

Abstract
Background
Chronic obstructive pulmonary disease (COPD) is characterized by expiratory flow limitation, causing air trapping and lung hyperinflation. Hyperinflation leads to reduced exercise tolerance and poor quality of life in COPD patients. Total lung capacity (TLC) is an indicator of hyperinflation particularly in subjects with moderate-to-severe airflow obstruction. The aim of our study was to identify genetic variants associated with TLC in COPD.
Methods
We performed genome-wide association studies (GWASs) in white subjects from three cohorts: the COPDGene Study; the Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE); and GenKOLS (Bergen, Norway). All subjects were current or ex-smokers with at least moderate airflow obstruction, defined by a ratio of forced expiratory volume in 1 second to forced vital capacity (FEV1/FVC) <0.7 and FEV1 < 80% predicted on post-bronchodilator spirometry. TLC was calculated by using volumetric computed tomography scans at full inspiration (TLCCT). Genotyping in each cohort was completed, with statistical imputation of additional markers. To find genetic variants associated with TLCCT, linear regression models were used, with adjustment for age, sex, pack-years of smoking, height, and principal components for genetic ancestry. Results were summarized using fixed-effect meta-analysis.
Results
Analysis of a total of 4,543 COPD subjects identified one genome-wide significant locus on chromosome 5p15.2 (rs114929486, β = 0.42L, P = 4.66 × 10−8).
Conclusions
In COPD, TLCCT was associated with a SNP in dynein, axonemal, heavy chain 5 (DNAH5), a gene in which genetic variants can cause primary ciliary dyskinesia. DNAH5 could have an effect on hyperinflation in COPD.
Electronic supplementary material
The online version of this article (doi:10.1186/s12931-014-0097-y) contains supplementary material, which is available to authorized users.
Keywords: Pulmonary disease, Chronic obstructive, Hyperinflation, Genome-wide association analysis, Total lung capacity, DNAH5
Introduction
Chronic obstructive pulmonary disease (COPD) is one of the leading causes of morbidity and mortality worldwide [1]. Even though cigarette smoking is an established risk factor, only a minority of smokers develop COPD, which suggests that genetic susceptibility may play a significant role [2]. The pathophysiologic hallmark of COPD is expiratory flow limitation, resulting in air trapping and lung hyperinflation [3]. Dyspnea is the primary symptom limiting exercise in patients with advanced COPD. However, the physiologic impairment, traditionally measured by FEV1, often does not correlate strongly with the degree of dyspnea [4]. Hyperinflation at rest and during exercise is common among COPD patients and may be one explanation for differing exercise tolerance in patients with similar FEV1 levels [5]. Hyperinflation provides a mechanistic link between expiratory flow limitation and dyspnea [5]. Reducing hyperinflation is a mechanism for symptom relief from inhaled bronchodilators even in COPD patients who do not demonstrate a post-bronchodilator improvement in FEV1 [6]. Hyperinflation is closely linked to reduced exercise tolerance, poor quality of life, and even increased mortality in COPD subjects [7].
Genome-wide association studies (GWASs) of hyperinflation in COPD subjects have not been previously reported. We used total lung capacity measured by chest computed tomography scans (TLCCT) as an indicator of hyperinflation [8]. Our hypothesis was that genetic variants would be associated with TLCCT in COPD.
Methods
Subjects
Subjects were current and former smokers from three COPD studies: the COPDGene Study (NCT00608764, http://www.copdgene.org); the Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE, NCT00292552, http://www.eclipse-copd.com); and GenKOLS (Bergen, Norway). All subjects had self-described European white ancestry, though African American subjects (AAs) from the COPDGene Study were included for an additional analysis. Study design and details of each study have been previously described [9–15]. Briefly, COPDGene is a multi-center genetic and epidemiologic investigation to study COPD. Subjects were between 45 and 80 years old and had at least 10 pack-years of cigarette smoking. ECLIPSE is a non-interventional, longitudinal prospective three-year study in cases with COPD and control subjects. The entry criteria for COPD cases and smoking controls were 40–75 years old with ≥10 pack-years of smoking. COPD was defined as FEV1 < 80% predicted and FEV1/FVC < 0.7. The control subjects were selected based on FEV1 ≥ 80% predicted and FEV1/FVC ≥ 0.7. In GenKOLS study, subjects for the case-control study were recruited from Bergen, Norway. Enrolment criteria were: 1) self-reported Caucasian; 2) aged > 40 years; 3) current or former smoker with ≥2.5 pack-years of smoking history; and 4) no severe α1-antitrypsin deficiency. The spirometry criteria were the same as ECLIPSE. Subjects with a history of lung volume reduction surgery were excluded from all three studies. The current analysis was approved by the Partners Healthcare Research Committee (COPDGene: 2007P000554; ECLIPSE: 2005P002467; GenKOLS: 2009P000790).
In this analysis, subjects with COPD were defined by having airflow obstruction of at least spirometry grade 2 (post-bronchodilator FEV1/FVC < 0.7 and FEV1 < 80% predicted), based on the Global initiative for chronic Obstructive Lung Disease (GOLD 2-4) [3]. Additional analyses included smokers with normal spirometry (post-bronchodilator FEV1/FVC ≥ 0.7 and FEV1 ≥ 80%).
Chest CT scans
In each study, volumetric CT scans acquired in supine position at suspended full inspiration without administration of intravenous contrast. TLCCT in liters was calculated from volumetric CT measurements.
In the COPDGene study, multi-detector CT scanners (at least 16 detector channels) were used. Detailed CT protocols have been previously published [10]. CT scans were subjected to a standard quality control procedure. Computerized image analysis was performed at the COPDGene Imaging Center at National Jewish Health and Brigham and Women’s Hospital using Slicer (Version 2, www.slicer.org). In the ECLIPSE study, multi-detector CT scanners (GE Healthcare or Siemens Healthcare) were used with a minimum of 4 detectors. Exposure settings were 120 kVp and 40 mAs and images were reconstructed at 1.0 mm (Siemens) or 1.25 mm (GE) contiguous slices, using a low spatial frequency reconstruction algorithm (GE: Standard, Siemens: b35f). CT scanners were calibrated regularly and a standard CT phantom was scanned by all participating centers to produce comparable data. All CT scans were analyzed using Pulmonary Workstation 2.0 software (VIDA Diagnostics, Iowa City, IA) [16]. In the GenKOLS study, a GE LightSpeed Ultra CT scanner (120 kVp, 200 mA; GE Healthcare, Milwaukee, WI, USA) was used with 1-mm slice thickness at 20-mm intervals. The CT scans were reconstructed using both a low spatial frequency reconstruction algorithm (standard) for density measurements, and a high spatial frequency algorithm (bone) for airway measurements. All ECLIPSE and GenKOLS images were transferred to the James Hogg iCAPTURE Centre (Vancouver, BC, Canada) for quantitative analysis as previously described [13].
