

Abstract
Purification of suitable quantity of homogenous protein is very often the bottleneck in protein structural studies. Overexpression of a desired gene and attachment of enzymatically cleavable affinity tags to the protein of interest made a breakthrough in this field. Here we describe the structure of Galleria mellonella silk proteinase inhibitor 2 (GmSPI-2) determined both by X-ray diffraction and NMR spectroscopy methods. GmSPI-2 was purified using a new method consisting in non-enzymatic His-tag removal based on a highly specific peptide bond cleavage reaction assisted by Ni(II) ions. The X-ray crystal structure of GmSPI-2 was refined against diffraction data extending to 0.98 Å resolution measured at 100 K using synchrotron radiation. Anisotropic refinement with the removal of stereochemical restraints for the well-ordered parts of the structure converged with R factor of 10.57% and R free of 12.91%. The 3D structure of GmSPI-2 protein in solution was solved on the basis of 503 distance constraints, 10 hydrogen bonds and 26 torsion angle restraints. It exhibits good geometry and side-chain packing parameters. The models of the protein structure obtained by X-ray diffraction and NMR spectroscopy are very similar to each other and reveal the same β2αβ fold characteristic for Kazal-family serine proteinase inhibitors.
Introduction
Despite significant methodological progress [1], structural studies of proteins still require significant amount of pure samples. To achieve this goal, the affinity tag methodology is commonly used. However, the presence of an affinity tag may affect the biological activity of a target protein and interfere with crystallization [2]. Therefore it is recommended to remove affinity tags from the purified protein. This has often been, however, the Achilles heel of this approach. The proteinase-mediated enzymatic cleavage commonly used for affinity tag removal poses serious risks, such as non-specific degradation of the target protein [3]–[5]. Moreover, costly preparative-scale purification of the cleavage products is necessary, including the proteinase inactivation and removal step. Chemical cleavage agents are suggested as inexpensive alternative to proteolytic enzymes [6]–[8]. However, none of them is commonly used due to their low specificity and harsh reaction conditions [9]–[11].
Our previous studies demonstrated that Ni(II) ions hydrolyze the peptide bond preceding the serine or threonine residue in (S/T)XHZ peptide sequences [12]. The specificity of the cleavage was confirmed for a range of peptides and the reaction mechanism was precisely elucidated [13]. Recently we have positively verified the biotechnological applicability of the Ni(II)-depended peptide bond cleavage reaction for the recombinant GmSPI-2 protein, which is the subject of our structural analysis in this work. The protein purification procedures in that study were performed on an analytical scale [14]. However those results indicated that the methodology could be easily scaled up for preparative purification of recombinant proteins for structural studies.
The GmSPI-2 protein is a structurally unique Kazal-family serine proteinase inhibitor identified in the silk of wax moth Galleria mellonella [15]. It is the shortest Kazal-family serine proteinase inhibitor in animals. Unlike most Kazal-family serine proteinase inhibitors, where each functional domain consists of 50–60 amino acid residues with six conserved cysteines, GmSPI-2 is a single domain inhibitor of 36 residues with only four cysteines (Fig. 1). Computer modeling suggested that, in contrast to typical Kazal-family serine proteinase inhibitors, the conformation of GmSPI-2 includes not three but only two loops which are stabilized and closed into rings by disulfide bridges between the four conserved cysteines [15]. The inhibitor exhibits high activity against subtilisin and proteinase K (proteases from Bacillus subtilis and the Tritirachium album, respectively) [15]. Recombinant GmSPI-2 activity is identical with the native protein [16]. Since GmSPI-2 is a much potent proteinase inhibitor than some commercially available inhibitors (e.g. AEBSF, 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride; [17]), it could be used as a replacement or supplement of available inhibitors or inhibitor cocktails. Additionally, when fused to a target protein, GmSPI-2 could protect the target protein against proteinase degradation [17], [18]. Thus, GmSPI-2 can be considered as a valuable and economically important protective tool in biotechnology for enhancing the yields and prolonging the life of desired protein products.
Figure 1. Sequence alignment of classical (OMTKY3), non-classical group 1 (CrSPI-1-D1), non-classical group 2 (LDTI) and GmSPI-2 Kazal-family serine proteinase inhibitors.
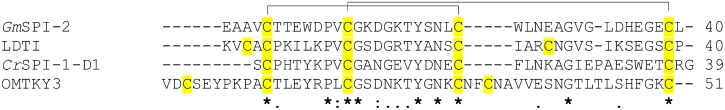
The alignment was calculated with ClustalW2 [46]. Cysteine residues are highlighted in yellow. Amino acids identical in all four proteins are marked with an asterisk (*), conservative substitutions with a colon (:), and semi-conservative substitutions with a period (.).
Here we discuss the application of the previously described nickel-based purification methodology, scaled up for this structural work, and demonstrate the usefulness of this innovative approach for structural studies. The determinations of the atomic resolution X-ray and high quality NMR structure of the GmSPI-2 protein, both critically dependent on large quantities of highly pure protein samples, were possible partially because of this protein purification method.
Materials and Methods
GmSPI-2-SRHWAP-H6 fusion protein expression and purification
The cDNA sequence encoding SPI-2 protein with modified C-terminal end was used as a template (Leu codon was added as described [16]). The primers were extended to introduce a PstI restriction site at the 5′ end of the amplified product and an XbaI restriction site at the 3′ end, followed nucleotides encoding SRHWAP and six histidyl residues. The alternative SPI2-SRHWAP-H6 fusion protein was designed in order to improve the yield of purification and the purity of the final product. The appropriate gene construct was successfully cloned under the control of AOX promoter in a pPICZαB vector (Invitrogen), using standard methods. As a result of the cloning procedure and the pre-protein processing in Pichia pastoris, GmSPI-2 was extended by the GluAlaAla- tripeptide at the N-terminus and by the -Leu40 residue at the C-terminus. The fusion protein secreted to the media was initially purified by affinity chromatography on Ni-NTA-agarose (Qiagen) in the presence of 20 mM phosphate buffer pH 7.4, containing 0.5 M NaCl. The fusion protein was then eluted from the column with 250 mM imidazole and dialyzed overnight against water in order to remove the excess of salts. Typically 2 ml of elution fraction was dialyzed against 2 L of water. Next, the protein was purified by HPLC on a Vydac C18 semipreparative column. The eluting solvent A was 0.1% TFA/water and solvent B was 0.1% TFA/90% acetonitrile/water. A linear gradient of acetonitrile from 10% to 40% in 30 min was applied at a flow rate of 2 mL/min, with detection at 220 nm and 280 nm. After elution, the fusion protein was frozen and lyophilized. The HPLC purification step was applied to assess the amount of GmSPI-2.
