

Abstract
Zinc ion homeostasis plays an important role in human cutaneous biology where it is involved in epidermal differentiation and barrier function, inflammatory and antimicrobial regulation, and wound healing. Zinc-based compounds designed for topical delivery therefore represent an important class of cutaneous therapeutics. Zinc pyrithione (ZnPT) is an FDA-approved microbicidal agent used worldwide in over-the-counter topical antimicrobials, and has also been examined as an investigational therapeutic targeting psoriasis and UVB-induced epidermal hyperplasia. Recently, we have demonstrated that cultured primary human skin keratinocytes display an exquisite sensitivity to nanomolar ZnPT concentrations causing induction of heat shock response gene expression and poly(ADP-ribose) polymerase (PARP)-dependent cell death (Cell Stress Chaperones 15:309–322, 2010). Here we demonstrate that ZnPT causes rapid accumulation of intracellular zinc in primary keratinocytes as observed by quantitative fluorescence microscopy and inductively coupled plasma mass spectrometry (ICP-MS), and that PARP activation, energy crisis, and genomic impairment are all antagonized by zinc chelation. In epidermal reconstructs (EpiDerm™) exposed to topical ZnPT (0.1–2% in Vanicream™), ICP-MS demonstrated rapid zinc accumulation, and expression array analysis demonstrated upregulation of stress response genes encoding metallothionein-2A (MT2A), heat shock proteins (HSPA6, HSPA1A, HSPB5, HSPA1L, DNAJA1, HSPH1, HSPD1, HSPE1), antioxidants (SOD2, GSTM3, HMOX1), and the cell cycle inhibitor p21 (CDKN1A). IHC analysis of ZnPT-treated EpiDerm™ confirmed upregulation of Hsp70 and TUNEL-positivity. Taken together our data demonstrate that ZnPT impairs zinc ion homeostasis and upregulates stress response gene expression in primary keratinocytes and reconstructed human epidermis, activities that may underlie therapeutic and toxicological effects of this topical drug.
Keywords: Zinc pyrithione, Keratinocyte, Reconstructed epidermis, Heat shock response, HSPA1A, ICP-MS
Introduction
Zinc is an essential trace element with important cellular functions dependent on its activity as metal-cofactor of various enzymes and structural component of transcription factors and other zinc finger-containing DNA binding proteins (Murakami and Hirano 2008; Plum et al. 2010). Zinc ion homeostasis plays an important role in cutaneous biology where it modulates epidermal barrier function (Takahashi et al. 2008), epithelial wound healing and tissue regeneration (Sharir et al. 2010), and inflammatory and antimicrobial regulation (Jarrousse et al. 2007).
Zinc-based compounds designed for topical delivery represent an important class of cutaneous therapeutics designed for photoprotective, antimicrobial, and anti-inflammatory intervention (Guthery et al. 2005; Bae et al. 2010; Beasley and Meyer 2010). Zinc pyrithione (ZnPT; CAS# 13463-41-7) is a 1:2 complex between a central zinc atom and the membrane permeable ionophore pyrithione (N-hydroxy-2-pyridinethione) (Barnett et al. 1977). This FDA-approved microbicidal agent is used worldwide in over-the-counter (OTC) topical antimicrobials and cosmetic consumer products including anti-dandruff shampoos where typical ZnPT concentrations are in the range of 1–2% (w/v) (Pierard-Franchimont et al. 2002; Bailey et al. 2003; Guthery et al. 2005). Following topical application in human skin, epidermal deposition and retention of the lipophilic metal chelate ZnPT has been demonstrated (Rutherford and Black 1969; Leyden et al. 1979). Moreover, percutaneous penetration of ZnPT has been observed in relevant animal models of human skin (Okamoto et al. 1967; Howes and Black 1975; Gibson and Calvin 1978; Guthery et al. 2005).
In the context of cutaneous pathology beyond antimicrobial intervention, ZnPT has been used as an investigational antipsoriatic drug and has also been examined as an experimental therapeutic for topical treatment of UVB-induced epidermal hyperplasia (Rowlands and Danby 2000; Cho et al. 2010). Accumulative evidence points towards a significant therapeutic potential of ZnPT and related zinc ionophores for antiviral and anticancer intervention (Magda et al. 2008; Ding and Lind 2009; Krenn et al. 2009). For example, systemic ZnPT and ZnPT-derived zinc ionophores target cancer cells by zinc-dependent induction of cell death as demonstrated in B cell lymphoma in vitro and A549 lung cancer in murine xenograft models (Magda et al. 2008). Recently, we have observed that exposure of primary human skin keratinocytes and melanocytes to submicromolar concentrations of ZnPT results in pronounced upregulation of heat shock response gene expression at the mRNA and protein level (Lamore et al. 2010a). Moreover, rapid loss of genomic integrity and induction of energy crisis occurred in response to ZnPT treatment. Further analysis revealed the causative role of poly(ADP-ribose) polymerase-1 (PARP-1) activation in ZnPT-induced ATP depletion and cell death in human keratinocytes, a finding independently confirmed using PARP-1 knockout mouse embryonic fibroblasts that were resistant to ATP depletion and cytotoxicity resulting from ZnPT exposure (Lamore et al. 2010a).
Given the significant therapeutic and toxicological potential of topical ZnPT in human skin, we felt that the molecular activity of ZnPT should be characterized further in a relevant model of human epidermis (Bause et al. 2009). First, using primary epidermal keratinocytes we demonstrate that ZnPT exposure causes rapid zinc ion dysregulation, impairment of genomic integrity, PARP activation, and PARP-dependent energy crisis in primary human skin keratinocytes, effects that are all antagonized by zinc ion chelation. Using reconstructed fully differentiated human epidermis as an organotypic tissue model we then provide first experimental evidence that topical ZnPT exposure at pharmaceutical doses causes epidermal zinc ion overload with dramatic upregulation of stress response gene expression.
Materials and methods
Chemicals
All chemicals including ZnPT (CAS Number: 13463-41-7) were from Sigma Chemical Co. A stock solution of ZnPT (1 mM) was prepared in DMSO. The PARP inhibitor PJ-34 was from Enzo Life Sciences Inc. Vanicream™ (FDA-NDC 45334-300-01) was obtained from Pharmaceutical Specialties, Inc.
General cell culture
Primary normal human epidermal keratinocytes (NHEK; neonatal HEKn-APF from Cascade Biologics) were cultured using Epilife medium supplemented with EDGS growth supplement. Cells were passaged using recombinant trypsin/EDTA and defined trypsin inhibitor and maintained in a humidified incubator (37°C, 5% CO2).
Epiderm™ skin equivalents: maintenance and sample preparation
After shipment refrigerated EpiDerm™ inserts (EPI-200, 9 mm diameter; MaTek, Corp.) were equilibrated in 6-well format (5% CO2, 37°C, 1 h; 0.9 ml of EPI-200-ASY media per well) followed by change of medium. EpiDerm™ tissues were topically treated by applying ZnPT [0.1–2% (w/w) in Vanicream™, 90 mg total] or Vanicream™ only (90 mg) and cultured at 37°C for the indicated amount of time after which cream was gently removed using a cotton swab, and the apical side of the tissue was rinsed with PBS (500 μl, ten times).
The following sample preparation regimens were applied: (I) For zinc determination by ICP-MS, tissues were treated with ZnPT [2% (w/w); 3 h exposure time). After cream removal, tissue was rinsed in PBS and then dissolved immediately in nitric acid (0.5 ml, 85°C, 3 h) followed by ICP-MS analysis. (II) For expression array analysis, tissues were treated with ZnPT [2% (w/w)]. ZnPT exposure time was 24 h (or 1 h exposure time followed by removal of ZnPT and further incubation for 23 h at 37°C, 5% CO2). The tissue equivalent was then rinsed in PBS and homogenized (300 μl RNeasy lysis buffer). Homogenates were centrifuged (16,000×g, 10 min) to pellet tissue debris, and the supernatant was retained for RNA isolation using the RNeasy kit (Qiagen) according to the manufacturer’s instructions. (III) For IHC and TUNEL analysis, tissues were treated with ZnPT [0.1 or 2% (w/w); 24 h exposure time). After cream removal tissues were fixed in neutral buffered formalin (10%, 24 h) and then placed in 70% ethanol followed by paraffin embedment.
Cell death analysis
Viability and induction of cell death (early and late apoptosis/necrosis) were examined by annexin-V-FITC/propidium iodide (PI) dual staining of cells followed by flow cytometric analysis using an apoptosis detection kit according to the manufacturer’s specifications (APO-AF, Sigma) as published previously (Lamore et al. 2010c).
Cellular ATP assay
Cells were seeded at 5,000 cells/well of an opaque 96-well plate. After 24 h, cells were treated with test compound. After 6 h ATP content per well was determined using the CellTiter-Glo luminescent assay (Promega) according to the manufacturer’s instructions (Lamore et al. 2010a). Data are normalized to ATP content in untreated cells and expressed as means ± SD (n = 3).