Genotyping quality control and imputation
Illumina platforms [HumanOmniExpress for COPDGene, HumanHap 550V3for ECLIPSE, and HumanHap 550 (V1, V3, and Duo) for GenKOLS; Illumina, Inc., San Diego, CA] were used for genotyping. Imputation in COPDGene was performed using MaCH [17] and minimac [18] using 1000 Genomes [19] Phase I v3 European (EUR) reference panels for non-Hispanic white subjects (NHWs). Details on genotyping quality control and imputation for the GenKOLS and ECLIPSE cohorts have been previously described [9,12,14,15,20,21]. Variants which passed genotyping or imputation quality control (R2 > 0.3) in all three cohorts, were included in the analysis.
Statistical analysis
In each study population, we performed linear regression analysis of single nucleotide polymorphisms (SNPs) under an additive model of inheritance with adjustment for age, gender, height, pack-years of cigarette smoking and genetic ancestry-based principal components using PLINK 1.07 [22], as previously described [9,14,15]. Imputed genotypes were analyzed in a similar manner, using SNP dosage data in PLINK 1.07 [22]. Fixed-effects meta-analysis [23] was performed using METAL (version 2011-03-25) [24] and R 3.0.2 (www.r-project.org) with the meta-package. Genome-wide significance was determined by P value < 5 × 10−8. We evaluated heterogeneity by calculating both I2 [25] and P values for Cochrane’s Q. In regions with evidence of genetic heterogeneity, we also applied a modified random-effects model optimized to detect associations under heterogeneity since the fixed-effects model is based on inverse-variance-weighted effect size [26]. Genomic inflation factors [27] were calculated using GenABEL [28]. LocusZoom [29] was used to create local association plots, using the 1000 Genomes EUR reference data to calculate linkage disequilibrium (LD).
To explore other SNPs independently associated with TLCCT in COPD, region-based conditional analyses were undertaken using linear regression with adjustment for the most significant (lead) SNP in a genome-wide significant region using genotyped or dosage data as appropriate. All SNPs within a 250 kb window on either side of the lead SNP were tested for association with TLCCT in COPD. For region-based analyses conditional on the top SNP, a P value of P < 5 × 10−4 was considered significant to reflect an approximate adjustment for a 500 kb interval [9,15].
Results
Baseline characteristics of each of the three cohorts are summarized in Table 1.
Table 1.
Baseline characteristics of COPD subjects included in the meta-analysis
| COPDGene | ECLIPSE | GenKOLS | |
|---|---|---|---|
| N | 2,653 | 1,464 | 426 |
| Age, yrs | 64.7 (8.2) | 63.5 (7.0) | 64.3 (9.3) |
| Sex, male % | 56.0 | 65.8 | 62.9 |
| Current smoker, % | 34.9 | 35.2 | 50.2 |
| Pack-years of cigarette smoking | 56.2 (27.9) | 49.7 (26.7) | 30.9 (18.2) |
| Height, cm | 169.7 (9.4) | 169.4 (9.0) | 170.7 (8.7) |
| Body mass index, kg/m2 | 28 (6.1) | 26.6 (5.6) | 25.6 (4.8) |
| FEV1 % predicted | 50 (17.9) | 47.4 (15.5) | 52.4 (17.0) |
| FVC % predicted | 76.5 (17.0) | 86.0 (19.9) | 80.0 (15.3) |
| FEV1/FVC | 0.49 (0.13) | 0.44 (0.11) | 0.52 (0.12) |
| Total lung capacity (TLC)CT, L | 6.19 (1.4) | 6.20 (1.44) | 5.57 (1.29) |
| TLC % predicted | 102.8 (16.6) | 101.7 (18.2) | 90.9 (18.8) |
Data are presented as mean (SD) or percentage, as appropriate.
In the meta-analysis of TLCCT in COPD, the combined GWAS of three cohorts included 4,543 subjects with COPD. A quantile-quantile (Q-Q) plot is displayed in Figure 1A (lambda = 1.03). Figure 1B shows a novel region on chromosome 5p15.2, which reached the genome-wide significance threshold. Results yielding a suggestive P value threshold of < 5×10−7 [30] are listed in Table 2. Figure 2 displays the regional association plots for the top five loci.
Figure 1.

The three-cohort meta-analysis including 1000 Genomes project imputed data for total lung capacity measured by chest CT in COPD subjects after adjustment for age, gender, height, pack-years of cigarette smoking and genetic ancestry-based principal components. (a) The quantile–quantile plot and (b) Manhattan plot of –log10 P values.
Table 2.
Meta-analysis results of three genome-wide association studies for total lung capacity measured by CT in COPD subjects *
| Locus | SNP | Nearest Gene | Distance (kb) | Risk/Non-risk Allele | FRQ | COPDGene NHW | ECLIPSE | GenKOLS | Overall | I 2 | Q | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| β | P | β | P | β | P | β | P | ||||||||
| 5p15.2 | rs114929486 | DNAH5 | 0 | A/G | 0.04 | 0.37† | 2.94 × 10−4 | 0.60† | 7.18 × 10−6 | 0.13† | 5.47 × 10−1 | 0.42 | 4.66 × 10−8 | 49 | 0.14 |
| 8q24.12 | rs10955930 | ENPP2 | 30 | T/A | 0.49 | 0.11† | 5.55 × 10−4 | 0.15† | 1.19 × 10−3 | 0.22† | 7.45 × 10−3 | 0.13 | 1.38 × 10−7 | 0 | 0.37 |
| 19p13.12 | rs590950 | CYP4F8 | 26 | T/C | 0.49 | 0.13† | 3.36 × 10−6 | 0.09† | 3.79 × 10−2 | 0.13† | 1.25 × 10−1 | 0.12 | 1.39 × 10−7 | 0 | 0.72 |
| 6q13 | rs72920744 | B3GAT2 | 6 | G/A | 0.99 | 0.40† | 8.05 × 10−3 | 0.83† | 6.32 × 10−6 | 0.61† | 1.11 × 10−1 | 0.58 | 2.18 × 10−7 | 39 | 0.19 |
| 8q24.3 | rs57629580 | LY6H | 35 | C/A | 0.91 | 0.30† | 1.47 × 10−5 | 0.27† | 4.02 × 10−3 | 0.10† | 6.05 × 10−1 | 0.28 | 2.56 × 10−7 | 0 | 0.66 |
Definition of abbreviations: FRQ = risk allele frequency; NHW = non-Hispanic white; SNP = single nucleotide polymorphism.
*Adjusted for age, sex, height, pack-years of cigarette smoking and genetic ancestry as summarized in the principal components.
†Imputed genotypes.
Figure 2.
