

Abstract
Background & Aims
Inflammation promotes the progression of non-alcoholic steatohepatitis (NASH). Toll-like receptor 4 (TLR4) and TLR9 activation through myeloid differentiation primary response gene 88 (MyD88) and production of mature interleukin-1β (IL-1β) via inflammasome activation contribute to steatohepatitis. Here, we investigated the inter-relationship between TLR signalling and inflammasome activation in dietary steatohepatitis.
Methods
Wild type (WT), TLR4- and MyD88-deficient (KO) mice received methionine-choline-deficient (MCD) or -supplemented (MCS) diets for 5 weeks and a subset was challenged with TLR9 ligand CpG-DNA.
Results
TLR4, TLR9, AIM2 (absent in melanoma 2) and NLRP3 (NLR family pyrin domain containing 3) inflammasome mRNA, and mature IL-1β protein levels were increased in MCD diet-induced steatohepatitis compared to MCS controls. TLR9 stimulation resulted in greater up-regulation of the DNA-sensing AIM2 expression and IL-1β production in livers of MCD compared to MCS diet-fed mice. High mobility group box 1 (HMGB1), a TLR9-activating danger molecule and phospho-HMGB1 protein levels were also increased in livers of MCD diet-fed mice. MyD88- but not TLR4-deficiency prevented up-regulation of AIM2, NLRP3 mRNA and IL-1β protein production in dietary steatohepatitis. Selective MyD88 deficiency either in bone marrow (BM)-derived or non-BM-derived cells attenuated hepatic up-regulation of inflammasome mRNA, caspase-1 activation and IL-1β protein production, but only BM-derived cell-specific MyD88-deficiency attenuated liver injury.
Conclusions
Our data demonstrate that both bone marrow-derived and non-BM-derived cells contribute to inflammasome activation in a MyD88-dependent manner in dietary steatohepatitis. We show that AIM2 inflammasome expression and activation are further augmented by TLR9 ligands in dietary steatohepatitis.
Keywords: AIM2, caspase-1, HMGB1, IL-1β, inflammasome, MyD88, NLRP3, non-alcoholic steatohepatitis, TLR4, TLR9
Non-alcoholic fatty liver disease (NAFLD) is increasingly common in the USA, Europe and world-wide (1). The progression of NAFLD to non-alcoholic steatohepatitis (NASH) is associated with necroinflammation leading to fibrosis and cirrhosis (1). The molecular mechanisms linking hepatocyte necrosis to inflammation and progressive liver damage are yet to be defined. Damage associated molecular patterns (DAMPs) released from injured cells (2) initiate inflammatory responses via activation of pattern recognition receptors, including Toll-like receptors (TLRs), Nod-like receptors (NLRs), and pyrin and HIN domain containing (PYHIN) inflammasome receptors. TLR4, a sensor of bacterial endotoxin, and TLR9, a sensor of modified self DNA (3) were shown to contribute to the pathogenesis of steatohepatitis via the common TLR signalling adapter molecule, MyD88 in animal models (4). Inflammasome-triggering sensors include AIM2, which senses damaged self DNA (5), and NLRP3, which senses potassium release and adenosine triphosphate (ATP) as well as lysosome destabilization and cathepsin release (6).
Inflammasomes are large, multiprotein complexes that assemble in response to exogenous or endogenous danger signals to activate caspase-1 leading to the processing and release of IL-1β and IL-18 (6). Inflammasome activation for IL-1β release is a multistep process. The first step involves TLR-mediated up-regulation of the mRNA and expression of the pro-IL-1β substrate. TLR-mediated signalling may also be required for expression of inflammasome components themselves. For example, NLRP3 expression can be up-regulated by TLR-mediated signalling in different cells (7). Assembly and activation of the inflammasome occurs upon a second triggering event, resulting in caspase-1 activation and IL-1β release (6, 8). AIM2 inflammasome activation is triggered by damaged self, bacterial or viral dsDNA (5, 6). Hepatocellular apoptosis leading to DNA fragmentation is a response to metabolic, ROS and other insults in NASH indicated by increased caspase-3 activity and plasma cytokeratin-18 fragments (9, 10). Recent reports suggest that mitochondrial DNA that contains cytidine phosphateguanosine (CpG)-DNA repeats owed to its saprophytic bacterial origin is released during tissue injury and induces activation of polymorphonuclear cells (PMNs) (11). Mitochondrial DNA sequences were found in nuclear DNA of rat hepatocytes (12) and TLR9 is activated by unmethylated CpG sequences of DNA (13). DAMPs released from damaged hepatocytes during necrosis, such as HMGB1, are recognized by TLR4 and TLR9 (14). In addition, HMGB1 can induce caspase-1 activation (15). TLR4 also senses the bacterial endotoxin (LPS) which could result from increased gut permeability described in NASH (16). Both TLR4 and TLR9 utilize the common TLR adaptor molecule, MyD88, and TLR4 and TLR9 deficiency attenuated experimental steatohepatitis in mice (3, 17). However, there is no consensus regarding the role of MyD88 in steatohepatitis in the literature. While the protective role of TLR4 has been shown in both the high-fat diet (18) and the MCD diet model of NASH (16), mice with MyD88 deficiency were protected against the choline- deficient amino acid defined (CDAA) diet-induced steatohepatitis (3), but not from the high-fat diet-induced liver injury (19). MyD88 deficiency has not yet been investigated in the MCD model of NASH.