Comet assay (alkaline single cell gel electrophoresis)
The alkaline comet assay was performed on according to the manufacturer’s instructions (Trevigen) as published recently (Wondrak et al. 2003; Cabello et al. 2009b). NHEKs (100,000) were seeded one day prior to experimentation. After treatment, cells were suspended in PBS, mixed 1:10 with low-melting-point agarose and spread on pretreated microscope slides. After drying, slides were immersed in icecold lysis solution containing 10% DMSO and incubated at 4°C for 45 min. To allow DNA unwinding and expression of alkali-labile sites, slides were exposed to alkaline buffer (1 mM EDTA, 300 mM NaOH, pH > 13) for 45 min. Electrophoresis was conducted in the same alkaline buffer for 20 min at 300 mA. Slides were rinsed in ddH2O, fixed in 70% ethanol and dried at 32°C. Nucleoids were stained with SYBR® Green and visualized (Olympus IX70 digital fluorescence microscope; fluorescein filter) followed by data analysis using the free CASP software (casplab.com). At least 100 tail moments for each treatment group were calculated (mean ± SEM, n = 3).
Poly(ADP-ribose) (PAR) immunoblot analysis
A recently published method was used (Lamore et al. 2010a). One day before treatment, 1 × 106 cells were seeded in a 100 mm dish. After treatment cells were washed with PBS, lysed in 1× SDS-PAGE sample buffer, and heated for 3 min at 95°C prior to separation by 4–12% SDS-PAGE (Bio-Rad). After separation, proteins were transferred electrophoretically to a nitrocellulose membrane. Equal protein loading was confirmed by Ponceau S stain (0.1% in 1% acetic acid). After blocking in 5% milk-TBST, mouse anti-PAR monoclonal antibody (Trevigen) was used 1:1000 in 5% milk-TBST overnight at 4°C. Incubation with HRP-conjugated goat anti-mouse antibody (Jackson Immunoresearch Laboratories) at 1:20,000 dilution was then followed by visualization using enhanced chemiluminescence reagents.
Zinquin fluorescence
Zinquin ethyl ester (Sigma) was used as a probe for intracellular free Zn2+ according to a published procedure (Pirev et al. 2008). NHEKs (10,000) were seeded on microcover glasses (VWR) one day prior to experimentation. After rinsing with PBS, Zinquin ethyl ester (10 μM in PBS; Alexis) was added followed by incubation in the dark (20 min, 37°C, 5% CO2). After exchange of PBS cells were then exposed to ZnPT for the indicated time, and rinsed coverslips were then mounted onto slides with glycerol. Formation of Zinquin–Zn2+ complexes was monitored by fluorescence microscopy (Olympus IX70 equipped with a 359/461 nm (λex/λem) filter. Quantitative image analysis was performed using ImageJ software (rsbweb.nih.gov/ij).
Quantitative total zinc analysis of NHEKs and EpiDerm™ tissue by inductively-coupled plasma mass spectrometry (ICP-MS)
Published ICP-MS protocols detecting total intracellular zinc were adapted as follows (Kondo et al. 2002; Rudolf and Cervinka 2010): After treatment, NHEKs (1 × 106) or EpiDerm™ tissue were rinsed with PBS and dried, followed by dissolution in concentrated nitric acid (0.5 ml, 85°C, 3 h). Solutions were analyzed using an ELAN DRC-II ICP-MS (Perkin Elmer). Instrument parameters were as follows: RF power: 1450 W; dwell time: 50 ms; sweeps per replicate: 40; number of replicates: 3; acquisition mode: peak hopping; argon flow rate (l/min): nebulizer flow (0.95); coolant (15); auxiliary (1.3). Calibration standards were prepared from multi-element stock solutions purchased from AccuStandard. The stocks were diluted in 1% nitric acid to provide a working calibration curve of at least 5 points. Samples were also diluted with 1% nitric acid until their response was determined to be within the calibration range. Internal standards (Rh) were added to both standards and samples prior to analysis.
Human stress and toxicity pathwayfinder™ RT2 profiler™ PCR expression array
After pharmacological exposure, total cellular RNA from EpiDerm™ was prepared using the RNeasy kit (Qiagen) according to the manufacturer’s instructions. Reverse transcription was performed using the RT2 First Strand kit (SA Biosciences) and 1 μg total RNA. The Human Stress and Toxicity PathwayFinder™ RT2 Profiler™ PCR Expression Array (Super-Array) profiling the expression of 84 stress- and toxicity-related genes was run as published recently (Lamore et al. 2010a, b), using the following PCR conditions: 95°C for 10 min, followed by 40 cycles of 95°C for 15 s alternating with 60°C for 1 min (Applied Biosystems 7000 SDS). Gene-specific product was normalized to GAPDH and quantified using the comparative (ΔΔCt) Ct method as described in the ABI Prism 7000 sequence detection system user guide. Expression values were averaged across three independent array experiments, and standard deviation was calculated for graphing.
Immunohistochemical detection of heat shock protein 70 (Hsp70)
Tissue sections (3 μm) from formalin fixed, paraffin embedded EpiDerm™ reconstructs were processed for standard hematoxylin and eosin (H&E) staining and Hsp70-immunohistochemistry. Immunohistochemistry was performed using the Discovery XT automated staining platform (VMSI, Ventana Medical Systems). All steps were performed using VMSI validated reagents, including deparaffinization, cell conditioning (antigen retrieval with a borate-EDTA buffer), primary antibody staining, detection and amplification using a biotinylated-streptavidin-HRP and DAB (3,3′-diaminobenzidine tetrahydrochloride) system, and hematoxylin counterstaining. Hsp70 was detected using a murine primary monoclonal antibody (Assay Designs, C92F3A-5; dilution: 1:100) followed by an anti-mouse biotinylated secondary antibody. Images were captured using an Olympus BX50 microscope equipped with an Olympus Dp72 camera and CellSense Digital Image software. Images were standardized for light intensity.
In situ-Terminal dUTP Nick End Labeling (TUNEL) Assay
Tissue sections (3 μm) from formalin fixed, paraffin embedded EpiDerm™ reconstructs were collected onto slides, deparrafinized, rehydrated, and analyzed for DNA fragments using the DermaTACS™ in situ terminal deoxynucleotidyltransferase (TdT) kit (Trevigen) according to the manufacturer’s instructions. Sections were treated with proteinase K and then incubated with TdT enzyme and brominated dNTP mixture (37°C, 30 min). Afterwards, samples were labeled with biotinylated anti-BrdU antibody (37°C, 30 min), followed by streptavidin-conjugated HRP and incubation with TACS Blue Label™ substrate and Red Counterstain C. Slides were dehydrated, clarified (ethanol, p-xylene), and mounted for viewing. Images were captured using an Olympus BX50 with an Olympus Dp72 camera and CellSense Digital Image software. The number of TUNEL-positive cells per viewing field (200×) was counted in six random fields, and percentage TUNEL positive cells was calculated.
Statistical analysis
Data were analyzed employing analysis of variance (ANOVA) with Tukey’s post hoc test using the Prism 4.0 software. Unless indicated differently the results are presented as means (±SD) of at least three independent experiments where means with common letter differ (P < 0.05). Selected data (Fig. 4c, e) were analyzed using the two-sided Student’s t test (*P < 0.05; **P < 0.01; ***P < 0.001).
Fig. 4.

ICP-MS and gene expression array analysis of ZnPT-treated human reconstructed epidermis (EpiDerm™). a Human reconstructed epidermis (EpiDerm™; 9 mm insert diameter) was treated topically and maintained in 6 well format. b H&E stained cross-section of formalin-fixed and paraffin-embedded terminally differentiated EpiDerm™ (SC stratum corneum, E viable epidermal keratinocytes). c ICP-MS analysis of zinc accumulation in EpiDerm™ in response to topical treatment {ZnPT [2% (w/w) in Vanicream™, 90 mg total] or Vanicream™ only; 3 h exposure time; mean + SD}. d RT2 Human Stress and Toxicity Pathway Finder™ PCR Expression Array analysis of differential gene expression in EpiDerm™ exposed to ZnPT [2% (w/w) in Vanicream™, 90 mg total; 24 h exposure) or Vanicream™ only. Changes in cycle threshold (Ct) for genes of interest relative to GAPDH for vehicle control (x-axis) versus ZnPT-treated (y-axis) EpiDerm™ are displayed as scatter blot. Upper and lower lines represent the cut-off indicating four fold up- or down-regulated expression, respectively. The arrows specify the genes with statistically significant upregulation greater than ten-fold (n = 3; P < 0.05). ZnPT-induced up- or downregulation of expression by at least twofold is summarized in Table 1. e Comparison of selected gene expression changes observed in ZnPT-exposed cultured NHEKs [500 nM, 24 h; values taken from Lamore et al. (2010a)] versus ZnPT-exposed EpiDerm™ (as specified in panel d)
Results
Zinc chelation antagonizes ZnPT-induced impairment of genomic integrity, PARP activation, PARP-dependent ATP depletion, and cell death in human epidermal keratinocytes
Recently, we have demonstrated that cultured primary human skin keratinocytes display an exquisite vulnerability to nanomolar concentrations of ZnPT causing rapid loss of genomic integrity with activation of poly(ADP-ribose) polymerase (PARP) and caspase-independent cell death (Lamore et al. 2010a).