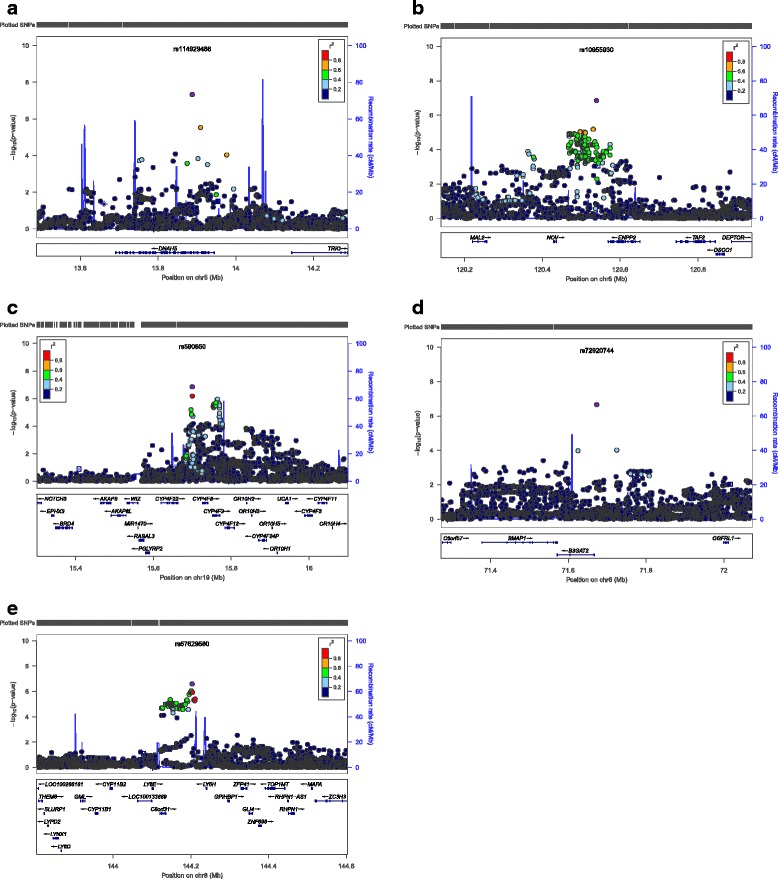
The meta-analysis for total lung capacity measured by chest CT in COPD subjects. Regional association plots for (a) one genome-wide significant locus and (b-e) the other four suggestive loci. The x-axis is chromosomal position, and the y-axis shows the -log10 P value. The most significant SNP at each locus is labeled in purple, with other SNPs colored by degress of linkage disequilibrium (r2). Plots were generated using LocusZoom.
The most significant SNP on chromosome 5p15.2 was rs114929486 (β = 0.42L, P = 4.66 × 10−8), which was located within the gene dynein, axonemal, heavy chain 5 (DNAH5). Although some evidence of heterogeneity was present (P = 0.14 for Cochrane’s Q, I2 = −1.1), a modified random-effects meta-analysis model revealed similar significance (P = 6.15 × 10−8). The second most significant SNP was rs10955930 (β = 0.13L, P = 1.38 × 10−7), near ectonucleotide pyrophosphatase/phosphodiesterase 2 (ENPP2) on chromosome 8q24.12.
To determine whether there was likely to be more than one functional genetic variant within the genome-wide significant region, we performed analyses conditioning on the top (lead) SNP from the meta-analysis. All SNPs within 250 kb flanking the top signal were examined. We found evidence suggestive of secondary associations on 5p15.2 (conditioning on rs114929486) in two SNPs (rs4701985, β = 0.28L, P = 4.11 × 10−4; rs1502044, β = 0.31L, P = 4.45 × 10−4) located within the same gene, DNAH5.
Additional analyses
There is significant difference in TLCCT by genotypes of rs114929486 among COPD subjects of our study (Table 3). Additionally we compared clinical and radiological characteristics of the COPDGene NHW subjects stratified by genotypes of rs114929486 (Additional file 1: Table S1). Thicker airway walls (higher Pi10) and lower FEV1 % predicted values were seen among carriers of the risk allele for higher TLC (P <0.05).
Table 3.
Mean total lung capacity measured by CT (TLC CT ) according to genotype of rs114929486 in DNAH5 (N = 4,543) *
| rs114929486 | GG | AG | P | |
|---|---|---|---|---|
| All | N | 4282 | 258 | |
| TLCCT, L | 6.12 (1.41) | 6.50 (1.52) | 0.0001 | |
| COPDGene | n | 2531 | 121 | |
| TLCCT, L | 6.18 (1.40) | 6.47 (1.47) | 0.042 | |
| ECLIPSE | N | 1364 | 99 | |
| TLCCT, L | 6.15 (1.42) | 6.84 (1.57) | 1.96 × 10-5 | |
| GenKOLS | N | 387 | 38 | |
| TLCCT, L | 5.56 (1.30) | 5.70 (1.25) | 0.486 |
Data are presented as mean (SD).
*Three subjects with AA genotype (each one from each cohort) were excluded from this analysis.
The top SNP, rs114929486, was very rare in African American subjects in the COPDGene Study (minor allele frequency = 0.0056) and therefore, was not analyzed further.
Hyperinflation may be due to different underlying mechanisms and genetic factors in different COPD patients, depending on their specific phenotypes. We performed additional GWASs and meta-analyses of TLCCT in COPD subjects with either emphysema-predominant COPD (Additional file 1: Tables S2 and S4) or chronic bronchitis (Additional file 1: Tables S3 and S5). The stratified analyses did not show any genome-wide significant associations (Additional file 1: Figures S1 and S2). Among COPD subjects with ≥10% emphysema, chromosome 11p15.5, which includes genes AP2A2, MUC6 and CHID1, was the most significant region (rs7483870, β = 0.16L, P = 3.93 × 10−7; rs4076950, β = 0.13L, P = 2.88 × 10−6; rs4963123, β = 0.13L, P =3.40 × 10−6). The top SNP from the original meta-analysis, rs114929486, located in DNAH5 maintained nominal significance in both subjects with emphysema-predominant COPD (β = 0.39L, P = 4.19 × 10−6) and those with chronic bronchitis (β = 0.56L, P = 1.74 × 10−4).
We also performed meta-analyses of three GWASs for TLCCT of smokers with normal spirometry (smoking controls) as well as combined analyses of both COPD and smoking control subjects. Additional file 1: Table S6 shows baseline characteristics of the included subjects. There were no genome-wide significant loci in the meta-analyses (Additional file 1: Figures S3 and S4) though three SNPs reached genome-wide significant thresholds in individual study GWAS. The results of the genome-wide significant SNPs were summarized in Additional file 1: Tables S7, S8 and S9.
Discussion
This study is the first GWAS of total lung capacity in COPD subjects. Our meta-analysis of three GWASs of TLCCT in subjects with moderate-to-severe COPD has found a genome-wide significant region on chromosome 5p15.2, containing the gene DNAH5 and several suggestive loci on chromosomes 8q24, 19p13, and 6q13, with P values < 5 × 10−7.
The genome-wide significant SNP was located within DNAH5, encoding a dynein protein, which is part of a microtubule-associated motor protein complex consisting of heavy, light, and intermediate chains. This protein is an axonemal heavy chain dynein. It functions as a force-generating protein of respiratory cilia with ATPase activity, where the release of ADP is thought to produce the force-producing power stroke. Mutations in this gene can cause primary ciliary dyskinesia (PCD) type 3, as well as Kartagener syndrome, another disease due to ciliary defects [31–33]. PCD is characterized by marked peripheral airway dysfunction [34] and small airways obstruction leading to air trapping and a consequent increase in residual volume (RV) and RV/TLC ratio [35,36]. Similarly, the major sites of obstruction in COPD are small airways [37–39]. Recently a study demonstrated that cigarette smoking induces epithelial epigenetic changes in the small airway epithelium, collected by fiberoptic bronchoscopic brushings [40]. DNAH5 was one of the most hyper-methylated genes in smokers compared to nonsmokers [40]. Therefore, DNAH5 variants may contribute to genetic susceptibility to develop hyperinflation in patients with COPD.