Recent reports indicate increased expression of the NLRP3 inflammasome, activation of the inflammasome complex and increased IL-1β production in choline-deficient (CD), methionine-choline-deficient (MCD) and high-fat (HF) diet-induced steatohepatitis in mice (3, 20, 21). However, the triggers for IL-1β and inflammasome up-regulation and activation are yet to be understood. Current reports are somewhat contradictory on whether protection from steatohepatitis is conferred by deficiency in inflammasome component expression or by caspase inhibitors (3, 21–25). Notably, these studies included total body knock-out mice that were lacking the different inflammasome components both in BM-derived and liver parenchymal cells. Henao-Mejia et al. suggested that inflammasomes in intestinal cells are important players in the progression of steatohepatitis, since inflammasome-deficiency worsens the hepatic steatosis and inflammation in diet-induced models of steatohepatitis because of changes in gut microbiota and therefore increased influx of TLR4 and TLR9 ligands into the portal circulation (21). This may suggest that the influence of inflammasomes on the development of steatohepatitis is not solely dependent on their activation in hepatic immune cells. In contrast, in alcoholic liver disease, a disease that shares common pathogenic features with NASH, Petrasek J et al. reported that the inflammasome-dependent IL-1β production was crucial for the pathogenesis, and it involved the liver-resident macrophages, Kupffer-cells (26).
In this study, we hypothesized that inflammasome activation in dietary steatohepatitis may involve TLR/MyD88 signalling as well as DAMPs, such as HMGB1, to amplify inflammation and liver damage. Our novel data indicate that steatohepatitis in the MCD diet model is associated with increased AIM2 expression and inflammasome activation which requires MyD88-dependent pathways in both hepatocytes and BM-derived cells. Our data demonstrated that inflammasome expression and liver damage in dietary steatohepatitis can be amplified by TLR9 stimulation.
Methods
Animal studies
Female C57Bl/6J wild type (WT) mice, and those deficient (knock-out, KO) in TLR4 or MyD88 were employed (n = 6–8/group). We also used WT mice transplanted with MyD88-deficient bone marrow (WT/MyD88) and MyD88-deficient mice transplanted with WT bone marrow (MyD88/WT). Bone marrow transplantation was performed as described earlier (27). This study was approved by the Institutional Animal Use and Care Committee (IACUC) at UMASS. Mice were fed with either a methionine-choline-deficient (MCD) diet or an identical, but D, L-methionine and choline-bitartrate supplemented (MCS) control diet (Dyets Inc., Bethlehem, PA, USA) for 5 weeks. TLR9 ligand CpG-ODN (5 mg/kg b.w., InvivoGen, San Diego, CA, USA) was injected intraperitoneally.
Serum was separated from whole blood. Livers were snap-frozen in liquid nitrogen, stored in RNAlater™ (Qiagen GmbH, Hilden, Germany) for RNA extraction, fixed in formalin for histopathological analysis, or fixed in OCT medium (Sakura Finetek Inc., Torrance, CA, USA) for Oil Red O staining.
Biochemical analysis and cytokine measurements
Serum alanine aminotransferase (ALT) was determined using a kinetic method (D-TEK, Bensalem, PA, USA). Liver triglyceride levels were assessed using the L-Type Triglyceride H kit (Wako Chemicals USA Inc. Richmond, VA, USA). Serum and liver IL-1β levels were determined by ELISA (R&D Systems, Minneapolis, MN, USA).
Histopathological analysis
Sections of formalin-fixed livers were stained with haematoxylin-eosin, while OCT frozen samples were stained with Oil Red O; all slides were analysed by microscopy. The slides were scored for steatosis, necrosis, hepatocyte ballooning and lobular and portal inflammation by a pathologist expert (Park JK) using the scoring system described by Kleiner DE, Brunt EM et al. (28).
RNA analysis
RNA was purified using the RNeasy™ kit (Qiagen Sciences, Germantown, MD, USA). cDNA was transcribed with Reverse Transcription System (Promega Corp., Madison, WI, USA). Real-time quantitative polymerase chain reaction was performed using iCycler (Bio-Rad Laboratories Inc., Hercules, CA, USA) and SYBRGreen. Primer sequences are available upon request.
Western blot
Whole cell lysates were extracted from liver. Samples with equal amounts of protein were separated in polyacrylamide gel, transferred and identified on nitrocellulose membrane with specific primary antibodies followed by HRP-labelled secondary antibodies. The following antibodies were applied: caspase-1 p10 (Santa Cruz Bio-technology Inc., Dallas, TX, USA), IL-1β (R&D). Beta tubulin (Abcam) was used as loading control.
Immunoprecipitation
Whole liver lysates were precleared with antirabbit IgG beads followed by overnight incubation with 5 μg primary antibody (HMGB1) and precipitated with IgG beads. The immunoprecipitates were lysed and denatured using β-mercaptoethanol containing buffer and heating. Proteins were separated on polyacrylamide gel and detected by specific antibodies (phosphoserine, HMGB1).
Statistical analysis
Statistical significance was determined using non-parametric Kruskal–Wallis or Mann–Whitney tests. Data are shown as mean ± standard error and were considered statistically significant at P ≤ 0.05.