In an attempt to further substantiate the role of intracellular zinc dysregulation in ZnPT effects on keratinocytes we first examined ZnPT-cytotoxicity and its potential antagonism by zinc chelation using flow cytometric analysis of annexinV/propidium iodide (AV/PI) stained cells (Fig. 1). A dose response (100–1000 nM ZnPT, 24 h) analysis indicated that induction of cell death required a threshold concentration of approximately 500 nM, and less than 10% viable cells were detected after 24 h exposure to concentrations as low as 1000 nM (Fig. 1). In contrast, cotreatment with diethylenetriaminepentaacetic acid (DTPA), a cell impermeable zinc chelator (Mackenzie et al. 2002; Yui et al. 2002; Hashemi et al. 2007), completely protected from ZnPT-induced cell death, and negligible cytotoxicity was observed upon exposure to ZnSO4 (25 μM; Fig. 1).
Fig. 1.
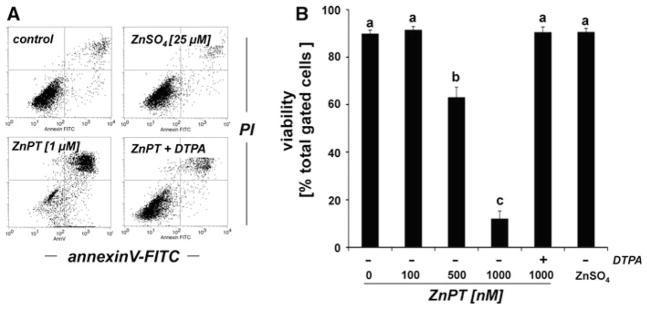
DTPA-antagonism of ZnPT-induced cell death in human skin keratinocytes. Cells were exposed to ZnSO4 (25 μM) or ZnPT (0.1–1 μM, 24 h) in the presence or absence of DTPA (60 μM) or left untreated (control), and viability was assessed by flow cytometric analysis of AV-FITC/PI-stained cells. a Representative flow cytometric scatter blots: control, ZnPT (1 μM), ZnPT (1 μM) with DTPA (60 μM), ZnSO4 (25 μM). b Quantitative analysis indicating percent viable cells (AV−, PI−, lower left quadrant) of total gated cells (mean + SD, n = 3)
Next, the integrity of cellular DNA in NHEKs treated with ZnPT (500 nM, 1 h exposure time) in the presence or absence of DTPA was examined using alkaline single cell gel electrophoresis (comet assay) as a sensitive genotoxicity assay (Fig. 2a). As a positive control, cells were exposed to H2O2, an established genotoxic agent. In agreement with our data published earlier (Lamore et al. 2010a), exposure to ZnPT significantly impaired NHEK genomic integrity within 1 h exposure time as evident from formation of nuclear comets with average tail moments that were increased approximately fivefold over untreated controls (Fig. 2a). In contrast, DTPA cotreatment completely suppressed ZnPT-dependent increase in average tail moment, and no comet formation was observed as a result of exposure to ZnSO4.
Fig. 2.

DTPA-antagonism of ZnPT-induced impairment of genomic integrity, PARP activation, and energy crisis in human skin keratinocytes. a NHEKs were exposed to ZnSO4 (1 μM) or ZnPT (1 μM; 1 h) in the presence or absence of DTPA (60 μM) or PJ34 (2 μM), and DNA damage was detected using the alkaline comet assay. As a positive control, cells were exposed to H2O2 (100 μM, 30 min). Representative comets as visualized by fluorescence microscopy are displayed. b Formation of poly(ADP-ribose) polymer (PAR)-modified cellular proteins as detected by immunoblotting of cellular extracts obtained from NHEKs exposed to ZnPT (5 μM; 15 min) in the absence or presence of DTPA (60 μM) or PJ-34 (2 μM). In addition, cells exposed to ZnSO4 (50 μM, 15 min) or left untreated were analyzed. c Cellular ATP depletion in NHEKs exposed to ZnPT (1 μM, 6 h) in the absence or presence of zinc chelators (DTPA, 60 μM; TPEN, 10 μM) or PJ-34 (2 μM). In addition, cells exposed to ZnSO4 (50 μM, 6 h) or left untreated were analyzed; (mean + SD, n = 3)
Next, we examined the possibility that DTPA treatment may antagonize ZnPT-induced activation of PARP-1. Indeed, DTPA-suppression of ZnPT-induced activation of PARP activity was confirmed by immunodetection of poly(ADP-ribose) polymer (PAR)-modified cellular proteins (Fig. 2b). Consistent with earlier observations (Lamore et al. 2010a), pronounced PAR-formation was observed in NHEKs exposed to ZnPT (5 μM, 15 min), a molecular event strongly suppressed upon coadministration of DTPA. In contrast, negligible PAR formation was detected as a result of exposure to ZnSO4 (50 μM, 15 min exposure), and ZnPT-induced PAR-formation was blocked by cotreatment with the PARP-1 inhibitor PJ34. However, pharmacological PARP inhibition using PJ34 did not suppress the ZnPT-induced increase in average tail moment shown above (Fig. 2a), consistent with activation of PARP-1 occurring downstream of early impairment of genomic integrity.
It is well established that PARP activation in response to genotoxic stress leads to rapid cellular NAD and ATP depletion (Ethier et al. 2007; Kehe et al. 2008), and our earlier research has demonstrated that early cellular energy crisis occurs in response to ZnPT-induced PARP activation (Lamore et al. 2010a). Indeed, ATP-depletion in response to ZnPT exposure was blocked by cotreatment with PJ34, and was also suppressed in the presence of DTPA or TPEN, an intracellular zinc-antagonist (Fig. 2c) (Hwang et al. 2010; Gurusamy et al. 2011).
Taken together these data strongly suggest a causative involvement of intracellular zinc dysregulation in ZnPT-induced genomic destabilization, PARP-1 activation, PARP-1 dependent induction of energy crisis, and loss of keratinocyte viability.
ZnPT induces rapid zinc overload in primary skin keratinocytes and reconstructed human epidermis
To further substantiate the role of intracellular zinc dysregulation in ZnPT-induced impairment of NHEK viability and function, we assessed the accumulation of free intracellular zinc ions in response to ZnPT exposure using Zinquin-based fluorimetric analysis and total zinc ion quantification by ICP-MS (Fig. 3). Quantitative analysis of Zinquin fluorescence revealed a rapid increase in intracellular free zinc concentrations by 2.5 fold in response to ZnPT treatment (1 μM, 5 min exposure), and elevated levels were maintained over the one hour observation period (Fig. 3a). Further dose–response analysis indicated that at higher concentrations (5 μM) ZnPT caused an almost 3 fold upregulation within 5 min exposure (Fig. 3b). In order to independently confirm these data using an alternative analytical methodology and to avoid potential inaccuracies associated with a non-linear dose response relationship of zinquin fluorescence we then determined total cellular zinc content by ICP-MS analysis (Fig. 3c). Consistent with the fluorescence data ZnPT exposure (1 μM, 1 h) caused a significant rise in total zinc content of NHEKs that reached a maximum of 207.1 ± 7.1 ng per 106 cells, an approximately twofold increase over untreated control cells. In addition, ICP-MS analysis indicated a significant elevation of intracellular zinc levels by almost 50% in response to submicromolar (500 nM) concentrations of ZnPT. Importantly, no increase in intracellular zinc levels in response to ZnSO4 exposure was detected, even at a 50-fold molar excess (50 μM) over ZnPT.
Fig. 3.