Affinity tag cleavage
The GmSPI-2 fusion protein after lyophilization was weighed (portions of 5–7 mg) and dissolved in 20 mM phosphate buffer pH 7.4, containing 0.5 M NaCl and incubated with Ni-NTA-agarose (Qiagen) for 2 h at 4°C. Then the GmSPI-2 fusion protein immobilized on Ni-NTA agarose was incubated in 100 mM Hepes buffer pH 8.2, containing 150 mM NaCl and 7.5 Mm NiCl2 at 50°C for 19 h. The GmSPI-2 protein obtained in the flow-through fraction was further purified using the Breeze HPLC system (Waters) on a Vydac C18 semipreparative column. The eluting solvent A was 0.1% TFA/water and solvent B was 0.1% TFA/90% acetonitrile/water. A linear gradient of solvent B from 10% to 40% in 30 min was used at a flow rate of 2 mL/min. The molecular mass of the collected HPLC single peak was measured on a Q-Tof1 ESI MS spectrometer (Micromass).
Crystallization
Screening for crystallization conditions was performed manually using Crystal Screen and Crystal Screen 2 [19] and the hanging-drop vapor-diffusion technique at 292 K, by mixing 1 µl protein (6.5 mg ml−1 in water) and 1 µl reservoir solution. Needle-like crystals grew to dimensions of 0.6×0.05×0.05 mm within one week over a reservoir solution consisting of 1.4 M sodium citrate and 0.1 M Hepes pH 7.5 (Crystal Screen condition no. 38). For cryoprotection, the crystal was transferred to a solution consisting of the reservoir solution supplemented with 20% (v/v) glycerol.
X-Ray data collection
Diffraction data were measured at 100 K on a Rayonics MX-225 CCD detector at beamline BL 14.1 of the Berliner Elektronenspeicherring-Gesellschaft für Synchrotronstrahlung m.b.H. (BESSY, Berlin). Integration, scaling and merging of the intensity data was accomplished using the XDS package [20]. The best crystal diffracted to 0.95 Å but due to a glitch of the data collection program, only 52° of the high-resolution pass were collected. Therefore, the low-resolution and truncated high-resolution passes were scaled together with the data collected for another crystal. This gave the complete data set at 0.98 Å, characterized in Table 1. An overall B-factor of 8.2 A2 was estimated from the Wilson plot using the XSCALE program from XDS package [20].
Table 1. X-ray data collection and model refinement statistics.
| Data Collection | |
| Crystal size (mm) | 0.7×0.05×0.05 |
| Space group | P212121 |
| Unit cell parameters (Å) | a = 27.27, b = 31.24, c = 35.74 |
| X-ray source | BESSY 14.1 |
| Temperature (K) | 100 |
| No. of images | 360a/180/52 |
| Oscillation angle (°) | 1.0a/2.0/1.0 |
| Wavelength (Å) | 0.91841a/0.80000/0.80000 |
| Resolution (Å) | 20.00–0.98 (1.01–0.98)b |
| R int c(%) | 7.2 (46.0) |
| <I/σI> | 22.52 (2.04) |
| Reflections | |
| measured | 222092 |
| unique | 17179 |
| in test set | 1031 |
| Mosaicity (°) | 0.13 |
| Completeness (%) | 94.8 (84.5) |
| Redundancy | 12.9 (2.3) |
| Wilson B-factor (Å2) | 8.2 |
| Refinement | |
| Resolution (Å) | 20.00–0.98 (1.02–0.98) |
| No. reflections | 17179 (1790) |
| R work/R free d | 10.57/12.91 |
| No. of atoms | |
| protein | 328 |
| solvent | 96 |
| B-factors (Å2) | |
| protein | 6.5 |
| solvent | 12.1 |
| Rmsd from ideal | |
| bond lengths (Å) | 0.023 |
| angle distances (Å) | 0.058 |
| Ramachandran statistics (%) | |
| favored | 90.6 |
| additional | 9.4 |
| PDB code | 4hgu |
Values for crystal no. 1.
Values in parentheses are for the highest resolution shell.
R int = ΣhklΣi|Ii(hkl)−<I(hkl)>|/ΣhklΣi I i(hkl), where I i(hkl) is the ith measurement of the intensity of reflection hkl and <I(hkl)> is the mean intensity of reflection hkl.
R = Σ||F o|−|F c||/Σ|F o|, where F o and F c are the observed and calculated structure factor amplitudes, respectively.
Data measured to high resolution with a synchrotron X-ray beam can be contaminated with effects of crystal radiation damage. However, the plot of decay R factor [21] against frame-number difference was around zero and scaling factors for the individual diffraction images fluctuated around 1 without any decreasing trend, indicating no or very little effect of radiation damage. The data were also checked for diffraction anisotropy [22]. A very low spread in values of the three principal components (0.48 Å2) indicated almost no anisotropy.