Results
MCD diet-induced steatohepatitis is associated with NLRP3- and AIM2- inflammasome up-regulation and activation
The mechanisms leading to liver inflammation in NASH are only partially understood. The methionine-choline-deficient (MCD) model of steatohepatitis results in macrovesicular steatosis (Fig. 1A, left panel) as well as increased hepatic triglyceride levels (Fig. 1A, middle panel), inflammatory cell infiltration (Fig. 1A, left panel), and elevated serum alanine aminotransferase (ALT) levels indicating liver injury (Fig. 1A, right panel) (17). In the MCD model of steatohepatitis, we found elevated serum and hepatic levels of the inflammatory cytokine IL-1β (Fig. 1B and C) (20). IL-1β production is dependent on processing of pro-IL-1β to mature IL-1β by inflammasomes. We found that MCD diet-induced elevated levels of mRNA for components of the multiprotein inflammasome complex, including the inflammasome sensors, AIM2 and NLRP3, the inflammasome adaptor molecule ASC and the effector enzyme pro-caspase-1 (Fig. 1D). Complex formation of either NLRP3 or AIM2 with ASC and pro-caspase-1 results in auto-activation and cleavage of the 45 kDa pro-caspase-1 to its enzymatically active form, a heterodimer of p20 and p10 subunits (6). Inflammasome activation in dietary steatohepatitis was indicated by increased levels of not only pro-caspase-1 (Fig. 1E) but also the active p10 subunit of caspase-1 (Fig. 1E) and increased cleaved IL-1β protein levels (Fig. 1B and F) in MCD compared to MCS diet-fed mice.
Fig. 1.
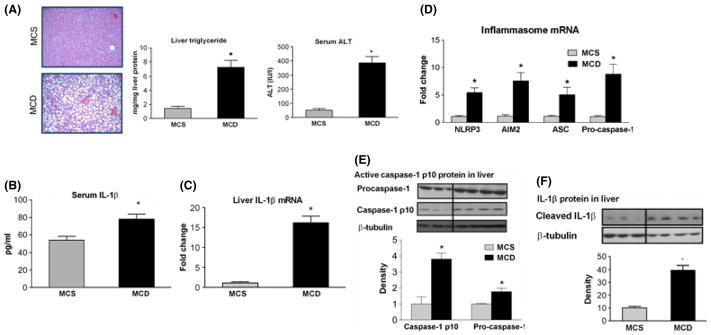
Methionine-choline-deficient (MCD) diet-induced steatohepatitis is associated with AIM2 and NLRP3 inflammasome up-regulation and activation. C57Bl/6J mice were fed a MCD or – methionine-choline-supplemented (MCS) diet. Liver histology (haematoxylin-eosin staining, 100×), liver triglycerides and serum ALT (A) shows that MCD diet-induced steatohepatitis. Serum IL-1β protein (B) and liver IL-1β mRNA (C) levels, as well as hepatic mRNA expression of AIM2 and NLRP3 inflammasome complexes (D) were measured. Pro-caspase-1 and caspase- 1 p10 active subunit (E) and IL-1β (F) protein expression were determined by Western blot in the liver. *indicates P italic> 0.05 MCS vs. corresponding MCD group.
Inflammasome gene up-regulation and IL-1β mRNA induction are MyD88-dependent in dietary steatohepatitis
TLR stimulation by pathogen-associated molecular patterns (PAMPs), such as LPS or bacterial DNA, can increase the expression of pro-IL-1β mRNA and protein as well as the expression of components of the inflammasome (6). We found increased mRNA expression of TLR4, TLR9 and their common adapter, MyD88, in MCD diet-fed livers compared to MCS controls (Fig. 2A). Thus, we hypothesized that TLR- and MyD88-dependent signalling could drive up-regulation of the inflammasome components and IL-1β induction in dietary steatohepatitis. We first asked if inflammasome genes were regulated by MyD88-dependent signalling in our model. We found that MyD88- but not TLR4-deficiency attenuated up-regulation of AIM2 (Fig. 2B and C), NLRP3 (Fig. 2B and C) and pro-IL-1β mRNA (Fig. 2D) as well as IL-1β protein levels (Fig. 2E) in steatohepatitis. The MCD diet-induced increased caspase-1 activation was prevented by MyD88 deficiency, but not by TLR4 deficiency (Fig. 2F). This data suggests that MyD88-dependent signalling is involved in increased inflammasome activation in dietary steatohepatitis.
Fig. 2.

Inflammasome up-regulation and IL-1β induction in MCD-steatohepatitis are MyD88-dependent. C57Bl/6J wild type (WT), and those deficient (KO) in MyD88 or TLR4 were fed a methionine-choline-deficient (MCD) or -supplemented (MCS) diet. TLR4, TLR9 and Myd88 mRNA expression was measured in the liver (A). AIM2 (B and C, left panels), NLRP3 (B and C, right panels) and IL-1β (D) mRNA expression was determined in the liver of WT, MyD88 KO (B and D) and TLR4 KO (C and D) mice. Liver IL-1β protein levels were measured by ELISA (E). Pro-caspase-1 and caspase-1 p10 active subunit protein expression was detected by western blot (Fig. 2F), the density graph represents the caspase-1 p10 active subunit. *indicates P bold> 0.05 MCS vs. corresponding MCD group.
In addition to protection from inflammasome activation, total body MyD88 KO mice showed decreased steatosis indicated by the liver histology (Fig. 3A) and the reduced liver triglyceride levels (Fig. 3B), as well as attenuated liver damage indicated by decreased ALT levels compared to WT mice (Fig. 3C). The liver necrosis score showed a similar trend as the serum ALT levels (Fig. 3A). This suggested that MyD88 was essential for both inflammasome expression and inflammasome activation for IL-1β production and liver damage. Surprisingly, thorough evaluation of liver histology showed that in contrast to the level of necrosis, the degree of hepatocyte ballooning was significantly higher in the MyD88 deficient livers (Fig. 3A).
Fig. 3.

MyD88 deficiency is partially protective against MCD-steatohepatitis. C57Bl/6J wild type (WT), and those deficient (KO) in MyD88 or TLR4 were fed a methionine-choline-deficient (MCD) or -supplemented (MCS) diet. Liver histology was evaluated by haematoxylin-eosin staining (200×, inserts 100×), representative slides are shown (A); black arrows show necrotic cells, black arrowheads show hepatocyte ballooning. Steatosis, necrosis and ballooning scores are shown below. Liver triglyceride (B) and serum alanine aminotransferase (ALT) (C) levels were determined. *indicates P < 0.05 MCS vs. corresponding MCD group.