Rapid induction of zinc dysregulation in ZnPT-treated in human skin keratinocytes. a Time course of increase in intracellular free zinc induced by ZnPT (1 μM) as detected by Zinquin fluorescence with representative fluorescence microscopy pictures: panel I: control; panel II: ZnPT (5 min); panel III: ZnPT (60 min). Quantitative analysis was performed as specified in “Materials and methods” (mean + SD). b Dose–response analysis of ZnPT-induced changes in Zinquin-fluorescence (0.1–5 μM; 5 min exposure). c Total cellular zinc content as analyzed by ICP-MS after ZnPT (0.5 and 1 μM, 1 h) and ZnSO4 (50 μM, 1 h) exposure (mean + SD; n = 3)
Next, we tested the possibility that topical exposure of human skin to ZnPT would induce a rapid increase in epidermal zinc content. To this end, quantitative ICP-MS assessment of zinc accumulation in response to topical ZnPT exposure was performed in human reconstructed epidermis (EpiDerm™), an established organotypic model of full differentiated human epidermis (Fig. 4a–c) (Kandarova et al. 2005; Bause et al. 2009). EpiDerm™ tissues were topically exposed to ZnPT [2% (w/w) in Vanicream™, 90 mg total; 3 h; Fig. 4a] or Vanicream™ followed by ICP-MS (Fig. 4c). An almost five fold increase in EpiDermal zinc ion content [ZnPT-Vanicream™: 348.2 ± 34.0 versus Vanicream™ control: 72.2 ± 5.1 (ng Zn/tissue reconstruct; mean ± SD; P < 0.01)] was measured in response to ZnPT topical application. In contrast, no statistically significant elevation of epidermal zinc content was observed following a control procedure where ICP-MS analysis was performed after the ZnPT-Vanicream preparation was topically applied followed by immediate removal and extensive rinsing (data not shown). This excludes artifactual ICP-MS detection of cutaneous zinc originating from residual ZnPT cream not removed during rinsing.
Gene array and IHC analyses demonstrate an upregulated stress response in reconstructed human epidermis exposed to topical ZnPT
Next, modulation of stress response gene expression was examined in reconstructed human epidermis exposed to topical ZnPT. To this end, EpiDerm™ tissue equivalents were first treated with a ZnPT-Vanicream™ formulation (2%, 24 h continuous exposure) shown to be effective in elevating tissue zinc ion content (Fig. 4c) and then subjected to expression profiling using the RT2 Human Stress and Toxicity Pathway Finder™ PCR Expression Array technology (Fig. 4d; Table 1a) (Cabello et al. 2009a; Lamore et al. 2010a, b). After 24 h continuous exposure of reconstructed epidermis to topical ZnPT, expression of the metallothionein encoding gene MT2A was dramatically upregulated by approximately 500-fold over Vanicream only-treated control, a finding consistent with cellular zinc ion overload and induction of a metal stress response (Andrews 2001; Plum et al. 2010).
Table 1.
Quantitative analysis of gene expression changes in EpiDerm™ exposed to topical ZnPT-treatment
| Gene symbol | Gene name | Fold change | P value |
|---|---|---|---|
| (a) | |||
| MT2A (NM_005953) | Metallothionein 2a | 499.4 | 0.000 |
| HSPA6 (NM_002155) | Heat shock 70 kDa protein 6 (HSP70B’) | 352.4 | 0.000 |
| TNF (NM_000594) | Tumor necrosis factor | 90.8 | 0.001 |
| HSPH1 (NM_006644) | Heat shock 105 kDa/110 kDa protein 1 | 72.6 | 0.002 |
| HSPA1A (NM_005345) | Heat shock 70 kDa protein 1A | 69.3 | 0.000 |
| HSPE1 (NM_002157) | Heat shock 10 kDa protein 1 (chaperonin 10) | 46.3 | 0.002 |
| DNAJB4 (NM_007034) | DnaJ (Hsp40) homolog, subfamily B, member 4 | 41.3 | 0.001 |
| SOD2 (NM_000636) | Superoxide dismutase 2, mitochondrial | 35.0 | 0.000 |
| EPHX2 (NM_001979) | Microsomal epoxide hydrolase | 22.7 | 0.010 |
| NOS2A (NM_000625) | Nitric oxide synthase 2, inducible | 19.5 | 0.007 |
| HSPD1 (NM_002156) | Heat shock 60 kDa protein 1 (chaperonin) | 18.1 | 0.006 |
| NFKB1 (NM_003998) | Nuclear factor of kappa light polypeptide gene enhancer in B-cells 1 | 17.5 | 0.001 |
| DNAJA1 (NM_001539) | DnaJ (Hsp40) homolog, subfamily A, member 1 | 12.1 | 0.002 |
| HSPA1L (NM_005527) | Heat shock 70 kDa protein 1-like | 12.0 | 0.008 |
| HSP90AA1 (NM_005348) | Heat shock protein 90 kDa alpha (cytosolic), class A member 1 | 11.6 | 0.011 |
| FMO1 (NM_002021) | Flavin containing monooxygenase 1 | 10.9 | 0.007 |
| GSTM3 (NM_000849) | Glutathione S-transferase mu 3 (brain) | 10.8 | 0.001 |
| CYP1A1 (NM_000499) | Cytochrome P450, family 1, subfamily A, polypeptide 1 | 8.4 | 0.032 |
| LTA (NM_000595) | Lymphotoxin alpha (TNF superfamily, member 1) | 8.4 | 0.032 |
| HSP90AB1 (NM_007355) | Heat shock protein 90 kDa alpha (cytosolic), class B member 1 | 7.3 | 0.002 |
| GDF15 (NM_004864) | Growth differentiation factor 15 | 7.2 | 0.027 |
| HMOX1 (NM_002133) | Heme oxygenase (decycling) 1 | 6.6 | 0.001 |
| HSPA8 (NM_006597) | Heat shock 70 kDa protein 8 | 6.4 | 0.005 |
| ANXA5 (NM_001154) | Annexin A5 | 6.2 | 0.001 |
| GSR (NM_000637) | Glutathione reductase | 5.8 | 0.034 |
| CDKN1A (NM_000389) | Cyclin-dependent kinase inhibitor 1A (p21, Cip1) | 5.6 | 0.010 |
| DDIT3 (NM_004083) | DNA-damage-inducible transcript 3 | 5.3 | 0.002 |
| MDM2 (NM_002392) | Mdm2 p53 binding protein homolog (mouse) | 4.9 | 0.009 |
| CASP1 (NM_033292) | Caspase 1, apoptosis-related cysteine peptidase (interleukin 1, beta, convertase) | 4.8 | 0.018 |
| HSPA4 (NM_002154) | Heat shock 70 kDa protein 4 | 4.4 | 0.005 |
| TP53 (NM_000546) | Tumor protein p53 | 4.1 | 0.028 |
| CASP8 (NM_001228) | Caspase 8, apoptosis-related cysteine peptidase | 3.9 | 0.001 |
| CHEK2 (NM_007194) | CHK2 checkpoint homolog (S. pombe) | 3.5 | 0.004 |
| GADD45A (NM_001924) | Growth arrest and DNA-damage-inducible, alpha | 3.5 | 0.045 |
| UNG (NM_003362) | Uracil-DNA glycosylase | 3.4 | 0.032 |
| SOD1 (NM_000454) | Superoxide dismutase 1, soluble | 3.4 | 0.036 |
| RAD50 (NM_005732) | RAD50 homolog (S. cerevisiae) | 3.2 | 0.032 |
| BAX (NM_004324) | BCL2-associated X protein | 3.2 | 0.022 |
| DDB1 (NM_001923) | Damage-specific DNA binding protein 1, 127 kDa | 3.1 | 0.049 |
| PRDX1 (NM_002574) | Peroxiredoxin 1 | 2.9 | 0.022 |
| FMO5 (NM_001461) | Flavin containing monooxygenase 5 | 2.8 | 0.004 |
| HSF1 (NM_005526) | Heat shock transcription factor 1 | 2.4 | 0.011 |
| HSPA5 (NM_005347) | Heat shock 70 kDa protein 5 (glucose-regulated protein, 78 kDa) | 2.3 | 0.045 |
| GPX1 (NM_000581) | Glutathione peroxidase 1 | 2.2 | 0.016 |
| RAD23A (NM_005053) | RAD23 homolog A (S. cerevisiae) | 2.0 | 0.035 |
| SERPINE1 (NM_000602) | Serpin peptidase inhibitor, clade E (nexin, plasminogen activator inhibitor type 1), member 1 | −2.4 | 0.014 |
| IGFBP6 (NM_002178) | Insulin-like growth factor binding protein 6 | −3.7 | 0.029 |
| BCL2L1 (NM_138578) | BCL2-like 1 | −3.8 | 0.017 |
| (b) | |||
| HSPA6 (NM_002155) | Heat shock 70 kDa protein 6 (HSP70B’) | 173.0 | 0.004 |
| MT2A (NM_005953) | Metallothionein 2a | 150.2 | 0.0003 |
| HSPA1A (NM_005345) | Heat shock 70 kDa protein 1A | 17.9 | 0.008 |
| DNAJB4 (NM_007034) | DnaJ (Hsp40) homolog, subfamily B, member 4 | 17.4 | 0.020 |
| LTA (NM_000595) | Lymphotoxin alpha (TNF superfamily, member 1) | 10.1 | 0.012 |