X-Ray structure solution and refinement
The structure was solved by molecular replacement using the MOLREP program from the CCP4 suite [23], [24] and the structure of leech-derived tryptase inhibitor (LDTI; PDB code 1an1; [25]) as the search model. The amino-acid sequence of the model shares 40% identity and 60% similarity (LALIGN; [26]) with GmSPI-2. The initial maximum-likelihood structure-factor refinement was carried out in REFMAC [27] using all data, with the exception of 1031 reflections (6%) flagged for cross-validation purposes. No σ cutoff was applied. The manual rebuilding of the model was performed in COOT [28]. The conjugate-gradient least-squares refinement in SHELXL [29] was used to refine the model at the later stages. The main steps of the refinement included (1) isotropic refinement with manually added water molecules and sodium ions, (2) anisotropic refinement, (3) addition of H atoms according to geometrical criteria implemented in SHELXL, (4) refinement of the occupancies of partially occupied/alternate conformations and solvent atoms, and finally (5) removal of the restraints for the well-ordered parts of the model. Six side-chains, namely Glu1, 8 and 38, Val4, Asp10 and Leu23, as well as two Cα atoms, of Val4 and Glu38, were modeled with alternate conformations. Additionally, the Cγ, Cδ, Oε1 and Oε2 atoms of Glu36, and the Cε and Nζ atoms of Lys15 and Lys18 were given partial occupancies. The stereochemical restraints were retained throughout refinement only for these side chains/atoms.
Sodium ions were found in the electron density map and identified on the basis of coordination number (6) and Na+ ⋅ ⋅ ⋅ O distances (2.33–2.58 Å), in agreement with the high concentration of sodium cations in the crystallization solution (1.4 M sodium citrate). In the final round, all data were used in the refinement, including the Rfree reflections, leading to the convergence with R values of 8.62% for the 14133 reflections with F o>4σ(F o) and 10.57% for all 17179 reflections (Table 1).
At the end of the refinement, one cycle of full-matrix minimization was calculated with all stereochemical restraints removed and with all parameter shifts damped to zero, which permitted the estimation of the standard uncertainties (s.u.) in all positional parameters. Full refinement statistics are given in Table 1.
NMR resonance assignment and structure determination
All NMR experiments were performed using an 18.8 T Varian DirectDrive 800 NMR spectrometer (operating 1H frequency 799.811 MHz). The NMR sample was obtained by diluting a GmSPI-2 protein sample in 90%/10% H2O/D2O, 20 mM phosphate buffer pH 4.5, with 50 mM NaCl, to a final concentration of 0.5 mM. Assignments of 1H, 13C, and 15N resonances were achieved utilizing standard methods on the basis of 2D TOCSY and NOESY data [30]. The homonuclear experiments were supplemented with 2D heteronuclear 1H-15N and 1H-13C HSQC spectra acquired at natural abundance of 15N and 13C nuclei. All NMR spectra were referenced using external DSS (sodium 2,2′-dimethyl-2-silapentane-5-sulfonate) [31] and processed by NMRPipe software [32]. The three-dimensional structure of the GmSPI-2 protein in solution was solved by standard 2D NMR techniques on the basis of 503 (141 intra-residue, 138 sequential, 84 medium, and 140 long range) distance constraints provided by the analysis of 2D homonuclear 1H-1H NOESY spectra acquired with 120 ms mixing time. 26 Restraints for the backbone φ and ψ torsion angles were defined using the analysis of chemical shifts with the program TALOS+ [33]. This procedure provided 58 restraints for the φ and ψ torsion angles for 29 residues, which were predicted as ‘good’ by TALOS+. Additionally, 20 distance constraints for 10 hydrogen bonds were defined as rHN-O = 1.5–2.8 and rN-O = 2.4–3.5 Å (Table 2). 200 structures were calculated by the CYANA (version 3.0) software [34]. Finally, 17 conformers, selected on the basis of low target function criteria, were subjected to a refinement procedure in a water shell using the YASARA program suite [35]. The statistics of NOE distance restraints together with the analysis of the ensemble of 17 lower energy structures evaluated on the basis of NMR data are presented in Table 2.
Table 2. NMR restraints and structural statistics for the ensemble of 17 lower energy of GmSPI-2 conformers.
| NOESY distance restraintsa | 506 |
| intraresidual (|i-j| = 0) | 135 |
| sequential (|i-j| = 1) | 135 |
| medium-range (1<|i-j| ≤5) | 77 |
| long-range (|i-j|>5) | 135 |
| Hydrogen bonds | 24 |
| Constraints per residue | 13.3 |
| Torsion angle restraints | |
| backbone (φ/ψ) | 26 |
| Structure Z-scores | |
| 1-st generation packing quality | −0.125±1.034 |
| 2-nd generation packing quality | 4.836±1.836 |
| Ramachandran plot appearance | −0.923±0.405 |
| χ1/χ2 rotamer normality | −2.538±0.874 |
| Backbone conformation | 0.189±0.470 |
| RMS Z-scoresb | |
| bond lengths | 1.174±0.008 |
| bond angles | 0.513±0.021 |
| omega angle restraints | 0.929±0.115 |
| side chain planarity | 0.673±0.117 |
| improper dihedral distribution | 0.878±0.099 |
| Ramachandran statisticsc (%) | |
| favored | 86.9 |
| additional | 13.1 |
| Rmsd from the mean structure | |
| backbone atoms (Å) | 0.63±0.27 |
| heavy atoms (Å) | 0.96±0.20 |
| PDB code | 2m5x |
| number of models | 17 |
Results and Discussion
Purification of GmSPI-2 protein for structural studies
The fusion construct designed for the affinity purification/tag removal approach according to our nickel-based method, contains the Ni(II)-specific SRHW sequence cloned between the GmSPI-2 target protein and its C-terminal His-tag. Two additional amino acids, Ala and Pro, were added after the crucial tetrapeptide sequence as a spacer in order to avoid unwanted interactions between this sequence and the tag. The purification procedure of the GmSPI-2-SRHWAP-H6 fusion protein produced in Pichia pastoris culture, included initial affinity chromatography on Ni-NTA agarose, followed by HPLC. The latter purification step was applied in order to assess the amount of the fusion protein for affinity tag removal, thus enabling a quantitative evaluation of this novel procedure at the preparative scale of 5–7 mg of protein. The purified GmSPI-2-SRHWAP-H6 protein was then reloaded on the Ni-NTA agarose column, and incubated with excess of Ni(II) ions. The cleavage conditions were based on our recently published analytical-scale paper [14]. The flow-through fraction collected contained only the pure GmSPI-2 protein, with the SRHWAP-H6 tag removed, as evidenced by the presence of a single peak on the HPLC chromatogram of this fraction (Fig. 2). A chromatogram of the wash buffer fraction indicated that a small amount of GmSPI-2 got stuck to the agarose column. The molecular masses of the collected peaks confirmed the absence of unspecific cleavage. The total yield of pure GmSPI-2 was 70% in repeated experiments. The efficiency of the cleavage reaction was calculated precisely using the values of HPLC peak areas corresponding to the substrate and products before and after protein incubation with Ni(II) ions.
