian carcinoma cell lines treated with ST1926, an atypical retinoid. Cell Death Differ. 2004;11:280–289. doi: 10.1038/sj.cdd.4401304. [DOI] [PubMed] [Google Scholar]
- 55.Valli C, Paroni G, Di Francesco AM, Riccardi R, Tavecchio M, et al. Atypical retinoids ST1926 and CD437 are S-phase-specific agents causing DNA double-strand breaks: significance for the cytotoxic and antiproliferative activity. Mol Cancer Ther. 2008;7:2941–2954. doi: 10.1158/1535-7163.MCT-08-0419. [DOI] [PubMed] [Google Scholar]
- 56.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, et al. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.WU-BLAST. San Diego, CA: http://www.ebi.ac.uk/blast2/ [Google Scholar]
- 59.Maestro, version 9.0. Schrödinger, LLC; New York, NY: 2009. [Google Scholar]
- 60.Halgren TA. Identifying and characterizing binding sites and assessing druggability. J Chem Inf Model. 2009;49:377–389. doi: 10.1021/ci800324m. [DOI] [PubMed] [Google Scholar]
- 61.Davis IW, Murray LW, Richardson JS, Richardson DC. MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004;32:W615–W619. doi: 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Luthy R, Bowie JU, Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;356:83–85. doi: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- 63.Morris AL, MacArthur MW, Hutchinson EG, Thornton JM. Stereochemical quality of protein structure coordinates. Proteins. 1992;12:345–364. doi: 10.1002/prot.340120407. [DOI] [PubMed] [Google Scholar]
- 64.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 65.Bowers KJ, Chow E, Xu H, Dror RO, Eastwood MP, et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. Proceedings of the ACM/IEEE Conference on Supercomputing (SC06); Florida (USA). 2006. [Google Scholar]
- 66.Desmond Molecular Dynamics System. Shaw DE Research; New York, NY: 2009. version 2.2. [Google Scholar]; Maestro-Desmond Interoperability Tools. Schrödinger; New York, NY: 2009. version 2.2. [Google Scholar]
- 67.MOE (The Molecular Operating Environment) Version 2009.10. Chemical Computing Group Inc; 1010 Sherbrooke Street West, Suite 910, Montreal, Canada H3A 2R7: [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.