| HMOX1 (NM_002133) | Heme oxygenase (decycling) 1 | 9.6 | 0.011 |
| HSPH1 (NM_006644) | Heat shock 105 kDa/110 kDa protein 1 | 9.6 | 0.033 |
| CYP7A1 (NM_000780) | Cytochrome P450, family 7, subfamily A, polypeptide 1 | 7.4 | 0.006 |
| HSP90AA1 (NM_005348) | Heat shock protein 90 kDa alpha (cytosolic), class A member 1 | 5.4 | 0.010 |
| HSPA1L (NM_005527) | Heat shock 70 kDa protein 1-like | 5.0 | 0.030 |
| HSPD1 (NM_002156) | Heat shock 60 kDa protein 1 (chaperonin) | 3.7 | 0.028 |
| HSP90AB1 (NM_007355) | Heat shock protein 90 kDa alpha (cytosolic), class B member 1 | 2.4 | 0.035 |
| NFKBIA (NM_020529) | Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha | −2.2 | 0.0147 |
| POR (NM_000941) | P450 (cytochrome) oxidoreductase | −2.7 | 0.0146 |
| CCNG1 (NM_004060) | Cyclin G1 | −2.878 | 0.0134 |
| PTGS1 (NM_000962) | Prostaglandin-endoperoxide synthase 1 (prostaglandin G/H synthase and cyclooxygenase) | −4.4 | 0.018 |
| FMO5 (NM_001461) | Flavin containing monooxygenase 5 | −4.9 | 0.027 |
RT2 Human Stress and Toxicity Pathway Finder™ PCR Expression Array analysis was performed as depicted in Fig. 4D (n = 3; P < 0.05). (a) Numerical gene expression changes induced by topical ZnPT-treatment (24 h continuous exposure; 2% (w/w) in Vanicream™, 90 mg total). (b) Numerical gene expression changes induced by topical ZnPT-treatment (1 h continuous exposure followed by removal and subsequent 23 h incubation period; 2% (w/w) in Vanicream™, 90 mg total)
Indicative of a massive ZnPT-induced cellular heat shock response, numerous genes encoding heat shock-related proteins (including HSPA6, HSPH1, HSPA1A, HSPE1, DNAJB4, HSPD1, DNAJA1, HSPA1L, HSP90AA1, HSP90AB1, HSPA8, HMOX1, HSPA4, HSPA5, and HSF1, the gene encoding the transcriptional regulator heat shock factor 1) were significantly upregulated at the mRNA level. Moreover, ZnPT-induced expression changes in EpiDerm™ involved upregulation of genes controlling inflammatory and irritation-response signaling (TNF, NFKB1, LTA, GDF15, ANXA5), xenobiotic metabolism (CYP1A1, EPHX2, FMO1, FMO5, GSTM3), and oxidative stress response and redox signaling (SOD2, NOS2A, HMOX1, SOD1, GSR, GPX1, PRDX1). Importantly, ZnPT-induced upregulation also involved genes with major roles in genotoxic stress response, DNA repair, and cell cycle progression (UNG, CDKN1A, DDIT3, MDM2, TP53, CHEK2, GADD45A, RAD50, DDB1, RAD23A).
A similar yet somewhat attenuated epidermal gene expression profile was observed when an alternative exposure regimen was followed where gene expression changes were assessed in response to short-term exposure to topical ZnPT-Vanicream preparation (2%, 1 h exposure time followed by rinse and 23 h postexposure incubation). Among various heat shock protein encoding genes upregulated by short term exposure to ZnPT, HSPA6 again displayed the most pronounced expression differential (173-fold upregulation), and massive induction of MT2A gene expression (150-fold upregulation) was observed. Interestingly, short term ZnPT exposure was not associated with transcriptional activation of DNA damage response gene expression, suggesting that ZnPT-associated impairment of epidermal genomic integrity only occurs under conditions of prolonged exposure.
These findings are consistent with earlier results indicating upregulation of gene expression in primary keratinocytes exposed to nanomolar concentrations of ZnPT (Lamore et al. 2010a). When comparing fold-induction of mRNA levels between ZnPT exposed NHEKs and EpiDerm™ (Fig. 4e), a consistent pattern of heat shock response gene (HSPA1A, HSPA6, HMOX1) upregulation was observed. Remarkably, upregulation of MT2A and TNF was dramatically increased in ZnPT treated EpiDerm™ over NHEKs (MT2A: 499.4 ± 44.9 versus 5.7 ± 2.0 fold upregulation; TNF: 90.8 ± 29.3 versus 2.2 ± 0.7 fold upregulation; mean ± SD, n = 3). A differential extent of ZnPT-induced upregulation of MT2A and TNF in EpiDerm™ over cultured NHEKs may be related to molecular changes associated with terminal differentiation that only occurs in the organotypic model. Indeed, it has been shown earlier that stress gene expression and signaling is altered in human skin reconstructs compared to cultured skin cells (Sun et al. 2004).
Next, IHC tissue analysis of EpiDerm™ reconstructs after topical exposure to ZnPT was used to examine heat shock protein expression at the protein level (Fig. 5a). Indeed, massive upregulation of cellular Hsp70 protein levels was observed in reconstructs that were exposed to topical ZnPT-Vanicream preparation (2%, 24 h), a finding already suggested by gene expression array analysis that indicated upregulation of genes encoding numerous members of the Hsp70 protein family (Table 1a). Vanicream treated control reconstructs did not stain positive for Hsp70, and a moderate upregulation of Hsp70 IHC staining was already observed in reconstructs exposed to a topical preparation containing considerably less ZnPT (0.1%, 24 h; Fig. 5a). Interestingly, Hsp70 upregulation in response to 2% ZnPT was most pronounced in keratinocytes residing in the basal layer of the reconstruct, consistent with an increased stress response in rapidly dividing cells that might not occur during later stages of terminal EpiDermal differentiation.
Fig. 5.
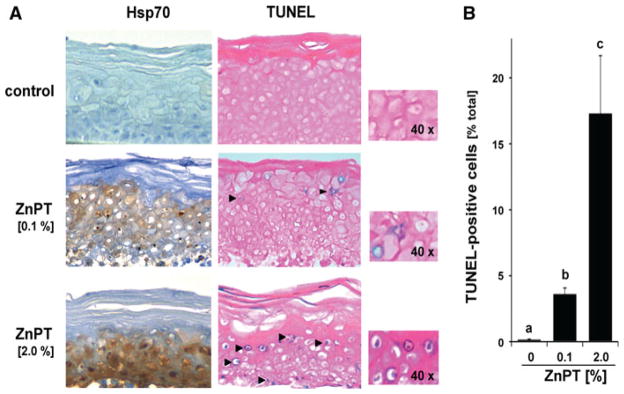
Immunohistochemical assessment of Hsp70 induction and TUNEL positivity in EpiDerm™ exposed to topical ZnPT. a EpiDerm™ was exposed to topical ZnPT (24 h continuous exposure; 0.1 and 2% (w/w) in Vanicream™, 90 mg total) or Vanicream control only (n = 3 per treatment group). Paraformaldehyde-fixed, paraffin-embedded 3 μm sections were then analyzed using Hsp70-IHC with hematoxylin counterstaining (left column). Alternatively, TUNEL-IHC was performed on duplicate tissue sections (right column). Per tissue section at least three high power fields were acquired; one representative photograph is shown (inset: 40× magnification). b Bar graph displaying quantitative analysis of TUNEL-IHC (percentage TUNEL positive nuclei of total nuclei per high power field; mean + S.D.; n = 3)
Our earlier published data demonstrated rapid induction of genotoxic stress associated with PARP activation and DNA fragmentation in NHEKs exposed to submicromolar concentrations of ZnPT (Lamore et al. 2010a). Based on pronounced induction of genotoxic stress documented in NHEKs (Lamore et al. 2010a) and observed here in reconstructed epidermis (Table 1), we assessed ZnPT-induced DNA fragmentation performing terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling in situ (TUNEL-IHC; Fig. 5a, b). Pronounced TUNEL-positivity (17.3 ± 4.4% positive cells) was detected in reconstructs that were exposed to topical ZnPT-Vanicream preparation (2%, 24 h). Even upon exposure to the less concentrated ZnPT formulation (0.1%, 24 h) a considerable percentage of cells (3.6 ± 0.5%) stained positive for fragmented DNA. However, no follow-up experimentation was performed that would allow to further define the mechanistic origin of ZnPT-induced epidermal DNA fragmentation.