The genome-wide significant SNP, rs114929486, and the other two significant SNPs from the region-based analysis conditional on the top SNP are located within intronic regions of DNAH5. These SNPs had minor allele frequency (MAF) of < 0.05 with relatively large effect sizes. In other diseases, an overlap between the genetic variants for Mendelian and complex diseases has been suggested [41,42]. Blair and colleagues [41] have found thousands of associations between Mendelian and complex diseases by mining the medical records of over 110 million patients. Using mathematical modeling, they also demonstrated that GWAS hits for common diseases are enriched in genes containing Mendelian variants [41]. Since the top GWAS SNP associated with TLCCT is located in a gene for a Mendelian disorder, possible connections between DNAH5 genetic variants and small airway disease should be investigated.
Although the second ranked SNP, rs10955930, did not reach genome-wide significance, the nearest gene, ENPP2, has been implicated in pulmonary fibrosis and allergic asthmatic inflammation [43]. The protein encoded by this gene, also known as autotaxin (ATX), functions as both a phosphodiesterase, which cleaves phosphodiester bonds at the 5′ end of oligonucleotides, and a phospholipase, which catalyzes production of lysophosphatidic acid (LPA) in extracellular fluids. LPA evokes growth factor-like responses including stimulation of cell proliferation and chemotaxis. ATX was highlighted as a therapeutic target for idiopathic pulmonary fibrosis since limiting LPA synthesis reduces fibrosis [44,45]. Recently, a study demonstrated marked and selective elevation of ATX and two of its LPA products, LPA 22:5 and LPA 22:6, in the bronchoalveolar lavage fluid of human patients with asthma in response to airway allergen challenge [46]. They also reported that ATX-overexpressing transgenic mice had a more severe asthmatic phenotype, whereas blocking ATX activity and knockdown of the LPA2 receptor in mice produced a marked attenuation of Th2 cytokines and allergic lung inflammation [46]. In the other study, Enpp2+/−mice, heterozygous for the autotaxin-encoding gene, showed not only diminished expression of autotaxin/lysophospholipase D and approximately half normal plasma LPA, but also exaggerated response to hypoxia-induced vasoconstriction and remodeling, as evidenced by increased right ventricular systolic pressure and an increased percentage of muscularized arterioles [47]. Even though there has been no direct evidence, ENPP2 (ATX) could play a role in COPD-associated hyperinflation through its influence on extracellular matrix and inflammation.
The current study has several limitations. First, whether the associated regions play a functional role has not been investigated. Second, a replication analysis has not been performed in additional populations even though this was a meta-analysis of three GWASs using the largest COPD cohorts to date. Third, TLC is only one possible indicator of hyperinflation. There is no standard marker of hyperinflation and each indicator has its advantages and disadvantages. Besides TLC, RV, the ratio of RV/TLC, functional residual capacity (FRC), inspiratory capacity (IC) and the ratio of IC/TLC are all used to assess the severity of hyperinflation [8]. Most of these measures are obtained by helium dilution testing or plethysmography, which are challenging to implement in a large population study. TLC may be normal until the late stages of COPD and therefore may not be the best marker for hyperinflation in mild airflow limitation. However, our primary analysis was limited to subjects with moderate-to-severe airway obstruction. Fourth, TLC was measured by CT, not plethysmography, though strong correlations between TLC measured by plethysmography and CT have been observed [48–51]. However, in subjects with airflow limitation, plethysmography systematically overestimates lung volume relative to CT despite adherence to recommendations for proper measurement technique [52]. TLCCT at full inspiration is a relatively standardized and widely used method [51] and may be easier for subjects to perform than plethysmography [52].
We have reported the first GWAS of TLCCT in COPD subjects and identified DNAH5 as a potential susceptibility gene associated with hyperinflation in COPD. The current study suggests that this gene for a Mendelian lung disease could also have an effect on hyperinflation in COPD, although future studies will be required for functional validation.
Acknowledgements
This work was supported by NIH R01 HL094635 (C.P.H.), R01 NR013377 (C.P.H.), R01 HL089856 (E.K.S.), P01 HL105339 (E.K.S.), and R01HL089897 (J.D.C.) The COPDGene study (NCT00608764) is also supported by the COPD Foundation through contributions made to an Industry Advisory Board comprised of AstraZeneca, Boehringer Ingelheim, Novartis, Pfizer, Siemens, GlaxoSmithKline, and Sunovion. The Norway GenKOLS study (Genetics of Chronic Obstructive Lung Disease, GSK code RES11080) and the ECLIPSE study (NCT00292552; GSK code SCO104960) were sponsored by GlaxoSmithKline.
We acknowledge and thank the COPDGene Core Teams:
Administrative Core: James D. Crapo, MD (PI); Edwin K. Silverman, MD, PhD (PI); Barry J. Make, MD; Elizabeth A. Regan, MD, PhD; Stephanie Bratschie, MPH; Rochelle Lantz; Sandra Melanson, MSW, LCSW; Lori Stepp
Executive Committee: Terri Beaty, PhD; Russell P. Bowler, MD, PhD; James D. Crapo, MD; Jeffrey L. Curtis, MD; Douglas Everett, PhD; MeiLan K. Han, MD, MS; John E. Hokanson, MPH, PhD; David Lynch, MB; Barry J. Make, MD; Elizabeth A. Regan, MD, PhD; Edwin K. Silverman, MD, PhD; E. Rand Sutherland, MD
External Advisory Committee: Eugene R. Bleecker, MD; Harvey O. Coxson, PhD; Ronald G. Crystal, MD; James C. Hogg, MD; Michael A. Province, PhD; Stephen I. Rennard, MD; Duncan C. Thomas, PhD
NHLBI: Thomas Croxton, MD, PhD; Weiniu Gan, PhD; Lisa Postow, PhD
COPD Foundation: John W. Walsh; Randel Plant; Delia Prieto
Biorepository Visit 1 (Baltimore): Homayoon Farzadegan, PhD; Samantha Bragan; Stacey Cayetano
Biorepository Visit 2 (Boston): Daniel Cossette; Roxanne K. Kelly, MBA