Inflammasome up-regulation and activation are dependent on MyD88 in BM-derived and non-BM-derived cells
TLRs and inflammasome genes are expressed in the liver both in BM-derived immune cells and in non-BM-derived cells (4, 20). Next, we aimed to evaluate which cell type was involved in the IL-1β production using chimaeras of WT mice transplanted with MyD88-deficient bone marrow (WT/MyD88-KO BM) and MyD88- deficient mice transplanted with WT bone marrow (MyD88-KO/WT BM). We found complete abrogation of AIM2 (Fig. 4A) and NLRP3 (Fig. 4B) mRNA up-regulation in both WT/MyD88-KO BM and in MyD88- KO/WT BM chimaeras compared to WT/WT BM controls in MCD diet-fed mice. Importantly, there was significant attenuation of IL-1β protein levels in both chimaeras (Fig. 4C) compared to WT/WT MCD diet-fed controls. Consistent with this, we found attenuated caspase-1 (p10) activation in both chimaeras (Fig. 4D) compared to the wild type controls. Together these results suggest that both hepatocytes and BM-derived cells contributed to inflammasome activation and IL-1β production in a MyD88-dependent manner in the MCD diet-induced steatohepatitis.
Fig. 4.

Inflammasome up-regulation and IL-1β induction involves both bone marrow-derived and non-bone marrow-derived cells. C57Bl/6J (WT/WT), and wild type (WT) mice transplanted with MyD88-deficient bone marrow (WT/MyD88) and MyD88-deficient mice transplanted with WT bone marrow (MyD88/WT) were fed a methionine-choline-deficient (MCD) or -supplemented (MCS) diet. Hepatic AIM2 (A) and NLRP3 (B) mRNA expression was determined by qPCR. Liver IL-1β protein levels were measured by ELISA (C). Procaspase- 1 and caspase-1 active p10 subunit were detected by Western blot (D), density graph represents the caspase-1 p10 active subunit. *indicates P < 0.05 MCS vs. corresponding MCD group.
Similar to the total body MyD88 KO mice (Fig. 3), WT/MyD88 KO BM mice showed attenuated liver damage (Fig. 5A and C) indicated by reduced ALT levels and slightly decreased steatosis (Fig. 5A and B) compared to WT/WT BM mice. Interestingly, MyD88-deficient mice with WT BM cells were not protected from liver damage and showed higher ALT levels (Fig. 5C) than WT/WT controls on MCD diet, as well as higher necrosis score on histology (Fig. 5A), although the difference in necrosis scores did not reach significance. Interestingly, liver triglyceride levels were also higher in the chimaeras with MyD88-deficient non-BM-derived with WT BM cells. The slight discrepancy between the steatosis score and liver triglyceride levels might be explained by the fact that in the MyD88 KO mice with WT BM the high number of necrotic cells interferes with the evaluation of steatosis on the histological slides. This suggested that the absence of MyD88 expression in non-BM-derived cells can accelerate liver injury and that the expression of MyD88 in BM-derived immune cells alone is sufficient to drive liver damage in dietary steatohepatitis.
Fig. 5.

Bone marrow-specific MyD88 deficiency is partially protective against MCD steatohepatitis. C57Bl/6J (WT/WT), and WT mice transplanted with MyD88-deficient bone marrow (WT/MyD88) and MyD88-deficient mice transplanted with WT bone marrow (MyD88/WT) were fed a methionine-choline-deficient (MCD) or -supplemented (MCS) diet. Liver histology was evaluated by haematoxylin-eosin staining (200×, inserts 100×), representative slides are shown (A); black arrows show necrotic cells, black arrowheads show hepatocyte ballooning. Steatosis, necrosis and ballooning scores are shown below. Liver triglyceride (B) and serum alanine aminotransferase (ALT) (C) levels were determined. *indicates P < 0.05 MCS vs. corresponding MCD group.
Increased levels of the alarmin and TLR9 ligand, HMGB1, in dietary steatohepatitis
TLR9 exclusively utilizes MyD88 as its signalling adapter and the lack of TLR9 has been shown to protect from choline-deficient (CD) diet-induced steatohepatitis (3). High mobility group box protein 1 (HMGB1), a highly conserved nuclear non-histone protein, serves as an endogenous danger signal in damaged cells via activation of TLRs including 2, 4 and 9 that all use MyD88 (29). In search of endogenous ligands that may activate MyD88, we analysed HMGB1 and found increased HMGB1 mRNA expression in livers of MCD diet-fed WT but not MyD88 KO mice compared to controls (Fig. 6A). We also found increased phosphorylated HMGB1 protein expression in MCD diet-fed mice compared to MCS controls (Fig. 6B).
Fig. 6.

Increased levels of the alarmin HMGB1 in steatohepatitis and sensitization of the liver to TLR9-ligand-induced up-regulation of AIM2 inflammasome. Liver HMGB1 mRNA (A) expression and phospho-HMGB1 protein (B) levels were measured by qPCR and Western blot, respectively in mice fed with methionine-choline-deficient (MCD) or -supplemented (MCS) diet. C57Bl/6J mice, after 5 weeks MCD or MCS diet-feeding, were challenged with TLR9 ligand CpG-ODN ip. for 6 h. Serum alanine aminotransferase (ALT) (C) levels were determined. Liver AIM2 (D), NLRP3 (E) and ASC (F) mRNA levels and liver IL-1β protein (G) expression were determined by qPCR and ELISA respectively. Caspase-1 active p10 subunit was detected by Western blot in liver whole cell lysates (H). *indicates P < 0.05 MCS vs. corresponding MCD group. #indicates P < 0.05 untreated vs. the corresponding CpG treated group.