Discussion
ZnPT is a widely-used cutaneous microbicidal OTC-drug (Pierard-Franchimont et al. 2002; Bailey et al. 2003; Smegal et al. 2004; Guthery et al. 2005), and topical safety and toxicity profile of ZnPT have been studied to some extent previously (Snyder et al. 1965; Brandrup and Menne 1985). However, cytotoxic and genotoxic effects of ZnPT have been substantiated in mouse lymphoma cells and teratogenicity was described in zebra fish (Goka 1999; Moller et al. 2002). Furthermore, ZnPT and another antifungal Cu/Zn-chelator and ionophore drug, clioquinol, have been shown to cause pathophysiologically relevant neuronal zinc disturbance that occurs through activation of TRPA1, a nociceptive ion channel and zinc-sensitive receptor for environmental irritants and oxidants (Andersson et al. 2009). As a consequence, clioquinol was withdrawn from the market when it was causatively linked to an epidemic of subacute myelo-optico-neuropathy (Konagaya et al. 2004).
Earlier experiments demonstrated that exposure of mammalian cells to various zinc salts (e.g. ZnSO4, ZnCl2, and Zn-acetate) is well tolerated at concentrations up to 200 μM without reduction of viability as also observed in this study (Figs. 1, 2) (Hatayama et al. 1993; Jourdan et al. 2002; Cortese et al. 2008), but potentiation of zinc effects and induction of cytotoxicity by zinc-specific ionophores including pyrithione has been documented earlier in non-cutaneous mammalian cells (Kim et al. 1999; Magda et al. 2008; Mann and Fraker 2005; Klein et al. 2006; Rudolf and Cervinka 2010). Recently, we have demonstrated that primary keratinocytes and melanocytes exposed to nanomolar concentrations of ZnPT undergo rapid loss of genomic integrity with PARP activation and heat shock response gene expression followed by non-apoptotic cell death (Lamore et al. 2010a).
This study documents for the first time ZnPT-induced dysregulation of intracellular zinc ion homeostasis in primary keratinocytes as observed by quantitative fluorescence microscopy (Fig. 3a–b) and ICP-MS (Fig. 3c), building on earlier ICP-MS data that determined total zinc ion accumulation in cultured cancer cell lines exposed to ZnPT (Kondo et al. 2002; Rudolf and Cervinka 2010). ZnPT-induced impairment of viability (Fig. 1), PARP activation (Fig. 2b), energy crisis (Fig. 2c), and genomic destabilization (Fig. 2a) were all antagonized by zinc chelation using either DTPA or TPEN, consistent with intracellular zinc ion accumulation as a causative factor in mediating the effects of nanomolar to low micromolar concentrations of ZnPT on cultured keratinocytes.
Remarkably, ZnPT activity against cultured NHEKs was observed in the nano- to micromolar range, whereas topical OTC-products contain much higher levels (up to 2%) corresponding to millimolar concentrations. However, in intact human skin, a multi-layered differentiated epidermis with stratum corneum serves as an essential barrier against environmental insults including ultraviolet radiation and chemical exposure, thereby potentially minimizing cellular effects of topical ZnPT (Chuong et al. 2002). We therefore examined molecular consequences of cutaneous ZnPT exposure in EpiDerm™, a 3-dimensional organotypic human skin model that incorporates normal epidermal keratinocytes undergoing terminal differentiation with intact stratum corneum and barrier function used extensively for transdermal drug delivery, cutaneous absorption, and skin irritation studies (Kandarova et al. 2005; Bause et al. 2009). In this epidermal model, topical application of ZnPT induced zinc dysregulation as substantiated by ICP-MS-based detection of rapid zinc accumulation (Fig. 4c). Expression array analysis indicated massive upregulation of the MT2A gene encoding metallothionein-2A (Fig. 4d; Table 1), shown earlier to be induced in the context of a cellular zinc stress response observed in Hep-2 cervical tumor cells exposed to ZnPT (Rudolf and Cervinka 2010). Further expression array analysis indicated upregulation of a wide range of stress response genes encoding heat shock proteins, antioxidants, and inflammatory factors (Fig. 4d; Table 1), and IHC analysis revealed upregulation of Hsp70 protein levels (Fig. 5a), confirming our earlier results obtained in cultured primary NHEKs (Lamore et al. 2010a).
These findings are consistent with the established role of zinc-ions as potent inducers of metal stress, heat shock, and antioxidant stress response pathways involved in cytoprotective as well as cytotoxic activities (Hatayama et al. 1993; Lee et al. 2000; Unoshima et al. 2001). Indeed, intracellular zinc ion availability has been shown to be involved in activation of stress response transcription factors including Nrf2 (electrophilic stress response), HSF (heat shock response), and MTF (heavy metal stress response) (Hatayama et al. 1993; Andrews 2001; Jourdan et al. 2002; Dinkova-Kostova et al. 2005; Cortese et al. 2008). Moreover, a mechanistic involvement of intracellular zinc in lipopolysaccharide-induced TNF-α upregulation has recently been demonstrated in monocytes (Haase et al. 2008), and it is therefore tempting to speculate that TNF upregulation observed in ZnPT-exposed Epiderm™ (that only contains primary keratinocytes undergoing terminal differentiation) may equally be zinc-dependent, a hypothesis to be tested by future experimentation.
PCR array expression analysis of ZnPT-exposed EpiDerm™ also revealed a significant upregulation of multiple genes involved in cellular response to genotoxic stress (Table 1a). This finding is consistent with ZnPT-induced PARP activation downstream of early loss of genomic integrity as observed in cultured primary NHEKs (Fig. 2), where PARP activation, energy crisis, and genomic impairment were all antagonized by zinc chelation using DTPA and TPEN. Thus, TUNEL-positivity in ZnPT-treated EpiDerm™ (as shown in Fig. 5) may indicate treatment-induced direct DNA fragmentation, but may also originate from DNA strand breaks introduced during later stages of apoptotic or necrotic cell death, a possibility that we have not further explored. It is interesting to note that disruption of intracellular zinc homeostasis has been identified as an important activator of nuclear endonucleases including DNase gamma (Shiokawa and Tanuma 1998), and high levels of intracellular zinc ions have been associated with formation of reactive oxygen species (Cabreiro et al. 2009), candidate molecular mechanisms that may be involved in ZnPT-effects on nuclear DNA to be addressed by future research.
Taken together our data demonstrate for the first time ZnPT-induced impairment of zinc ion homeostasis and upregulation of stress response gene expression in primary keratinocytes and reconstructed human epidermis, molecular activities that may underlie therapeutic and toxicological effects of this topical drug. The important question of whether cutaneous application of ZnPT formulations at OTC concentrations alters keratinocyte function and zinc ion homeostasis in intact human skin is currently investigated in our laboratory employing gene expression profiling and spatial resolution zinc ICP-MS of human skin biopsies.
Acknowledgments
Supported in part by grants from the National Institutes of Health (Award Number R01CA122484 from the National Cancer Institute, ES007091, ES06694, Arizona Cancer Center Support Grant CA023074). Immunohistochemical analysis was performed by the TACMASS Core (Tissue Acquisition and Cellular/Molecular Analysis Shared Service, AZCC).