Figure 2. Preparative HPLC chromatograms of protein samples after 19 h of incubation at 50°C.

Incubation buffer (top) and wash buffer (bottom). The peak labels denote the reaction substrate and products, identified using ESI-MS: P, pure GmSPI-2 protein (4310 Da); T, the SRHWAP-H6 peptide (the extended His-tag, which is the C-terminal hydrolysis product, 1574 Da); S, substrate (fusion protein, 5868 Da).
The quality of the crystal structure
The final crystallographic GmSPI-2 model (PDB code 4hgu) consists of 328 protein-atom sites for 300 non-hydrogen atoms, 92 water molecules and 4 sodium ions. The electron density for all atoms of the main-chain and fully occupied atoms of side-chains is exceptionally good. The number of reflections per parameter in the final cycle of refinement was 4.5, sufficient to justify refinement without any stereochemical restraints for the well-ordered parts of the molecule. Despite this radical approach, 90.6% of the residues are located in the most favored regions and 9.4% in the additionally allowed regions of the Ramachandran plot [36]. This, together with very good refinement statistics, confirmed that this refinement approach was correct.
The average value of the atomic displacement parameter (B eq) for GmSPI-2 is 6.5 Å2. The N/C termini in protein structures are very often disordered. The pattern of B eq values along the polypeptide chain shows that the whole main chain is well ordered (Fig. 3). Especially the residues from 11 to 14 (forming β-strand 1) and from 19 to 33 (forming β-strand 2 and α-helix) have slightly lower than average B eq value (with B eq values of 3.53 and 3.75 Å2 for residues 11–14 and 19–33, respectively). The mean B eq values of the main-chain, side-chain and solvent atoms of GmSPI-2 are 4.8, 8.2 and 12.1 Å2, respectively. There are only two structures of Kazal-family serine proteinase inhibitors determined at a comparable resolution which are available in the PDB, namely 1r0r at 1.10 Å and 2 gkr at 1.17 Å. The corresponding values of B eq for those structures are much higher, 16.7, 19.3 and 38.2 Å2 for 1r0r and 14.4, 17.0 and 32.6 Å2 for 2 gkr, respectively.
Figure 3. Plot of B eq averaged over main-chain (green) and side-chain (orange) atoms of the crystallographic model.

Values of zero in the lower plot correspond to glycines.
The estimated values of s.u. in all positional parameters for the fully occupied main-chain atoms range from 0.012 Å to 0.033 Å (Fig. 4). It is evident from Fig. 4 that coordinate errors have smaller values for ‘heavier’ atoms, e.g. oxygen, and slightly higher for nitrogen and carbon atoms. The s.u. values for the major conformation of the two main-chain atoms refined in two conformations are much higher, 0.048 Å for the Cα atom of Ala4 and 0.054 Å for the Glu38. A similar pattern was observed for the coordinate errors estimated for the structure of lysozyme refined at 0.65 Å resolution [37] or BPTI refined at 0.86 Å [38]. The quality of the crystallographic model can also be assessed using the statistics of the derived geometrical parameters. For instance, the peptide C = O bond lengths (range from 1.19 to 1.28 Å, with a mean of 1.23 Å) are characterized by standard uncertainties between 0.01 and 0.03 Å, with a mean of 0.02 Å. These statistics are almost identical to those reported for squash trypsin inhibitor (CMTI-I) studied at a comparable resolution of 1.03 Å [39]. However, the model of CMTI-I was refined with the BUMP and geometrical restraints retained on main-chain segments with excessive displacement parameters (with B eq>15 Å).
Figure 4. Coordinate errors for main-chain atoms estimated from the inversion of the least-squares matrix.

For dual-occupancy atoms, only the major component is plotted.
Crystal packing and intermolecular contacts
The GmSPI-2 molecules are densely packed with only 30.2% volume being occupied by solvent (the corresponding Matthews coefficient is 1.76 Å3/Da). All other Kazal-family serine proteinase inhibitors are more loosely packed, with the solvent content ranging from 31.9% (for N-terminally truncated turkey ovomucoid third domain, OMTKY3; PDB code 2 gkr) to 54.0% (for infestin 4; 2erw). The GmSPI-2 molecules are more solvent exposed along the crystallographic b axis (Fig. 5). There are 7 intermolecular hydrogen bonds, listed in Table 3. Two of them, linking molecules related by the 21 screw along [001] involve atoms with partial occupancy. The hydrogen bond involving the Cys24 N atom should be regarded as week due to an unfavorable angle and the presence of another hydrogen acceptor from the preceding Asn22 Oδ1 atom. Besides direct hydrogen bonds, water molecules play a profound role in mediating intermolecular contacts. An example of this is the N-terminus where the Glu1 N atom is anchored to two symmetry related GmSPI-2 molecules by hydrogen bonds through three well-ordered water molecules 203, 204 and 226 with B-factors (Å2)/donor-acceptor distances (Å) of 7.30/2.85, 7.99/2.82 and 6.27/2.70, respectively. Moreover, the first three N-terminal residues, which in fact are artificial to the native GmSPI-2 sequence, form only indirect intermolecular interactions through water molecules and a sodium ion. The first direct intermolecular hydrogen bond is made by the Val4 O atom (Table 3).