Steatohepatitis sensitizes to TLR9-induced up-regulation of NLRP3- and AIM2- inflammasomes in the liver
Based on our observation of MyD88-dependent inflammasome up-regulation in dietary steatohepatitis we hypothesized that TLR9-induced signals contribute to inflammasome induction in fatty liver. We have previously reported that TLR9 ligand stimulation results in inflammatory cell recruitment, cytokine induction and liver injury (30–32). We asked if TLR9 signalling was altered in our model. In vivo activation of TLR9 with CpG-DNA exacerbated MCD diet-induced liver injury as indicated by increased ALT levels (Fig. 6C). TLR9 ligand-induced up-regulation of inflammasome components, including AIM2, NLRP3, ASC (Fig. 6D–F), as well as IL-1β protein levels (Fig. 6G) and caspase-1 activation (Fig. 6H) were higher in mice with steatohepatitis compared to the controls. In contrast, TLR9 stimulation induced no increase in AIM2 and ASC mRNA expression in MCS control mice. These data suggested that TLR9 ligands induce inflammasome activation and that the dietary steatohepatitis is sensitized to up-regulation of the inflammasome components by TLR9.
Discussion
NAFLD is one of the most common liver diseases world-wide (1). Although its pathogenesis is only partially understood, there is accumulating evidence that innate immune pathways are activated in the metabolic syndrome and play a crucial role in the pathogenesis of NASH (33).
Here we report novel findings on the role of inflammasomes, including the DNA-sensing AIM2 inflammasome and the NLRP3 inflammasome, in the pathogenesis of dietary steatohepatitis. First, we show that AIM2 mRNA (as well as NLRP3, ASC and caspase- 1 mRNA) expression is up-regulated in MCD diet-induced steatohepatitis and that there is functional activation of inflammasomes resulting in increased IL-1β production. Second, we show that up-regulation of AIM2 and NLRP3 inflammasome sensors and inflammasome activation are MyD88-dependent and involve both BM-derived and non-BM-derived cells in the liver. Third, we identified increased levels of phospho-HMGB1, a DAMP, in the liver in MCD diet-induced steatohepatitis. Finally, we demonstrated that MCD-steatohepatitis sensitizes the liver to TLR9 ligand-induced up-regulation of the AIM2 and NLRP3 inflammasomes suggesting amplification between TLR9 and inflammasome pathways.
Increased circulating levels of the Toll-like receptor-4 (TLR4) ligand, LPS (20), and enhanced susceptibility to LPS-induced liver damage (34) brought attention to TLR4 in NASH. We and others have previously reported that LPS along with the saturated fatty acid, palmitic acid can induce the activation of the multiprotein signalling complex inflammasome (20). Inflammasomes can sense exogenous or endogenous danger signals via Nod-like receptors (NLRs) such as NLRP3 or via the PYHIN family proteins such as AIM2. Ligand engagement by NLRs or AIM2 leads to caspase-1 activation that mediates the processing of pro-inflammatory cytokines, including pro-IL-1β and pro-IL-18 (7). Here we show for the first time that the expression of the DNA-sensing inflammasome, AIM2, is up-regulated in the liver of MCD diet-fed mice. Up-regulation of the NLRP3 inflammasome, its adaptor molecule ASC and pro-caspase-1 was also present in the liver in steatohepatitis (20). We found that the inflammasomes were significantly more active in steatohepatitis than in controls, indicated by increased caspase-1 activity and increased cleaved IL-1β levels.
Inflammasome activation is a two-step process; the initial step involves TLR-mediated up-regulation of the mRNA expression of inflammasome components and the pro-IL-1β substrate (7). The role of TLRs in diet-induced up-regulation of the inflammasome is unknown. The role of TLR4 deficiency has been reported in several animal models of NASH, however, TLR4 deficiency only partially protects from diet-induced steatohepatitis (17) suggesting a role for other TLRs in the pathogenesis of steatohepatitis. Indeed, deficiency in TLR9 is protective in the CDAA model of steatohepatitis (3). Almost all TLRs, including TLR4 and TLR9 use MyD88 as a downstream signalling adaptor molecule, and in addition MyD88 is the sole adaptor in the TLR9 signalling pathway. Here, we found that deficiency in MyD88 fully protected from MCD diet-induced up-regulation of the inflammasome sensors, AIM2 and NLRP3. Our data demonstrates that enhanced expression of pro-IL-1β and inflammasome genes is a prerequisite for inflammasome activation, since MyD88 deficiency prevented both up-regulation of the AIM2 and NLRP3 inflammasomes (priming) and IL-1β production (inflammasome activation). Furthermore, TLR9 stimulation increased the expression of all the examined inflammasome components, suggesting an amplification loop between MyD88/TLR9 signalling and inflammasome up-regulation in dietary steatohepatitis.
We also found that MyD88 deficiency was partially protective from MCD diet-induced steatosis and liver injury. Similarly, Miura et al. found that MyD88 deficiency along with TLR9 deficiency protects from CDAA diet-induced steatosis, liver injury, inflammation and fibrosis (3). However, in contrast to these findings in the high fat diet model of steatohepatitis Hosoi et al. found increased liver injury (indicated by the serum ALT, JNK activation, PARP cleavage) along with more severe diabetes in MyD88 deficient mice (19). These opposite findings in the different animal models of non-alcoholic steatohepatitis show the complexity of the findings and underline the importance of the various animal models including our study.