Abbreviations
- AV
AnnexinV
- FITC
Fluorescein isothiocyanate
- DTPA
Diethylenetriaminepentaacetic acid
- HSP
Heat shock protein
- ICP-MS
Inductively coupled plasma mass spectrometry
- IHC
Immunohistochemistry
- NHEK
Normal human epidermal keratinocyte
- OTC
Over-the-counter
- PAR
Poly(ADP-ribose) polymer
- PARP
Poly(ADP-ribose) polymerase
- PBS
Phosphate buffered saline
- PI
Propidium iodide
- SDS-PAGE
Sodium dodecylsulfate polyacrylamide gel electrophoresis
- TPEN
N,N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediamine
- TUNEL
Terminal dUTP nick end labeling
- ZnPT
Zinc pyrithione
Footnotes
The content is solely the responsibility of the author and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
References
- Andersson DA, Gentry C, Moss S, Bevan S. Clioquinol and pyrithione activate TRPA1 by increasing intracellular Zn2+ Proc Natl Acad Sci USA. 2009;106:8374–8379. doi: 10.1073/pnas.0812675106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews GK. Cellular zinc sensors: MTF-1 regulation of gene expression. Biometals. 2001;14:223–237. doi: 10.1023/a:1012932712483. [DOI] [PubMed] [Google Scholar]
- Bae YS, Hill ND, Bibi Y, Dreiher J, Cohen AD. Innovative uses for zinc in dermatology. Dermatol Clin. 2010;28:587–597. doi: 10.1016/j.det.2010.03.006. [DOI] [PubMed] [Google Scholar]
- Bailey P, Arrowsmith C, Darling K, Dexter J, Eklund J, Lane A, Little C, Murray B, Scott A, Williams A, Wilson D. A double-blind randomized vehicle-controlled clinical trial investigating the effect of ZnPTO dose on the scalp vs antidandruff efficacy and antimycotic activity. Int J Cosmet Sci. 2003;25:183–188. doi: 10.1046/j.1467-2494.2003.00183.x. [DOI] [PubMed] [Google Scholar]
- Barnett BL, Kretschmar HC, Hartman FA. Structural characterization of bis(N-oxopyridine-2-thionato)zinc(II) Inorg Chem. 1977;16:1834–1838. [Google Scholar]
- Bause SM, Lamore SD, Wondrak GT. More than skin deep: the human skin tissue equivalent as an advanced drug discovery tool. Front Drug Des Discov. 2009;4:135–161. [Google Scholar]
- Beasley DG, Meyer TA. Characterization of the UVA protection provided by avobenzone, zinc oxide, and titanium dioxide in broad-spectrum sunscreen products. Am J Clin Dermatol. 2010;11:413–421. doi: 10.2165/11537050-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Brandrup F, Menne T. Zinc pyrithione (Zinc Omadine) allergy. Contact Dermatitis. 1985;12:50. doi: 10.1111/j.1600-0536.1985.tb01041.x. [DOI] [PubMed] [Google Scholar]
- Cabello CM, Bair WB, III, Lamore SD, Ley S, Bause AS, Azimian S, Wondrak GT. The cinnamon-derived Michael acceptor cinnamic aldehyde impairs melanoma cell proliferation, invasiveness, and tumor growth. Free Radic Biol Med. 2009a;46:220–231. doi: 10.1016/j.freeradbiomed.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabello CM, Bair WB, III, Ley S, Lamore SD, Azimian S, Wondrak GT. The experimental chemotherapeutic N(6)-furfuryladenosine (kinetin-riboside) induces rapid ATP depletion, genotoxic stress, and CDKN1A (p21) upregulation in human cancer cell lines. Biochem Pharmacol. 2009b;77:1125–1138. doi: 10.1016/j.bcp.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabreiro F, Picot CR, Perichon M, Friguet B, Petropoulos I. Overexpression of methionine sulfoxide reductases A and B2 protects MOLT-4 cells against zinc-induced oxidative stress. Antioxid Redox Signal. 2009;11:215–225. doi: 10.1089/ars.2008.2102. [DOI] [PubMed] [Google Scholar]
- Cho YS, Lee KH, Park JW. Pyrithione-zinc prevents UVB-induced epidermal hyperplasia by inducing HIF-1alpha. Korean J Physiol Pharmacol. 2010;14:91–97. doi: 10.4196/kjpp.2010.14.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuong CM, Nickoloff BJ, Elias PM, Goldsmith LA, Macher E, Maderson PA, Sundberg JP, Tagami H, Plonka PM, Thestrup-Pederson K, Bernard BA, Schroder JM, Dotto P, Chang CM, Williams ML, Feingold KR, King LE, Kligman AM, Rees JL, Christophers E. What is the ‘true’ function of skin? Exp Dermatol. 2002;11:159–187. doi: 10.1034/j.1600-0625.2002.00112.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortese MM, Suschek CV, Wetzel W, Kroncke KD, Kolb-Bachofen V. Zinc protects endothelial cells from hydrogen peroxide via Nrf2-dependent stimulation of glutathione biosynthesis. Free Radic Biol Med. 2008;44:2002–2012. doi: 10.1016/j.freeradbiomed.2008.02.013. [DOI] [PubMed] [Google Scholar]
- Ding WQ, Lind SE. Metal ionophores—an emerging class of anticancer drugs. IUBMB Life. 2009;61:1013–1018. doi: 10.1002/iub.253. [DOI] [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Holtzclaw WD, Wakabayashi N. Keap1, the sensor for electrophiles and oxidants that regulates the phase 2 response, is a zinc metalloprotein. Biochemistry. 2005;44:6889–6899. doi: 10.1021/bi047434h. [DOI] [PubMed] [Google Scholar]
- Ethier C, Labelle Y, Poirier GG. PARP-1-induced cell death through inhibition of the MEK/ERK pathway in MNNG-treated HeLa cells. Apoptosis. 2007;12:2037–2049. doi: 10.1007/s10495-007-0127-z. [DOI] [PubMed] [Google Scholar]
- Gibson WB, Calvin G. Percutaneous absorption of zinc pyridinethione in monkeys. Toxicol Appl Pharmacol. 1978;43:425–437. doi: 10.1016/s0041-008x(78)80002-7. [DOI] [PubMed] [Google Scholar]
- Goka K. Embryotoxicity of zinc pyrithione, an anti-dandruff chemical, in fish. Environ Res. 1999;81:81–83. doi: 10.1006/enrs.1998.3944. [DOI] [PubMed] [Google Scholar]
- Gurusamy KS, Farooqui N, Loizidou M, Dijk S, Taanman JW, Whiting S, Farquharson MJ, Fuller BJ, Davidson BR. Influence of zinc and zinc chelator on HT-29 colorectal cell line. Biometals. 2011;24:143–151. doi: 10.1007/s10534-010-9382-5. [DOI] [PubMed] [Google Scholar]
- Guthery E, Seal LA, Anderson EL. Zinc pyrithione in alcohol-based products for skin antisepsis: persistence of antimicrobial effects. Am J Infect Control. 2005;33:15–22. doi: 10.1016/j.ajic.2004.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase H, Ober-Blöbaum JL, Engelhardt G, Hebel S, Heit A, Heine H, Rink L. Zinc signals are essential for lipopolysaccharide-induced signal transduction in monocytes. J Immunol. 2008;181:6491–6502. doi: 10.4049/jimmunol.181.9.6491. [DOI] [PubMed] [Google Scholar]
- Hashemi M, Ghavami S, Eshraghi M, Booy EP, Los M. Cytotoxic effects of intra and extracellular zinc chelation on human breast cancer cells. Eur J Pharmacol. 2007;557:9–19. doi: 10.1016/j.ejphar.2006.11.010. [DOI] [PubMed] [Google Scholar]
- Hatayama T, Asai Y, Wakatsuki T, Kitamura T, Imahara H. Regulation of hsp70 synthesis induced by cupric sulfate and zinc sulfate in thermotolerant HeLa cells. J Biochem. 1993;114:592–597. doi: 10.1093/oxfordjournals.jbchem.a124222. [DOI] [PubMed] [Google Scholar]
- Howes D, Black JG. Comparative percutaneous absorption of pyrithiones. Toxicology. 1975;5:209–220. doi: 10.1016/0300-483x(75)90118-3. [DOI] [PubMed] [Google Scholar]
- Hwang JJ, Kim HN, Kim J, Cho DH, Kim MJ, Kim YS, Kim Y, Park SJ, Koh JY. Zinc(II) ion mediates tamoxifen-induced autophagy and cell death in MCF-7 breast cancer cell line. Biometals. 2010;23:997–1013. doi: 10.1007/s10534-010-9346-9. [DOI] [PubMed] [Google Scholar]
- Jarrousse V, Castex-Rizzi N, Khammari A, Charveron M, Dreno B. Zinc salts inhibit in vitro Toll-like receptor 2 surface expression by keratinocytes. Eur J Dermatol. 2007;17:492–496. doi: 10.1684/ejd.2007.0263. [DOI] [PubMed] [Google Scholar]
- Jourdan E, Emonet-Piccardi N, Didier C, Beani JC, Favier A, Richard MJ. Effects of cadmium and zinc on solar-simulated light-irradiated cells: potential role of zinc-metallothionein in zinc-induced genoprotection. Arch Biochem Biophys. 2002;405:170–177. doi: 10.1016/s0003-9861(02)00401-0. [DOI] [PubMed] [Google Scholar]
- Kandarova H, Liebsch M, Gerner I, Schmidt E, Genschow E, Traue D, Spielmann H. The EpiDerm test protocol for the upcoming ECVAM validation study on in vitro skin irritation tests—an assessment of the performance of the optimised test. Altern Lab Anim. 2005;33:351–367. doi: 10.1177/026119290503300408. [DOI] [PubMed] [Google Scholar]
- Kehe K, Raithel K, Kreppel H, Jochum M, Worek F, Thiermann H. Inhibition of poly(ADP-ribose) polymerase (PARP) influences the mode of sulfur mustard (SM)-induced cell death in HaCaT cells. Arch Toxicol. 2008;82:461–470. doi: 10.1007/s00204-007-0265-7. [DOI] [PubMed] [Google Scholar]