Figure 5. Crystal packing of GmSPI-2 molecules viewed down the crystallographic b axis.

Table 3. Direct protein-protein intermolecular hydrogen bonds in the GmSPI2 crystal structure.
| No | Residue, atom | Residue, atom | Distance(Å) |
| 1 | Val4, O | Gly32(a), N | 2.83 |
| 2 | Thr6, N | Gly30(a), O | 2.90 |
| 3 | Cys24, N | Ala29(a), O | 3.06 |
| 4 | Ser21, N | Leu40(b), OXT | 2.77 |
| 5 | Ser21, Oγ | Leu40(b), O | 2.58 |
| 6 | Glu8, Oε2 | Lys15(c), Nζ | 2.42 |
| 7 | Asn27, Nδ2 | Glu38(c), Oε1 | 3.07 |
x +½, −y + ½, −z.
−x, y + ½, −z+½.
−x+½, −y, z+½.
The crystal structure includes four partially occupied sodium ions. Two of them, namely Na101 and Na102 have complete octahedral coordination spheres. The coordination number of Na103 and Na104 is 5 and 4, respectively. These sodium ions are coordinated by oxygen atoms belonging to the GmSPI-2 molecule (Thr7 Oγ1, Asp16 O and Asp34 O and Oδ1), as well as by water molecules. It has not escaped our notice that two sodium ions, namely Na103 and symmetry related Na104 (−x, ½+y, ½−z), are 3.14 Å apart sharing two symmetry related water molecules 257 and 258 (−x, ½+y, ½−z) in their coordination spheres. Similar arrangement is often found in small molecule structures. In the structure of catena-(hexakis(µ2-Aqua)-di-sodium 2,5-dibenzoylterephthalate tetrahydrate) (CSD reference code HAYQUU) [40] Na1 and Na2 ions are 3.14 Å apart and share three water molecules (O4, O5 and O6) in their coordination spheres.
Description of the GmSPI2 structure
The overall structure of GmSPI-2 (Fig. 6A) resembles that of other Kazal-family serine proteinase inhibitors. Residues Val12-Gly14, Thr19-Tyr20 and Leu33-Glu36 form an anti-parallel β-sheet while residues Leu23-Ala29 form the central α-helix of the characteristic Kazal-family serine proteinase inhibitor β2αβ fold. GmSPI-2 shows, however, features of non-classical Kazal-family serine proteinase inhibitors, harboring an unusual pattern of disulfide bridges. Only two intradomain disulfide bridges formed between Cys residues 5 and 24 and Cys residues 13 and 39 are present. GmSPI-2 has been classified as a non-classical Kazal-family serine proteinase inhibitor group 1 [41]. This group of inhibitors is characterized by the shift of the first and the fifth half-cystine residues towards the C-terminus with respect to classical Kazal-family serine proteinase inhibitors [42]. A superposition of the X-ray structure of GmSPI-2 with a classical Kazal-family serine proteinase inhibitor (OMTKY3, 2 gkr), a group 1 non-classical Kazal-family serine proteinase inhibitor (CrSPI-1-D1, 3pis) and a group 2 non-classical Kazal-family serine proteinase inhibitor (LDTI, 1an1) shows that it resembles the structure of LDTI (Fig. 7) with a root-mean-square deviation (rmsd) of 0.92 Å for 37 superimposed Cα atoms [43]. CrSPI-1-D1 like GmSPI-2 has only two disulfide bridges. Both structures are similar up to Asp34 of GmSPI-2, but very different from there, till the very C-terminus. Residues Trp34-Cys37 of CrSPI-1-D1 form a 310-helix, which is not present in the structure of GmSPI-2.
Figure 6. The overall X-ray (A) and NMR (B) models of GmSPI-2.

(A) β-stands and disulfides are shown in yellow, α-helix in red and loops in green. (B) The ensemble of 17 lowest energy conformers are shown in different colors, with disulfides in yellow. The figure was prepared using PyMOL [47].
Figure 7. Stereo Cα tracing of the crystallographic model of GmSPI-2 (yellow) superimposed with the NMR structure of GmSPI-2 in solution (conformer 11, magenta), CrSPI-1-D1 (red, PDB code 3pis), OMTKY3 (blue, 2 gkr) and LDTI (green, 1an1).

The superposition was calculated in Coot (Emsley & Cowtan, 2004) using the SSM algorithm and displayed in PyMOL [47].
The fully exposed reactive site loop (RSL) of the inhibitor presents the P1 site with the Thr7 residue. The size of the GmSPI-2 RSL is typical for Kazal-family serine proteinase inhibitors, with seven amino acids between Cys5 and Cys13. It is generally thought that the rigidity of the RSL together with its specific sequence are the key factors conferring high potency on Kazal-family serine proteinase inhibitors [41]. There are usually eight hydrogen bonds stabilizing the RSL of Kazal-family serine proteinase inhibitors [25], [44]. The GmSPI-2 RSL has an additional strong (2.86 Å) hydrogen bond stabilizing the RSL between Thr6 (O) and Trp25 (Nε). Amongst Kazal-family serine proteinase inhibitors a tryptophan residue in this position is present only in GmSPI-2 [45].
The His-tag cleavage site was located between Leu40, and Ser41 of the fusion protein. The 2F o-F c electron density map for Leu40 is excellent for both the main-chain and side-chain atoms including the C-terminal oxygen atoms O and OXT (Fig. 8). The C-terminus is anchored by three strong hydrogen bonds involving both carboxylic oxygen atoms. Two of them are listed in Table 3. The third one involves the Leu40 OXT and Wat209 (x, y-1, z) atoms. This further confirms the sequence specificity of the presented tag cleavage procedure.
Figure 8. The C-terminus of the crystallographic model of GmSPI-2, residues Cys39 and Leu40, and symmetry-related Tyr20 and Ser21 residues.