In our study we further dissected the cell-associated role of MyD88 and showed that both liver non-BM-derived cells and BM-derived cells participated in IL-1β induction. It is unclear whether parenchymal cells contributed to IL-1β production directly or indirectly. The role of non-BM-derived hepatic stellate cells and sinusoidal endothelial cells in IL-1β production is also unclear. Previous reports indicate that hepatocytes can up-regulate and activate inflammasome and secrete IL-1β after saturated fatty acid and LPS stimulation in small quantity (15, 20, 35). However, the fact that MyD88 deficiency in non-BM-derived cells attenuated IL-1β production almost as much as the BM-derived cell-specific MyD88 deficiency, suggests that we cannot exclude the indirect effect of non-BM-derived cell-specific MyD88 deficiency on the BM-derived cells. Previously we have shown that hepatocyte-derived DAMPs can induce inflammasome activation in hepatic immune cells (20). However, here we found increased liver injury in MyD88 deficient mice with WT bone marrow. This raises the possibility that other mechanisms than reduced release of DAMPs from injured hepatocytes might contribute to attenuated IL-1β production in these mice. Notably, MyD88-KO/WT BM mice have impaired MyD88 expression not only in the hepatocytes, but also in other non-BM-derived cells. Inflammasome expression and activation has been reported in liver sinusoidal endothelial cells and hepatic stellate cells as well (36). Furthermore, NLRP3 inflammasome expression in intestinal epithelial cells has been shown to be important for the maintenance of the intestinal barrier and protection against colitis (37) and similar mechanism may contribute in this dietary steatohepatitis model (21).
Our data suggests that both non-BM-derived and BM-derived cells contributed to IL-1β production, yet we found differences in liver injury. While MyD88 deficiency in BM-derived cells protected against MCD diet-induced steatosis and liver injury, non-BM-derived cell-specific MyD88 deficiency accelerated the MCD diet-induced liver damage and steatosis. This suggests a potentially protective role of MyD88 in parenchymal cell injury. These data also allows us to speculate that the lack of protection from MCD diet-induced liver injury in caspase-1 KO mice (21) might be due to a possibly differential role of inflammasomes in different cell types. Although loss of intestinal barrier function in MyD88-KO/WT BM mice may exacerbate liver injury mediated by MyD88-expressing immune cells in the liver, the enhanced liver injury in non-BM-derived cell-specific MyD88 deficiency cannot be fully explained by enhanced inflammation caused by increased influx of TLR ligands from the gut, since we found significantly lower pro-inflammatory cytokine, IL-1β levels in both chimaeras compared to WTs.
Caspase-1 has been shown to activate the Sterol Regulatory Element Binding Proteins (SREBPs) and promote cell survival upon toxin exposure in Chinese Hamster Ovary (CHO) and HeLa cell lines (38). Since hepatocytes are specialized epithelial cells, it is tempting to speculate that the observed attenuation of caspase-1 activation may contribute to the enhanced liver injury in MyD88-deficient mice with WT bone marrow. In support of this, we found that SREBP2 cleavage was not enhanced in livers of MCD diet-fed caspase-1 KO mice compared to MCS diet-fed controls. In contrast, SREBP2 was increased after MCD diet-feeding in WT mice (unpublished observation). Caspase-1 was also suggested to play hepatoprotective role during haemorrhagic shock-induced liver injury (39).
Tissue damage and programmed cell death leads to DNA fragmentation (apoptosis). TLR9 recognizes extracellular DNA and CpG motif-containing DNA such as bacterial DNA (3,40). Furthermore, it has been shown that both nuclear and mitochondrial DNA can be released during tissue injury and induce inflammatory response (11). Mitochondrial DNA contains CpG-DNA repeats due to the saprophytic bacterial origin (11). Here we show that the TLR9-ligand CpG-DNA injection in vivo induced inflammasome activation and that steatohepatitis sensitized to up-regulation of the AIM2 inflammasome components (AIM2 and ASC) by the CpG-DNA. These data suggest that the initial liver injury and release of DAMPs including apoptotic DNA and/or mitochondrial DNA leads to amplification of the initial insult.
High mobility group box (HMGB) proteins are ‘universal sentinels’ for nucleic acids. Their binding to nucleic acids provides more efficient recognition of nucleic acids by TLRs and NLRs (41). Our data show increased expression of HMGB1 in MCD-steatohepatitis and increased phosphorylation of the HMGB1 protein. HMGB1 is a nuclear protein and its phosphorylation is necessary for nucleo-cytoplasmic shuttling (42) prior to secretion. HMGB1 has been shown to interact and pre-associate with TLR9 in the endoplasmic reticulum-Golgi intermediate compartment (ERGIC) that also needs the shuttle from the nucleus to the cytoplasm. Once preassociated with TLR9, HMGB1 hastens the TLR9 redistribution to early endosomes in response to CpG-ODN, accelerates the delivery of CpG-ODNs to its receptor and leads to TLR9-dependent augmentation of IL-6, IL-12 and TNFα production (43). Based on our data, we speculate that HMGB1 may be, at least partly, responsible for the increased sensitivity to TLR9 ligand in steatohepatitis. In addition to TLR activation, HMGB1 was shown to activate caspase-1 presumably in association with NLRP3 (15). Thus, increased HMGB1 in dietary steatohepatitis liver may contribute to caspase-1 activation in a MyD88-dependent manner.