- Kim CH, Kim JH, Moon SJ, Chung KC, Hsu CY, Seo JT, Ahn YS. Pyrithione, a zinc ionophore, inhibits NF-kappaB activation. Biochem Biophys Res Commun. 1999;259:505–509. doi: 10.1006/bbrc.1999.0814. [DOI] [PubMed] [Google Scholar]
- Klein C, Creach K, Irintcheva V, Hughes KJ, Blackwell PL, Corbett JA, Baldassare JJ. Zinc induces ERK-dependent cell death through a specific Ras isoform. Apoptosis. 2006;11:1933–1944. doi: 10.1007/s10495-006-0089-6. [DOI] [PubMed] [Google Scholar]
- Konagaya M, Matsumoto A, Takase S, Mizutani T, Sobue G, Konishi T, Hayabara T, Iwashita H, Ujihira T, Miyata K, Matsuoka Y. Clinical analysis of longstanding subacute myelo-optico-neuropathy: sequelae of clioquinol at 32 years after its ban. J Neurol Sci. 2004;218:85–90. doi: 10.1016/j.jns.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Kondoh M, Tasaki E, Araragi S, Takiguchi M, Higashimoto M, Watanabe Y, Sato M. Requirement of caspase and p38MAPK activation in zinc-induced apoptosis in human leukemia HL-60 cells. Eur J Biochem. 2002;269:6204–6211. doi: 10.1046/j.1432-1033.2002.03339.x. [DOI] [PubMed] [Google Scholar]
- Krenn BM, Gaudernak E, Holzer B, Lanke K, Van Kuppeveld FJ, Seipelt J. Antiviral activity of the zinc ionophores pyrithione and hinokitiol against picornavirus infections. J Virol. 2009;83:58–64. doi: 10.1128/JVI.01543-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamore SD, Cabello CM, Wondrak GT. The topical antimicrobial zinc pyrithione is a heat shock response inducer that causes DNA damage and PARP-dependent energy crisis in human skin cells. Cell Stress Chaperones. 2010a;15:309–322. doi: 10.1007/s12192-009-0145-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamore SD, Qiao S, Horn D, Wondrak GT. Proteomic identification of cathepsin B and nucleophosmin as novel UVA-targets in human skin fibroblasts. Photochem Photobiol. 2010b;86:1307–1317. doi: 10.1111/j.1751-1097.2010.00818.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamore SD, Azimian S, Horn D, Anglin BL, Uchida K, Cabello CM, Wondrak GT. The malondialdehyde-derived fluorophore DHP-lysine is a potent sensitizer of UVA-induced photooxidative stress in human skin cells. J Photochem Photobiol B. 2010c;101:251–264. doi: 10.1016/j.jphotobiol.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Park J, Kim YH, Kim DH, Kim CG, Koh JY. Induction by synaptic zinc of heat shock protein-70 in hippocampus after kainate seizures. Exp Neurol. 2000;161:433–441. doi: 10.1006/exnr.1999.7297. [DOI] [PubMed] [Google Scholar]
- Leyden JJ, Stewart R, Kligman AM. Updated in vivo methods for evaluating topical antimicrobial agents on human skin. J Invest Dermatol. 1979;72:165–170. doi: 10.1111/1523-1747.ep12676347. [DOI] [PubMed] [Google Scholar]
- Mackenzie GG, Keen CL, Oteiza PI. Zinc status of human IMR-32 neuroblastoma cells influences their susceptibility to iron-induced oxidative stress. Dev Neurosci. 2002;24:125–133. doi: 10.1159/000065691. [DOI] [PubMed] [Google Scholar]
- Magda D, Lecane P, Wang Z, Hu W, Thiemann P, Ma X, Dranchak PK, Wang X, Lynch V, Wei W, Csokai V, Hacia JG, Sessler JL. Synthesis and anticancer properties of water-soluble zinc ionophores. Cancer Res. 2008;68:5318–5325. doi: 10.1158/0008-5472.CAN-08-0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann JJ, Fraker PJ. Zinc pyrithione induces apoptosis and increases expression of Bim. Apoptosis. 2005;10:369–379. doi: 10.1007/s10495-005-0811-9. [DOI] [PubMed] [Google Scholar]
- Moller M, Adam W, Saha-Moller CR, Stopper H. Studies on cytotoxic and genotoxic effects of N-hydroxypyridine-2-thione (Omadine) in L5178Y mouse lymphoma cells. Toxicol Lett. 2002;136:77–84. doi: 10.1016/s0378-4274(02)00279-5. [DOI] [PubMed] [Google Scholar]
- Murakami M, Hirano T. Intracellular zinc homeostasis and zinc signaling. Cancer Sci. 2008;99:1515–1522. doi: 10.1111/j.1349-7006.2008.00854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Ito T, Hasegawa A. Percutaneous absorption of zin bis-(2-pyridylthio)-1,1′-dioxide and residual amounts left on the surface of the skin. Eisei Kagaku. 1967;13:323–329. [Google Scholar]
- Pierard-Franchimont C, Goffin V, Decroix J, Pierard GE. A multicenter randomized trial of ketoconazole 2% and zinc pyrithione 1% shampoos in severe dandruff and seborrheic dermatitis. Skin Pharmacol Appl Skin Physiol. 2002;15:434–441. doi: 10.1159/000066452. [DOI] [PubMed] [Google Scholar]
- Pirev E, Calles C, Schroeder P, Sies H, Kroncke KD. Ultraviolet-A irradiation but not ultraviolet-B or infrared-A irradiation leads to a disturbed zinc homeostasis in cells. Free Radic Biol Med. 2008;45:86–91. doi: 10.1016/j.freeradbiomed.2008.04.011. [DOI] [PubMed] [Google Scholar]
- Plum LM, Rink L, Haase H. The essential toxin: impact of zinc on human health. Int J Environ Res Public Health. 2010;7:1342–1365. doi: 10.3390/ijerph7041342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowlands CG, Danby FW. Histopathology of psoriasis treated with zinc pyrithione. Am J Dermatopathol. 2000;22:272–276. doi: 10.1097/00000372-200006000-00013. [DOI] [PubMed] [Google Scholar]
- Rudolf E, Cervinka M. Zinc pyrithione induces cellular stress signaling and apoptosis in Hep-2 cervical tumor cells: the role of mitochondria and lysosomes. Biometals. 2010;23:339–354. doi: 10.1007/s10534-010-9302-8. [DOI] [PubMed] [Google Scholar]
- Rutherford T, Black JG. The use of autoradiography to study the localization of germicides in skin. Br J Dermatol. 1969;81:75–86. [Google Scholar]
- Sharir H, Zinger A, Nevo A, Sekler I, Hershfinkel M. Zinc released from injured cells is acting via the Zn2+-sensing receptor, ZnR, to trigger signaling leading to epithelial repair. J Biol Chem. 2010;285:26097–26106. doi: 10.1074/jbc.M110.107490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiokawa D, Tanuma S. Molecular cloning and expression of a cDNA encoding an apoptotic endonuclease DNase gamma. Biochem J. 1998;332(Pt 3):713–720. doi: 10.1042/bj3320713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smegal D, McMahon TF, Aviado D, Montague K, Shamim N, Mostaghimi S. [EPA AD (2004 (9–30)] Zinc Pyrithione (Zinc Omadine): AD Risk Assessment for the Reregistration Eligibility Decision (RED) Document. 2004:1–51. Chemical No. 088002. Case No. 2480. DP Barcode: D308704. ID: EPA-HQ-OPP-2004-0147-0023. www.regulations.gov.
- Snyder FH, Buehler EV, Winek CL. Safety evaluation of zinc 2-pyridinethiol 1-oxide in a shampoo formulation. Toxicol Appl Pharmacol. 1965;7:425–437. doi: 10.1016/0041-008x(65)90144-4. [DOI] [PubMed] [Google Scholar]
- Sun T, Haycock JW, Szabo M, Hill RP, Macneil S. Measurement of NF-kappaB in normal and reconstructed human skin in vitro. J Mater Sci Mater Med. 2004;15:743–749. doi: 10.1023/b:jmsm.0000032812.18703.a9. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Nakazawa M, Takahashi K, Aihara M, Minami M, Hirasawa T, Ikezawa Z. Effects of zinc deficient diet on development of atopic dermatitis-like eruptions in DS-Nh mice. J Dermatol Sci. 2008;50:31–39. doi: 10.1016/j.jdermsci.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Unoshima M, Nishizono A, Takita-Sonoda Y, Iwasaka H, Noguchi T. Effects of zinc acetate on splenocytes of endotoxemic mice: enhanced immune response, reduced apoptosis, and increased expression of heat shock protein 70. J Lab Clin Med. 2001;137:28–37. doi: 10.1067/mlc.2001.111514. [DOI] [PubMed] [Google Scholar]
- Wondrak GT, Roberts MJ, Cervantes-Laurean D, Jacobson MK, Jacobson EL. Proteins of the extracellular matrix are sensitizers of photo-oxidative stress in human skin cells. J Invest Dermatol. 2003;121:578–586. doi: 10.1046/j.1523-1747.2003.12414.x. [DOI] [PubMed] [Google Scholar]
- Yui S, Nakatani Y, Hunter MJ, Chazin WJ, Yamazaki M. Implication of extracellular zinc exclusion by recombinant human calprotectin (MRP8 and MRP14) from target cells in its apoptosis-inducing activity. Mediators Inflamm. 2002;11:165–172. doi: 10.1080/09622935020138208. [DOI] [PMC free article] [PubMed] [Google Scholar]