The 2F o-F c electron density map was contoured at 1.4 σ. Hydrogen bonds are shown as dash lines.
The structure of the GmSPI-2 protein in solution determined by nmr spectroscopy
The three-dimensional structure of GmSPI-2 in solution was determined by NMR spectroscopy using 503 distance constraints derived from the analysis of 2D 1H-1H NOESY data sets. The ensemble of 17 lowest-energy structures (PDB code 2m5x) selected from a total of 200 calculated models is characterized by good convergence and low rmsd from ideal geometry (Table 2). The NMR-evaluated 3D structure is similar to that determined by X-ray crystallography, and contains the same α (Leu23-Ala29) and β (Val12-Gly14, Thr1-Tyr20, Leu33-Glu36) elements folded into the β2αβ motif of the hydrophobic core of the protein (Fig. 6B). A least-squares superposition of the 34 Cα atoms of the Cys5-Glu38 segment of the crystallographic model and the NMR conformer 11, representing the NMR ensemble, is characterized by an rmsd of 0.89 Å. Upon detailed inspection, the indole group of Trp25 and the imidazole ring of His35 are found to be in different orientations in the two models. Specifically, the χ2 torsion angle of Trp25 differs by 30° between the X-ray and NMR structures. In effect, the Trp25 Nε⋅ ⋅ ⋅Thr6 O hydrogen bond observed in the crystal structure is not present in solution. Likewise, the His35 Hδ1⋅ ⋅ ⋅ Glu36 O hydrogen bond is broken in solution due to reorientation of the His35 ring from χ1 of −40° (gauche−) to +60° (gauche+). Some minor conformational differences observed at the N-termini can be attributed to crystallographic packing.
Methodological remarks
The applicability of our method is based on the assumption that Ni(II) ions can interact with -SRHW-like sequences only at solvent exposed tags. Otherwise we would be posed with unspecific protein cleavage by Ni(II) ions. In previous study we verified the crucial assumption that Ni(II) ions would not be able to penetrate protein interiors or distort secondary structure elements using human ubiquitin. This protein naturally possesses the potentially active Thr-Leu-His-Leu sequence. However, despite prolonged incubations in the presence of Ni(II) ions at elevated temperatures no cleavage was observed for the natively folded protein, while Ubi denaturated by GuHCl was hydrolyzed [14].
The results obtained by X-ray crystallography and NMR spectroscopy consistently demonstrate that the molecular structure of GmSPI-2 is highly similar in solution and in the crystalline state. However, several small differences could be still detected. For instance, the exact conformation of the two disulfide bridges could not be unambiguously determined from the NMR data due to insufficient concentration of the GmSPI-2 protein in solution. The conformation of the first four N-terminal residues is different in the two structures as a result of crystal packing interactions, i.e. absence of intermolecular hydrogen bonds in solution.
His-tag and other small affinity tags are routinely used to obtain pure recombinant proteins, and structural studies in solution are often conducted without tag removal. This is, however, often impossible in the solid state because the crystal packing can lead to non-native interactions between the tag and the rest of the molecule. Therefore, the quality of X-ray data strongly depends on the homogeneity of the protein material, that is on the efficacy of the tag removal procedure and on the absence of non-specific cleavage products, which are usually generated by proteolytic enzymes. In this perspective, the high resolution of the X-ray diffraction data obtained in this work can be related to the truly perfect removal of the affinity tag afforded by the nickel-based methodology. Furthermore, the high yield of this method on the preparative scale (conversion of 70% of the starting material to the final product, with 100% homogeneity) makes it a good tool for obtaining pure thermostable proteins for structural studies.
The short GmSPI-2 gene is a promising target for mutagenesis directed toward engineering novel variants of the protein, specific for selected serine proteinases (study in progress). Such a study must be based on the precise knowledge of the starting polypeptide structure. Only with such knowledge one can carry out rational modeling and docking studies of GmSPI-2 derivatives to identify suitable hits for overexpression and activity evaluation. A clear understanding of the relation between the polypeptide structures in the crystal and in solution is also a prerequisite for the validity of such an approach. In this perspective, the accurate structure of this unique polypeptide belonging to non-classical Kazal-family serine proteinase inhibitors, by two methods, has to be regarded as setting the stage for further studies. It is worth mentioning that in a set of biologically interesting proteins, short domains with varying homology to GmSPI-2 seem to be frequently present at either the N- or the C-terminus (Kaczanowski & Zagórski, unpublished). Prediction of the probable function of such domains will be facilitated by the present results, which have defined the structural properties of a bona-fide inhibitor.
Conclusions
In the present study, the GmSPI-2 protein sequence was extended C-terminally by an -SRHWAP-H6 dodecapeptide, which comprises the underlined Ni(II)-sensitive tetrapeptide linked to the His-tag domain. The fusion protein was expressed in Pichia pastoris and affinity purified on Ni-NTA columns. The cleavage of the tag directly on the Ni-NTA column enabled us to combine the affinity purification and the tag removal into one step. The GmSPI-2 protein obtained in flow-through fractions exhibited 100% homogeneity. The absolute sequence specificity of the cleavage, observed previously in analytical scale purifications, has been preserved on the preparative scale as well. No protein impurities whatsoever could be detected in the protein fractions tested by HPLC and ESI-MS. The efficiency of cleavage was 70% on the preparative scale. The resulting GmSPI-2 protein was fully active. The results obtained by X-ray crystallography and NMR spectroscopy show that the structure of GmSPI-2 is highly similar in solution and in the crystalline state. The resolution of the crystal structure of 0.98 Å is the highest for the Kazal-type serine protease inhibitors deposited in the PDB. The number of reflections per parameter justified refinement without any stereochemical restraints for the well-ordered parts of the inhibitor. The refinement converged with R = 10.57% for all reflections. One cycle of full-matrix minimization permitted the estimation of the standard uncertainties in all positional parameters which, for example, for the fully occupied main-chain atoms range from 0.012 Å to 0.033 Å. The 2F o-F c electron density map for Leu40, the last residue of the mature inhibitor, is excellent for both the main-chain and side-chain atoms including the C-terminal oxygen atoms O and OXT. This clearly confirms the sequence specificity of the presented tag cleavage procedure.