Our study should be interpreted with the understanding that different animal models of steatohepatitis might be different in certain steps of the pathogenesis, since there is not a single ‘ideal’ animal model for the human NASH. For example, while the MCD diet rapidly induces steatohepatitis, it is not associated with obesity and peripheral insulin resistance (44). However, choline and methionine deficiency might be relevant in certain population such as pregnant and lactating women, patients with parenteral nutrition or malnutrition (45). Notably, choline and methionine deficiency affects phosphatidylcholine that is major component of cell membranes and critical for the packaging of very low density lipoprotein. Therefore in this specific model, we cannot exclude the role of altered lipid membrane components in the activation of inflammasome.
In summary, our data indicate that up-regulation and activation of the AIM2 and NLRP3 inflammasomes via TLR9-MyD88 pathways involve both BM-derived and non-BM-derived cells in dietary steatohepatitis. In addition, we propose that HMGB1 could be one of the factors responsible for sensitization to nucleic acids and nucleic acid-induced inflammasome up-regulation.
Acknowledgments
This work was supported by NIH grant: AA017729.
Abbreviations
- AIM2
absent in melanoma 2
- ASC
apoptosis associated speck-like protein containing a CARD
- BM
bone marrow
- CD
choline deficient
- CpG-ODN
cytidine phosphateguanosine oligonucleotide
- DAMP
damage associated molecular patterns
- HMGB1
high mobility group box 1
- IL-1β
interleukin-1 beta
- LPS
lipopolysaccharide
- MCD
methionine-choline deficient
- MCS
methionine-choline supplemented
- MyD88
myeloid differentiation factor primary response gene 88
- NAFLD
non-alcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- NLRP3
NLR family pyrin domain containing 3
- PAMP
pathogen associated molecular patterns
- PYHIN
pyrin and HIN domain containing protein
- ROS
reactive oxygen species
- SREBP
sterol regulatory element binding protein
- TLR
toll-like receptor
Footnotes
Conflict of interest: The authors do not have any disclosures to report.
References
- 1.Tiniakos DG, Vos MB, Brunt EM. Nonalcoholic fatty liver disease: pathology and pathogenesis. Annu Rev Pathol. 2010;5:145–71. doi: 10.1146/annurev-pathol-121808-102132. [DOI] [PubMed] [Google Scholar]
- 2.Rubartelli A, Lotze MT. Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 2010;28:429–36. doi: 10.1016/j.it.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 3.Miura K, Kodama Y, Inokuchi S, et al. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1 beta in mice. Gastroenterology. 2010;139:e7. doi: 10.1053/j.gastro.2010.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seki E, Brenner A. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology. 2008;48:322–35. doi: 10.1002/hep.22306. [DOI] [PubMed] [Google Scholar]
- 5.Muruve DA, Pétrilli V, Zaiss AK, et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers innate immune response. Nature. 2008;452:103–8. doi: 10.1038/nature06664. [DOI] [PubMed] [Google Scholar]
- 6.Bauernfeind F, Ablasser A, Bartok E, et al. Inflammasomes: current understanding and open questions. Cell Mol Life Sci. 2011;68:765–83. doi: 10.1007/s00018-010-0567-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13:397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bauernfeind FG, Horvath G, Stutz A, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183:787–91. doi: 10.4049/jimmunol.0901363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kitazumi I, Tsukahara M. Regulation of DNA fragmentation: the role of caspases and phosphorylation. FEBS J. 2011;278:427–41. doi: 10.1111/j.1742-4658.2010.07975.x. [DOI] [PubMed] [Google Scholar]
- 10.Wieckowska A, Zein NN, Yerian LM, et al. In vivo assessment of liver cell apoptosis as a novel biomarker of disease severity in nonalcoholic fatty liver disease. Hepatology. 2006;44:27–33. doi: 10.1002/hep.21223. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Q, Raoof M, Chen Y, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–7. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caro P, Gómez J, Arduini A, et al. Mitochondrial DNA sequences are present inside nuclear DNA in rat tissues and increase with age. Mitochondrion. 2010;10:479–86. doi: 10.1016/j.mito.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 13.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–50. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 14.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–5. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 15.Yan W, Chang Y, Liang X, et al. High mobility group box 1 activates caspase-1 and promotes hepatocellular carcinoma invasiveness and metastases. Hepatology. 2012;55:1863–75. doi: 10.1002/hep.25572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szabo G, Bala S, Petrasek J, Gattu A. Gut-liver axis and sensing microbes. Dig Dis. 2010;28:737–44. doi: 10.1159/000324281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Csak T, Velayudham A, Hritz I, et al. Deficiency in myeloid differentiation factor-2 and toll-like receptor 4 expression attenuates nonalcoholic steatohepatitis and fibrosis in mice. Am J Physiol Gastrointest Liver Physiol. 2011;300:G433–41. doi: 10.1152/ajpgi.00163.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li L, Chen L, Hu L, et al. Nuclear factor high-mobility group box1 mediating the activation of Toll-like receptor 4 signaling in hepatocytes in the early stage of nonalcoholic fatty liver disease in mice. Hepatology. 2011;54:1620–30. doi: 10.1002/hep.24552. [DOI] [PubMed] [Google Scholar]