Data Coordinating Center: Douglas Everett, PhD; Andre Williams, PhD; Ruthie Knowles; Carla Wilson, MS
Epidemiology Core: John Hokanson, MPH, PhD; Jennifer Black-Shinn, MPH; Gregory Kinney, MPH
Genetic Analysis Core: Terri Beaty, PhD; Peter J. Castaldi, MD, MSc; Michael Cho, MD; Dawn L. DeMeo, MD, MPH; Marilyn G. Foreman, MD, MS; Nadia N. Hansel, MD, MPH; Megan E. Hardin, MD; Craig Hersh, MD, MPH; Jacqueline Hetmanski, MS; John E. Hokanson, MPH, PhD; Nan Laird, PhD; Christoph Lange, PhD; Sharon M. Lutz, MPH, PhD; Manuel Mattheisen, MD; Merry-Lynn McDonald, MSc, PhD; Margaret M. Parker, MHS; Elizabeth A. Regan, MD, PhD; Stephanie Santorico, PhD; Edwin K. Silverman, MD, PhD; Emily S. Wan, MD; Jin Zhou, PhD
Genotyping Cores: Genome-Wide Core: Terri Beaty, PhD; Candidate Genotyping Core: Craig P. Hersh, MD, MPH; Edwin K. Silverman, MD, PhD
Imaging Core: David Lynch, MB; Mustafa Al Qaisi, MD; Jaleh Akhavan; Christian W. Cox, MD; Harvey O. Coxson, PhD; Deanna Cusick; Jennifer G. Dy, PhD; Shoshana Ginsburg, MS; Eric A. Hoffman, PhD; Philip F. Judy, PhD; Alex Kluiber; Alexander McKenzie; John D. Newell, Jr., MD; John J. Reilly, Jr., MD; James Ross, MSc; Raul San Jose Estepar, PhD; Joyce D. Schroeder, MD; Jered Sieren; Arkadiusz Sitek, PhD; Douglas Stinson; Edwin van Beek, MD, PhD, MEd; George R. Washko, MD; Jordan Zach
PFT QA Core: Robert Jensen, PhD; E. Rand Sutherland, MD
Biological Repository, Johns Hopkins University, Baltimore, MD: Homayoon Farzadegan, PhD: Samantha Bragan; Stacey Cayetano
We further wish to acknowledge the COPDGene Investigators from the participating Clinical Centers:
Ann Arbor VA: Jeffrey Curtis, MD, Ella Kazerooni, MD
Baylor College of Medicine, Houston, TX: Nicola Hanania, MD, MS, Philip Alapat, MD, Venkata Bandi, MD, Kalpalatha Guntupalli, MD, Elizabeth Guy, MD, Antara Mallampalli, MD, Charles Trinh, MD, Mustafa Atik, MD, Hasan Al-Azzawi, MD, Marc Willis, DO, Susan Pinero, MD, Linda Fahr, MD, Arun Nachiappan, MD, Collin Bray, MD, L. Alexander Frigini, MD, Carlos Farinas, MD, David Katz, MD, Jose Freytes, MD, Anne Marie Marciel, MD
Brigham and Women’s Hospital, Boston, MA: Dawn DeMeo, MD, MPH, Craig Hersh, MD, MPH, George Washko, MD, Francine Jacobson, MD, MPH, Hiroto Hatabu, MD, PhD, Peter Clarke, MD, Ritu Gill, MD, Andetta Hunsaker, MD, Beatrice Trotman-Dickenson, MBBS, Rachna Madan, MD
Columbia University, New York, NY: R. Graham Barr, MD, DrPH, Byron Thomashow, MD, John Austin, MD, Belinda D’Souza, MD
Duke University Medical Center, Durham, NC: Neil MacIntyre, Jr., MD, Lacey Washington, MD, H Page McAdams, MD
Reliant Medical Group, Worcester, MA: Richard Rosiello, MD, Timothy Bresnahan, MD, Joseph Bradley, MD, Sharon Kuong, MD, Steven Meller, MD, Suzanne Roland, MD
Health Partners Research Foundation, Minneapolis, MN: Charlene McEvoy, MD, MPH, Joseph Tashjian, MD
Johns Hopkins University, Baltimore, MD: Robert Wise, MD, Nadia Hansel, MD, MPH, Robert Brown, MD, Gregory Diette, MD, Karen Horton, MD
Los Angeles Biomedical Research Institute at Harbor UCLA Medical Center, Los Angeles, CA: Richard Casaburi, MD, Janos Porszasz, MD, PhD, Hans Fischer, MD, PhD, Matt Budoff, MD, Mehdi Rambod, MD
Michael E. DeBakey VAMC, Houston, TX: Amir Sharafkhaneh, MD, Charles Trinh, MD, Hirani Kamal, MD, Roham Darvishi, MD, Marc Willis, DO, Susan Pinero, MD, Linda Fahr, MD, Arun Nachiappan, MD, Collin Bray, MD, L. Alexander Frigini, MD, Carlos Farinas, MD, David Katz, MD, Jose Freytes, MD, Anne Marie Marciel, MD
Minneapolis VA: Dennis Niewoehner, MD, Quentin Anderson, MD, Kathryn Rice, MD, Audrey Caine, MD
Morehouse School of Medicine, Atlanta, GA: Marilyn Foreman, MD, MS, Gloria Westney, MD, MS, Eugene Berkowitz, MD, PhD
National Jewish Health, Denver, CO: Russell Bowler, MD, PhD, David Lynch, MB, Joyce Schroeder, MD, Valerie Hale, MD, John Armstrong, II, MD, Debra Dyer, MD, Jonathan Chung, MD, Christian Cox, MD
Temple University, Philadelphia, PA: Gerard Criner, MD, Victor Kim, MD, Nathaniel Marchetti, DO, Aditi Satti, MD, A. James Mamary, MD, Robert Steiner, MD, Chandra Dass, MD, Libby Cone, MD
University of Alabama, Birmingham, AL: William Bailey, MD, Mark Dransfield, MD, Michael Wells, MD, Surya Bhatt, MD, Hrudaya Nath, MD, Satinder Singh, MD
University of California, San Diego, CA: Joe Ramsdell, MD, Paul Friedman, MD
University of Iowa, Iowa City, IA: Alejandro Cornellas, MD, John Newell, Jr., MD, Edwin JR van Beek, MD, PhD
University of Michigan, Ann Arbor, MI: Fernando Martinez, MD, MeiLan Han, MD, Ella Kazerooni, MD
University of Minnesota, Minneapolis, MN: Christine Wendt, MD, Tadashi Allen, MD
University of Pittsburgh, Pittsburgh, PA: Frank Sciurba, MD, Joel Weissfeld, MD, MPH, Carl Fuhrman, MD, Jessica Bon, MD, Danielle Hooper, MD
University of Texas Health Science Center at San Antonio, San Antonio, TX: Antonio Anzueto, MD, Sandra Adams, MD, Carlos Orozco, MD, Mario Ruiz, MD, Amy Mumbower, MD, Ariel Kruger, MD, Carlos Restrepo, MD, Michael Lane, MD
We acknowledge and thank the ECLIPSE investigators:
Investigators — Bulgaria: Y. Ivanov, Pleven; K. Kostov, Sofia. Canada: J. Bourbeau, Montreal; M. Fitzgerald, Vancouver, BC; P. Hernandez, Halifax, NS; K. Killian, Hamilton, ON; R. Levy, Vancouver, BC; F. Maltais, Montreal; D. O'Donnell, Kingston, ON. Czech Republic: J. Krepelka, Prague. Denmark: J. Vestbo, Hvidovre. The Netherlands: E. Wouters, Horn-Maastricht. New Zealand: D. Quinn, Wellington. Norway: P. Bakke, Bergen. Slovenia: M. Kosnik, Golnik. Spain: A. Agusti, J. Sauleda, P. de Mallorca. Ukraine: Y. Feschenko, V. Gavrisyuk, L. Yashina, Kiev; N. Monogarova, Donetsk. United Kingdom: P. Calverley, Liverpool; D. Lomas, Cambridge; W. MacNee, Edinburgh; D. Singh, Manchester; J. Wedzicha, London. United States: A. Anzueto, San Antonio, TX; S. Braman, Providence, RI; R. Casaburi, Torrance CA; B. Celli, Boston; G. Giessel, Richmond, VA; M. Gotfried, Phoenix, AZ; G. Greenwald, Rancho Mirage, CA; N. Hanania, Houston; D. Mahler, Lebanon, NH; B. Make, Denver; S. Rennard, Omaha, NE; C. Rochester, New Haven, CT; P. Scanlon, Rochester, MN; D. Schuller, Omaha, NE; F. Sciurba, Pittsburgh; A. Sharafkhaneh, Houston; T. Siler, St. Charles, MO; E. Silverman, Boston; A. Wanner, Miami; R. Wise, Baltimore; R. ZuWallack, Hartford, CT.