These exceptionally high grade of the protein purification product was reflected in the high quality of the structural determinations, both in the solid state and in solution. The high resolution of these structures was certainly facilitated by the perfect homogeneity of the protein sample after affinity tag removal.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. The atomic coordinates and structure factors have been deposited in the Protein Data Bank under the accession codes 4hgu & 2m5x.
Funding Statement
This research was supported in part by the project “Metal-dependent peptide hydrolysis. Tools and mechanisms for biotechnology, toxicology and supramolecular chemistry” (TEAM/2009-4/1) to WB from the Foundation for Polish Science, and by the project POIG.01.01.02-14-007/08-00 to WOZ, both co-financed from European Regional Development Fund resources within the framework of Operational Program Innovative Economy. The equipment used was sponsored in part by the Centre for Preclinical Research and Technology (CePT), a project co-sponsored by European Regional Development Fund and Innovative Economy, The National Cohesion Strategy of Poland, to IBB PAS. This work was also partly supported by Slovenian Research Agency and Ministry of Higher Education, Science, and Technology of Republic of Slovenia (www.arrs.gov.si), Program P1-242, to MLŽ and by the EN-FIST Center of Excellence (www.enfist.si), to IZ. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Wlodawer A, Minor W, Dauter Z, Jaskolski M (2013) Protein crystallography for aspiring crystallographers or how to avoid pitfalls and traps in macromolecular structure determination. FEBS J 280: 5705–5736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wojtkowiak A, Witek K, Hennig J, Jaskolski M (2012) Two high-resolution structures of potato endo-1,3-β-glucanase reveal subdomain flexibility with implications for substrate binding. Acta Crystallogr D Biol Crystallogr 68: 713–723. [DOI] [PubMed] [Google Scholar]
- 3. Choi SI, Song HW, Moon JW, Seong BL (2001) Recombinant enterokinase light chain with affinity tag: expression from Saccharomyces cerevisiae and its utilities in fusion protein technology. Biotechnol Bioeng 75: 718–24. [DOI] [PubMed] [Google Scholar]
- 4. Jenny RJ, Mann KG, Lundblad RL (2003) A critical review of the methods for cleavage of fusion proteins with thrombin and factor Xa. Protein Expr Purif 31: 1–11. [DOI] [PubMed] [Google Scholar]
- 5. Waugh DS (2011) An overview of enzymatic reagents for the removal of affinity tags. Protein Expr Purif 80: 283–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Milović NM, Dutca LM, Kostić NM (2003) Transition-metal complexes as enzyme-like reagents for protein cleavage: complex cis-[Pt(en)(H2O)2]2+ as a new methionine-specific protease. Chem Eur J 9: 5097–5106. [DOI] [PubMed] [Google Scholar]
- 7. Allen G, Campbell O (1996) Specific cleavage of histidine-containing peptides by copper(II). Int J Peptide Protein Res 48: 265–273. [DOI] [PubMed] [Google Scholar]
- 8. Yashiro M, Yamamura A, Takarada T, Komiyama M (1997) Sequence-specific hydrolysis of peptides by metal assisted autocatalysis of an internal hydroxyl group. J Inorg Biochem 67: 225. [Google Scholar]
- 9. Humphreys DP, Smith BJ, King LM, West SM, Reeks DG, et al. (1999) Efficient site-specific removal of C-terminal FLAG fusion from Fab’ using copper(II) ion catalyzed protein cleavage. Protein Eng 12: 179–184. [DOI] [PubMed] [Google Scholar]
- 10. Humphreys DP, King LM, West SM, Chapman AP, Sehdev M, et al. (2000) Improved efficiency of site-specific copper(II) ion-catalyzed protein cleavage effected mutagenesis of cleavage site. Protein Eng 13: 201–206. [DOI] [PubMed] [Google Scholar]
- 11. Dou F, Qiao F, Hu J, Zhu T, Xu X, et al. (2000) Preliminary study on the cleavage of fusion protein GST-CMIV with palladium(II) complex. Prep Biochem Biotechnol 30: 69–78. [DOI] [PubMed] [Google Scholar]
- 12. Krężel A, Kopera E, Protas AM, Poznański J, Wysłouch-Cieszyńska A, et al. (2010) Sequence-specific Ni(II)-dependent peptide bond hydrolysis for protein engineering. Combinatorial library determination of optimal sequences. J Am Chem Soc 132: 3355–3366. [DOI] [PubMed] [Google Scholar]
- 13. Kopera E, Krężel A, Protas AM, Belczyk A, Bonna A, et al. (2010) Sequence-specific Ni(II)-dependent peptide bond hydrolysis for protein engineering: reaction conditions and molecular mechanism. Inorg Chem 49: 6636–6645. [DOI] [PubMed] [Google Scholar]
- 14. Kopera E, Belczyk-Ciesielska A, Bal W (2012) Application of Ni(II)-assisted peptide bond hydrolysis to non-enzymatic affinity tag removal. PLoS One 7: e36350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nirmala X, Kodrik D, Zurovec M, Sehnal F (2001) Insect silk contains both a Kunitz-type and a unique Kazal-type proteinase inhibitor. Eur J Biochem 268: 2064–2073. [DOI] [PubMed] [Google Scholar]
- 16. Kludkiewicz B, Kodrik D, Grzelak K, Nirmala X, Sehnal F (2005) Structurally unique recombinant Kazal-type proteinase inhibitor retains activity when terminally extended and glycosylated. Protein Expr Purif 43: 94–102. [DOI] [PubMed] [Google Scholar]
- 17. Milner M, Chroboczek J, Zagórski W (2007) Engineered resistance against proteinases. Acta Biochim Pol 54: 523–536. [PubMed] [Google Scholar]