- 19.Hosoi T, Yokoyama S, Matsuo S, Akira S, Ozawa K. Myeloid differentiation factor 88 (MyD88)-deficiency increases risk of diabetes in mice. PLoS ONE. 2010;5:e12537. doi: 10.1371/journal.pone.0012537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Csak T, Ganz M, Pespisa J, et al. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology. 2011;54:133–44. doi: 10.1002/hep.24341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henao-Mejia J, Elinav E, Jin C, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–85. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Witek RP, Stone WC, Karaca FG, et al. Pan-Caspase inhibitor VX-166 reduces firbosis in an animal model of nonalcoholic steatoehaptitis. Hepatology. 2009;50:1421–30. doi: 10.1002/hep.23167. [DOI] [PubMed] [Google Scholar]
- 23.Anstee QM, Concas D, Kudo H, et al. Impact of pan-caspase inhibition in animal models of established steatosis and non-alcoholic steatohepatitis. J Hepatol. 2010;53:542–50. doi: 10.1016/j.jhep.2010.03.016. [DOI] [PubMed] [Google Scholar]
- 24.Dixon LJ, Berk M, Thapaliya S, Papouchado BG, Feldstein AE. Caspase-1-mediated regulation of fibrogenesis in diet-induced steatohepatitis. Lab Invest. 2012;92:713–23. doi: 10.1038/labinvest.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stienstra R, van Diepen JA, Tack CJ, et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc Natl Acad Sci U S A. 2011;108:15324–9. doi: 10.1073/pnas.1100255108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petrasek J, Bala S, Csak T, et al. Il-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steato-hepatitis in mice. J Clin Invest. 2012;122:3476–89. doi: 10.1172/JCI60777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petrasek J, Dolganiuc A, Csak T, et al. Interferon regulatory factor 3 and type I interferons are protective in alcoholic liver injury in mice by way of crosstalk of parenchymal and myeloid cells. Hepatology. 2011;53:649–60. doi: 10.1002/hep.24059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kleiner DE, Brunt EM, Natta MV, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–21. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 29.Tang D, Kang R, Zeh HJ, III, Lotze MT. High mobility group box 1, oxidative stress and disease. Antioxid Redox Signal. 2011;14:1315–35. doi: 10.1089/ars.2010.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petrasek J, Dolganiuc A, Csak T, Kurt-Jones EA, Szabo G. Type I interferons protect from toll-like receptor 9- associated liver injury and regulate IL-1 receptor antagonist in mice. Gastroenterology. 2011;140:697–708. doi: 10.1053/j.gastro.2010.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Romics L, Jr, Dolganiuc A, Velyudham A, et al. Toll-like receptor 2 mediates inflammatory cytokine induction but not sensitization for liver injury by Propionibacterium acnes. J Leukoc Biol. 2005;78:1255–64. doi: 10.1189/jlb.0804448. [DOI] [PubMed] [Google Scholar]
- 32.Hritz I, Velayudham A, Dolganiuc A, et al. Bone marrow-derived immune cells mediate sensitization to liver injury in a Myeloid Differentiation Factor 88-dependent fashion. Hepatology. 2008;48:1342–7. doi: 10.1002/hep.22557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52:1836–46. doi: 10.1002/hep.24001. [DOI] [PubMed] [Google Scholar]
- 34.Szabo G, Velayudham A, Romics L, Jr, Mandrekar P. Modulation of non-alcoholic steatohepatitis by pattern recognition receptors in mice: the role of toll-like receptors 2 and 4. Alcohol Clin Exp Res. 2005;29:140S–5S. doi: 10.1097/01.alc.0000189287.83544.33. [DOI] [PubMed] [Google Scholar]
- 35.Burdette D, Haskett A, Presser L, et al. Hepatitis C virus activates interleukin-1{beta} via caspase-1-inflammasome complex. J Gen Virol. 2012;93:235–46. doi: 10.1099/vir.0.034033-0. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36.Szabo G, Csak T. Inflammasome in liver diseases. J Hepatol. 2012;57:642–54. doi: 10.1016/j.jhep.2012.03.035. [DOI] [PubMed] [Google Scholar]
- 37.Zaki MH, Boyd KL, Vogel P, et al. The NLRP3 inflammasome protects against loss of epithelial intergrity and mortality during experimental colitis. Immunity. 2010;32:379–91. doi: 10.1016/j.immuni.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gurcel L, Abrami L, Girardin S, Tschopp J, van der Goot FG. Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell. 2006;126:1135–45. doi: 10.1016/j.cell.2006.07.033. [DOI] [PubMed] [Google Scholar]
- 39.Menzel CL, Sun Q, Loughran PA, et al. Caspase-1 is hepatoprotective during trauma and hemorrhagic shock by reducing liver injury and inflammation. Mol Med. 2011;17:1031–8. doi: 10.2119/molmed.2011.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Imaeda AB, Watanabe A, Sohail MA, et al. Acetaminophen- induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest. 2009;119:305–14. doi: 10.1172/JCI35958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yanai H, Ban T, Wang Z, et al. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature. 2009;462:99–103. doi: 10.1038/nature08512. [DOI] [PubMed] [Google Scholar]
- 42.Zhang X, Wheeler D, Tang Y, et al. Calcium/calmodulin-dependent protein kinase (CaMK) IV mediates nucleocytoplasmic shuttling and release of HMGB1 during lipopolysaccharide stimulation of macrophages. J Immunol. 2008;181:5015–23. doi: 10.4049/jimmunol.181.7.5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ivanov S, Dragoi AM, Wang X, et al. A novel role for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood. 2007;110:1970–81. doi: 10.1182/blood-2006-09-044776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Larter CZ, Yeh MM. Animal models of NASH: getting both pathology and metabolic context right. J Gastroenterol Hepatol. 2008;23:1635–48. doi: 10.1111/j.1440-1746.2008.05543.x. [DOI] [PubMed] [Google Scholar]
- 45.Al-Humaidi H, Zarros A, Kyriakaki A, Al-Saigh R, Liapi C. Choline deprivation: an overview of the major hepatic metabolic response pathways. Scand J Gastroenterol. 2012;47:874–86. doi: 10.3109/00365521.2012.685755. [DOI] [PubMed] [Google Scholar]