Steering Committee: H. Coxson (Canada), C. Crim (GlaxoSmithKline, USA), L. Edwards (GlaxoSmithKline, USA), D. Lomas (UK), W. MacNee (UK), E. Silverman (USA), R. Tal Singer (Co-chair, GlaxoSmithKline, USA), J. Vestbo (Co-chair, Denmark), J. Yates (GlaxoSmithKline, USA).
Scientific Committee: A. Agusti (Spain), P. Calverley (UK), B. Celli (USA), C. Crim (GlaxoSmithKline, USA), B. Miller (GlaxoSmithKline, USA), W. MacNee (Chair, UK), S. Rennard (USA), R. Tal-Singer (GlaxoSmithKline, USA), E. Wouters (The Netherlands), J. Yates (GlaxoSmithKline, USA).
Abbreviations
- AA
African American
- ECLIPSE
Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints
- GOLD
Global Initiative for chronic Obstructive Lung Disease
- GWAS
Genome-wide association study
- NHW
non-Hispanic white
- SNP
Single nucleotide polymorphism
- TLC
Total lung capacity
Additional file
Methods – Variable definitions. Table S1. Clinical and radiological characteristics according to genotypes of rs114929486 among non-Hispanic white COPDGene subjects with COPD (GOLD 2-4) (N = 2,652). Table S2. Baseline characteristics of subjects with emphysema-predominant COPD. Table S3. Meta-analysis of total lung capacity measured by CT in emphysema-predominant COPD. Table S4. Baseline characteristics of COPD subjects with chronic bronchitis. Table S5. Meta-analysis results of total lung capacity measured by CT in COPD subjects with chronic bronchitis. Table S6. Baseline characteristics of COPD subjects and smokers with normal spirometry. Table S7. Results of a genome-wide association study (GWAS) for total lung capacity measured by CT in smokers with normal spirometry of ECLIPSE study. Table S8. Results of a GWAS for total lung capacity measured by CT in both of COPD subjects and smokers with normal spirometry of COPDGene Study. Table S9. Results of a GWAS for total lung capacity measured by CT in both of COPD subjects and smokers with normal spirometry of GenKOLS study. Figure S1. (A) The quantile–quantile plot and (B) Manhattan plot of –log10 P values for the three-cohort meta-analysis for total lung capacity measured by CT in COPD subjects with ≥10% emphysema. Figure S2. (A) The quantile–quantile plot and (B) Manhattan plot of –log10 P values for the three-cohort meta-analysis for total lung capacity measured by CT in COPD subjects with chronic bronchitis. Figure S3. (A) The quantile–quantile plot and (B) Manhattan plot of –log10 P values for the three-cohort meta-analysis for total lung capacity measured by CT in smokers with normal spirometry. Figure S4. (A) The quantile–quantile plot and (B) Manhattan plot of –log10 P values for the three-cohort meta-analysis for total lung capacity measured by CT in both of smokers with normal spirometry and COPD subjects.
Footnotes
Competing interests
In the past 3 years, Drs Lee, McDonald, Wan, Castaldi, Hunninghake, Marchetti, Lynch, Crapo, Lomas, Coxson and Bakke have no competing interests related to the subject of the manuscript. In the past 3 years, Dr Silverman reports grants from NIH, grants and other support from COPD Foundation, grants and personal fees from GlaxoSmithKline, during the conduct of the study; personal fees from Merck and travel support from Novartis, outside the submitted work. In the past 3 years, Dr Cho has received NIH grant and consulted for Merck (outside the submitted work). In the past 3 years, Dr Hersh has received NIH grant and consulted for Novartis, CSL Behring (outside the submitted work).
Authors’ contributions
Study concept and design: JHL, EKS, CPH. Data acquisition and quality control: MHC, PJC, CPH, ESW, JDC, NM, DAL, DAL, EKS, PSB. Analysis and interpretation: JHL, MHC, MNM, PJC, CPH, ESW, GMH, HOC, PSB. Critical revision of manuscript: all authors. All authors read and approved the final manuscript.
Contributor Information
Jin Hwa Lee, Email: jinhwalee@ewha.ac.kr.
Merry-Lynn N McDonald, Email: remrm@channing.harvard.edu.
Michael H Cho, Email: remhc@channing.harvard.edu.
Emily S Wan, Email: reesw@channing.harvard.edu.
Peter J Castaldi, Email: repjc@channing.harvard.edu.
Gary M Hunninghake, Email: GHUNNINGHAKE@partners.org.
Nathaniel Marchetti, Email: nathaniel.marchetti@tuhs.temple.edu.
David A Lynch, Email: lynchd@njhealth.org.
James D Crapo, Email: CrapoJ@njhealth.org.
David A Lomas, Email: d.lomas@ucl.ac.uk.
Harvey O Coxson, Email: harvey.coxson@vch.ca.
Per S Bakke, Email: Per.Bakke@k2.uib.no.
Edwin K Silverman, Email: ed.silverman@channing.harvard.edu.
Craig P Hersh, Email: craig.hersh@channing.harvard.edu.