- 18. Redkiewicz P, Więsyk A, Góra-Sochacka A, Sirko A (2012) Transgenic tobacco plants as production platform for biologically active human interleukin 2 and its fusion with proteinase inhibitors. Plant Biotechnol J 10: 1–9. [DOI] [PubMed] [Google Scholar]
- 19. Jancarik J, Kim SH (1991) Sparse matrix sampling: a screening method for crystallization of proteins. J Appl Crystallogr 24: 409–411. [Google Scholar]
- 20. Kabsch W (1993) Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallogr 26: 795–800. [Google Scholar]
- 21. Diederichs K (2006) Some aspects of quantitative analysis and correction of radiation damage. Acta Crystallogr D Biol Crystallogr 62: 96–101. [DOI] [PubMed] [Google Scholar]
- 22. Strong M, Sawaya MR, Wang S, Phillips M, Cascio D (2006) Toward the structural genomics of complexes: crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 103: 8060–8065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vagin A, Teplyakov A (1997) MOLREP: an Automated Program for Molecular Replacement. J Appl Crystallogr 30: 1022–1025. [Google Scholar]
- 24. Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67: 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee T-W, Qasim MA Jr, Laskowski M, James NG (2007) Structural insights into the non-additivity effects in the sequence-to-reactivity algorithm for serine peptidases and their inhibitors. J Mol Biol 367: 527–546. [DOI] [PubMed] [Google Scholar]
- 26. Huang X, Miller W (1991) A time-efficient linear-space local similarity algorithm. Adv Appl Math 12: 337–357. [Google Scholar]
- 27. Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53: 240–255. [DOI] [PubMed] [Google Scholar]
- 28. Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132. [DOI] [PubMed] [Google Scholar]
- 29. Sheldrick GM (2008) A short history of SHELX. Acta Crystallogr A Found Adv 64: 112–122. [DOI] [PubMed] [Google Scholar]
- 30.Wüthrich K (1986) NMR of Proteins and Nucleic Acids. Wiley, New York. 166 p.
- 31. Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, et al. (1995) 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J Biomol NMR 6: 135–140. [DOI] [PubMed] [Google Scholar]
- 32. Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, et al. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6: 277–293. [DOI] [PubMed] [Google Scholar]
- 33. Shen Y, Delaglio F, Cornilescu G, Bax A (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR 44: 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Güentert P, Mumenthaler K, Wüthrich K (1997) Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol 273: 283–298. [DOI] [PubMed] [Google Scholar]
- 35. Krieger E, Koraimann G, Vriend G (2002) Increasing the precision of comparative models with YASARA NOVA–a self-parameterizing force field. Proteins 47: 393–402. [DOI] [PubMed] [Google Scholar]
- 36. Ramachandran GN, Sasisekharan V (1968) Conformation of polypeptides and proteins. Adv Protein Chem 23: 283–437. [DOI] [PubMed] [Google Scholar]
- 37. Wang J, Dauter M, Alkire R, Joachimiak A, Dauter Z (2007) Triclinic lysozyme at 0.65 Å. Acta Crystallogr D Biol Crystallogr. 63: 1254–1268. [DOI] [PubMed] [Google Scholar]
- 38. Addlagatta A, Czapinska H, Krzywda S, Otlewski J, Jaskolski M (2001) Ultrahigh-resolution structure of a BPTI mutant. Acta Crystallogr D Biol Crystallogr 57: 649–663. [DOI] [PubMed] [Google Scholar]
- 39. Thaimattam R, Tykarska E, Bierzynski A, Sheldrick GM, Jaskolski M (2002) Atomic resolution structure of squash trypsin inhibitor: unexpected metal coordination. Acta Crystallogr D Biol Crystallogr 58: 1448–61. [DOI] [PubMed] [Google Scholar]
- 40. Wang D-D, Zhu H-J, Song G-L, Wang J-T (2005) Bis(triaquasodium) 2,5-dibenzoylterephthalate tetrahydrate. Acta Crystallogr Sect E Struct Rep Online 61: m2610–m2612. [Google Scholar]
- 41. Shenoy RT, Thangamani S, Velazquez-Campoy A, Ho B, Ding JL (2011) Structural basis for dual-inhibition mechanism of a non-classical Kazal-type serine protease inhibitor from horseshoe crab in complex with subtilisin. PloS One 6: e18838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hemmi H, Kumazaki Yoshizawa-Kumagaye K, Nishiuchi Y, Yoshida T, Ohkubo T, et al. (2005) Structural and functional study of an Anemonia elastase inhibitor, a “nonclassical” Kazal-type inhibitor from Anemonia sulcata . Biochemistry 44: 9626–9636. [DOI] [PubMed] [Google Scholar]
- 43. Cohen GH (1997) ALIGN: a program to superimpose protein coordinates, accounting for insertions and deletions. J Appl Crystallogr 30: 1160–1161. [Google Scholar]
- 44. Di Marco S, Priestle JP (1997) Structure of the complex of leech-derived tryptase inhibitor (LDTI) with trypsin and modeling of the LDTI-tryptase system. Structure 5: 1465–1474. [DOI] [PubMed] [Google Scholar]
- 45. Rimphanitchayakit V, Tassanakajon A (2010) Structure and function of invertebrate Kazal-type serine proteinase inhibitors. Dev Comp Immunol 34: 377–386. [DOI] [PubMed] [Google Scholar]
- 46. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, et al. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948. [DOI] [PubMed] [Google Scholar]
- 47.DeLano WL (2002) DeLano Scientific, San Carlos, CA, USA.
- 48. Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR 8: 477–486. [DOI] [PubMed] [Google Scholar]
- 49. Vriend G (1990) WHAT IF: A molecular modeling and drug design program. J Mol Graph 8: 52–56. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. The atomic coordinates and structure factors have been deposited in the Protein Data Bank under the accession codes 4hgu & 2m5x.