References
- 1.The state of US health, 1990-2010: burden of diseases, injuries, and risk factors.JAMA 2013, 310:591–608. [DOI] [PMC free article] [PubMed]
- 2.Hallberg J, Dominicus A, Eriksson UK, Gerhardsson de Verdier M, Pedersen NL, Dahlback M, Nihlen U, Higenbottam T, Svartengren M. Interaction between smoking and genetic factors in the development of chronic bronchitis. Am J Respir Crit Care Med. 2008;177:486–490. doi: 10.1164/rccm.200704-565OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vestbo J, Hurd SS, Agusti AG, Jones PW, Vogelmeier C, Anzueto A, Barnes PJ, Fabbri LM, Martinez FJ, Nishimura M, Stockley RA, Sin DD, Rodriguez-Roisin R. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2013;187:347–365. doi: 10.1164/rccm.201204-0596PP. [DOI] [PubMed] [Google Scholar]
- 4.Agusti A, Calverley PM, Celli B, Coxson HO, Edwards LD, Lomas DA, MacNee W, Miller BE, Rennard S, Silverman EK, Tal-Singer R, Wouters E, Yates JC, Vestbo J, Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) investigators Characterisation of COPD heterogeneity in the ECLIPSE cohort. Respir Res. 2010;11:122. [Google Scholar]
- 5.O'Donnell DE. Hyperinflation, dyspnea, and exercise intolerance in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2006;3:180–184. doi: 10.1513/pats.200508-093DO. [DOI] [PubMed] [Google Scholar]
- 6.Casaburi R, Porszasz J. Reduction of hyperinflation by pharmacologic and other interventions. Proc Am Thorac Soc. 2006;3:185–189. doi: 10.1513/pats.200508-095DO. [DOI] [PubMed] [Google Scholar]
- 7.Casanova C, Cote C, de Torres JP, Aguirre-Jaime A, Marin JM, Pinto-Plata V, Celli BR. Inspiratory-to-total lung capacity ratio predicts mortality in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;171:591–597. doi: 10.1164/rccm.200407-867OC. [DOI] [PubMed] [Google Scholar]
- 8.Bancalari E, Clausen J. Pathophysiology of changes in absolute lung volumes. Eur Respir J. 1998;12:248–258. doi: 10.1183/09031936.98.12010248. [DOI] [PubMed] [Google Scholar]
- 9.Cho MH, Castaldi PJ, Wan ES, Siedlinski M, Hersh CP, DeMeo DL, Himes BE, Sylvia JS, Klanderman BJ, Ziniti JP, Lange C, Litonjua AA, Sparrow D, Regan EA, Make BJ, Hokanson JE, Murray T, Hetmanski JB, Pillai SG, Kong X, Anderson WH, Tal-Singer R, Lomas DA, Coxson HO, Edwards LD, MacNee W, Vestbo J, Yates JC, Agusti A, Calverley PM, et al. A genome-wide association study of COPD identifies a susceptibility locus on chromosome 19q13. Hum Mol Genet. 2012;21:947–957. doi: 10.1093/hmg/ddr524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Regan EA, Hokanson JE, Murphy JR, Make B, Lynch DA, Beaty TH, Curran-Everett D, Silverman EK, Crapo JD. Genetic epidemiology of COPD (COPDGene) study design. COPD. 2010;7:32–43. doi: 10.3109/15412550903499522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vestbo J, Anderson W, Coxson HO, Crim C, Dawber F, Edwards L, Hagan G, Knobil K, Lomas DA, MacNee W, Silverman EK, Tal-Singer R, ECLIPSE investigators Evaluation of COPD Longitudinally to Identify Predictive Surrogate End-points (ECLIPSE) Eur Respir J. 2008;31:869–873. doi: 10.1183/09031936.00111707. [DOI] [PubMed] [Google Scholar]
- 12.Zhu G, Warren L, Aponte J, Gulsvik A, Bakke P, Anderson WH, Lomas DA, Silverman EK, Pillai SG. The SERPINE2 gene is associated with chronic obstructive pulmonary disease in two large populations. Am J Respir Crit Care Med. 2007;176:167–173. doi: 10.1164/rccm.200611-1723OC. [DOI] [PubMed] [Google Scholar]
- 13.Grydeland TB, Dirksen A, Coxson HO, Pillai SG, Sharma S, Eide GE, Gulsvik A, Bakke PS. Quantitative computed tomography: emphysema and airway wall thickness by sex, age and smoking. Eur Respir J. 2009;34:858–865. doi: 10.1183/09031936.00167908. [DOI] [PubMed] [Google Scholar]
- 14.Cho MH, Boutaoui N, Klanderman BJ, Sylvia JS, Ziniti JP, Hersh CP, DeMeo DL, Hunninghake GM, Litonjua AA, Sparrow D, Lange C, Won S, Murphy JR, Beaty TH, Regan EA, Make BJ, Hokanson JE, Crapo JD, Kong X, Anderson WH, Tal-Singer R, Lomas DA, Bakke P, Gulsvik A, Pillai SG, Silverman EK. Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat Genet. 2010;42:200–202. doi: 10.1038/ng.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho MH, McDonald ML, Zhou X, Mattheisen M, Castaldi PJ, Hersh CP, Demeo DL, Sylvia JS, Ziniti J, Laird NM, Lange C, Litonjua AA, Sparrow D, Casaburi R, Barr RG, Regan EA, Make BJ, Hokanson JE, Lutz S, Dudenkov TM, Farzadegan H, Hetmanski JB, Tal-Singer R, Lomas DA, Bakke P, Gulsvik A, Crapo JD, Silverman EK, Beaty TH, NETT Genetics, ICGN, ECLIPSE and COPDGene Investigators Risk loci for chronic obstructive pulmonary disease: a genome-wide association study and meta-analysis. Lancet Respir Med. 2014;2:214–225. doi: 10.1016/S2213-2600(14)70002-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gietema HA, Edwards LD, Coxson HO, Bakke PS. Impact of emphysema and airway wall thickness on quality of life in smoking-related COPD. Respir Med. 2013;107:1201–1209. doi: 10.1016/j.rmed.2013.04.016. [DOI] [PubMed] [Google Scholar]
- 17.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34:816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet. 2012;44:955–959. doi: 10.1038/ng.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kong X, Cho MH, Anderson W, Coxson HO, Muller N, Washko G, Hoffman EA, Bakke P, Gulsvik A, Lomas DA, Silverman EK, Pillai SG, ECLIPSE Study NETT Investigators Genome-wide association study identifies BICD1 as a susceptibility gene for emphysema. Am J Respir Crit Care Med. 2011;183:43–49. doi: 10.1164/rccm.201004-0541OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pillai SG, Ge D, Zhu G, Kong X, Shianna KV, Need AC, Feng S, Hersh CP, Bakke P, Gulsvik A, Ruppert A, Lødrup Carlsen KC, Roses A, Anderson W, Rennard SI, Lomas DA, Silverman EK, Goldstein DB, ICGN Investigators A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet. 2009;5:e1000421. doi: 10.1371/journal.pgen.1000421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Bakker PI, Ferreira MA, Jia X, Neale BM, Raychaudhuri S, Voight BF. Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum Mol Genet. 2008;17:R122–R128. doi: 10.1093/hmg/ddn288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ. 2003;327:557–560. doi: 10.1136/bmj.327.7414.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han B, Eskin E. Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am J Hum Genet. 2011;88:586–598. doi: 10.1016/j.ajhg.2011.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341X.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 28.Aulchenko YS, Ripke S, Isaacs A, van Duijn CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007;23:1294–1296. doi: 10.1093/bioinformatics/btm108. [DOI] [PubMed] [Google Scholar]
- 29.Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, Abecasis GR, Willer CJ. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–2337. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.The Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Failly M, Bartoloni L, Letourneau A, Munoz A, Falconnet E, Rossier C, de Santi MM, Santamaria F, Sacco O, DeLozier-Blanchet CD, Lazor R, Blouin JL. Mutations in DNAH5 account for only 15% of a non-preselected cohort of patients with primary ciliary dyskinesia. J Med Genet. 2009;46:281–286. doi: 10.1136/jmg.2008.061176. [DOI] [PubMed] [Google Scholar]
- 32.Hornef N, Olbrich H, Horvath J, Zariwala MA, Fliegauf M, Loges NT, Wildhaber J, Noone PG, Kennedy M, Antonarakis SE, Blouin JL, Bartoloni L, Nüsslein T, Ahrens P, Griese M, Kuhl H, Sudbrak R, Knowles MR, Reinhardt R, Omran H. DNAH5 mutations are a common cause of primary ciliary dyskinesia with outer dynein arm defects. Am J Respir Crit Care Med. 2006;174:120–126. doi: 10.1164/rccm.200601-084OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leigh MW, Pittman JE, Carson JL, Ferkol TW, Dell SD, Davis SD, Knowles MR, Zariwala MA. Clinical and genetic aspects of primary ciliary dyskinesia/Kartagener syndrome. Genet Med. 2009;11:473–487. doi: 10.1097/GIM.0b013e3181a53562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Green K, Buchvald FF, Marthin JK, Hanel B, Gustafsson PM, Nielsen KG. Ventilation inhomogeneity in children with primary ciliary dyskinesia. Thorax. 2012;67:49–53. doi: 10.1136/thoraxjnl-2011-200726. [DOI] [PubMed] [Google Scholar]
- 35.Stanescu D. Small airways obstruction syndrome. Chest. 1999;116:231–233. doi: 10.1378/chest.116.1.231. [DOI] [PubMed] [Google Scholar]
- 36.Pifferi M, Bush A, Pioggia G, Caramella D, Tartarisco G, Di CM, Zangani M, Chinellato I, Maggi F, Tezza G, Macchia P, Boner A. Evaluation of pulmonary disease using static lung volumes in primary ciliary dyskinesia. Thorax. 2012;67:993–999. doi: 10.1136/thoraxjnl-2011-200137. [DOI] [PubMed] [Google Scholar]
- 37.Hogg JC, Macklem PT, Thurlbeck WM. Site and nature of airway obstruction in chronic obstructive lung disease. N Engl J Med. 1968;278:1355–1360. doi: 10.1056/NEJM196806202782501. [DOI] [PubMed] [Google Scholar]
- 38.McDonough JE, Yuan R, Suzuki M, Seyednejad N, Elliott WM, Sanchez PG, Wright AC, Gefter WB, Litzky L, Coxson HO, Paré PD, Sin DD, Pierce RA, Woods JC, McWilliams AM, Mayo JR, Lam SC, Cooper JD, Hogg JC. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med. 2011;365:1567–1575. doi: 10.1056/NEJMoa1106955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yanai M, Sekizawa K, Ohrui T, Sasaki H, Takishima T. Site of airway obstruction in pulmonary disease: direct measurement of intrabronchial pressure. J Appl Physiol (1985) 1992;72:1016–1023. doi: 10.1152/jappl.1992.72.3.1016. [DOI] [PubMed] [Google Scholar]
- 40.Buro-Auriemma LJ, Salit J, Hackett NR, Walters MS, Strulovici-Barel Y, Staudt MR, Fuller J, Mahmoud M, Stevenson CS, Hilton H, Ho MW, Crystal RG. Cigarette smoking induces small airway epithelial epigenetic changes with corresponding modulation of gene expression. Hum Mol Genet. 2013;22:4726–4738. doi: 10.1093/hmg/ddt326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blair DR, Lyttle CS, Mortensen JM, Bearden CF, Jensen AB, Khiabanian H, Melamed R, Rabadan R, Bernstam EV, Brunak S, Jensen LJ, Nicolae D, Shah NH, Grossman RL, Cox NJ, White KP, Rzhetsky A. A nondegenerate code of deleterious variants in Mendelian loci contributes to complex disease risk. Cell. 2013;155:70–80. doi: 10.1016/j.cell.2013.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Flintoft L. Disease genetics: A Mendelian code for complex disease. Nat Rev Genet. 2013;14:746. doi: 10.1038/nrg3599. [DOI] [PubMed] [Google Scholar]
- 43.Knowlden S, Georas SN. The Autotaxin-LPA Axis Emerges as a Novel Regulator of Lymphocyte Homing and Inflammation. J Immunol. 2014;192:851–857. doi: 10.4049/jimmunol.1302831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oikonomou N, Mouratis MA, Tzouvelekis A, Kaffe E, Valavanis C, Vilaras G, Karameris A, Prestwich GD, Bouros D, Aidinis V. Pulmonary autotaxin expression contributes to the pathogenesis of pulmonary fibrosis. Am J Respir Cell Mol Biol. 2012;47:566–574. doi: 10.1165/rcmb.2012-0004OC. [DOI] [PubMed] [Google Scholar]
- 45.Tager AM. Autotaxin emerges as a therapeutic target for idiopathic pulmonary fibrosis: limiting fibrosis by limiting lysophosphatidic acid synthesis. Am J Respir Cell Mol Biol. 2012;47:563–565. doi: 10.1165/rcmb.2012-0235ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park GY, Lee YG, Berdyshev E, Nyenhuis S, Du J, Fu P, Gorshkova IA, Li Y, Chung S, Karpurapu M, Deng J, Ranjan R, Xiao L, Jaffe HA, Corbridge SJ, Kelly EA, Jarjour NN, Chun J, Prestwich GD, Kaffe E, Ninou I, Aidinis V, Morris AJ, Smyth SS, Ackerman SJ, Natarajan V, Christman JW. Autotaxin production of lysophosphatidic acid mediates allergic asthmatic inflammation. Am J Respir Crit Care Med. 2013;188:928–940. doi: 10.1164/rccm.201306-1014OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheng HY, Dong A, Panchatcharam M, Mueller P, Yang F, Li Z, Mills G, Chun J, Morris AJ, Smyth SS. Lysophosphatidic acid signaling protects pulmonary vasculature from hypoxia-induced remodeling. Arterioscler Thromb Vasc Biol. 2012;32:24–32. doi: 10.1161/ATVBAHA.111.234708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zaporozhan J, Ley S, Eberhardt R, Weinheimer O, Iliyushenko S, Herth F, Kauczor HU. Paired inspiratory/expiratory volumetric thin-slice CT scan for emphysema analysis: comparison of different quantitative evaluations and pulmonary function test. Chest. 2005;128:3212–3220. doi: 10.1378/chest.128.5.3212. [DOI] [PubMed] [Google Scholar]
- 49.Coxson HO, Nasute Fauerbach PV, Storness-Bliss C, Muller NL, Cogswell S, Dillard DH, Finger CL, Springmeyer SC. Computed tomography assessment of lung volume changes after bronchial valve treatment. Eur Respir J. 2008;32:1443–1450. doi: 10.1183/09031936.00056008. [DOI] [PubMed] [Google Scholar]
- 50.Gierada DS, Hakimian S, Slone RM, Yusen RD. MR analysis of lung volume and thoracic dimensions in patients with emphysema before and after lung volume reduction surgery. Am J Roentgenol. 1998;170:707–714. doi: 10.2214/ajr.170.3.9490958. [DOI] [PubMed] [Google Scholar]
- 51.Kauczor HU, Heussel CP, Fischer B, Klamm R, Mildenberger P, Thelen M. Assessment of lung volumes using helical CT at inspiration and expiration: comparison with pulmonary function tests. Am J Roentgenol. 1998;171:1091–1095. doi: 10.2214/ajr.171.4.9763003. [DOI] [PubMed] [Google Scholar]
- 52.O'Donnell CR, Bankier AA, Stiebellehner L, Reilly JJ, Brown R, Loring SH. Comparison of plethysmographic and helium dilution lung volumes: which is best for COPD? Chest. 2010;137:1108–1115. doi: 10.1378/chest.09-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]