

Abstract
Purpose
Familial exudative vitreoretinopathy (FEVR) is a group of inherited blinding eye diseases characterized by defects in the development of the retinal vessels. Recent studies have identified genetic variants in tetraspanin 12 (TSPAN12) as a cause of FEVR. The purpose of this study was to identify novel TSPAN12 mutations in Chinese patients with FEVR and to describe the associated phenotypes.
Methods
Mutation screening was performed by directly sequencing PCR products of genomic DNA with primers designed to amplify the seven coding exons and adjacent intronic regions of the FEVR-causing gene TSPAN12. Clinical phenotypes of the patients with TSPAN12 mutations were documented. Wild-type and mutant TSPAN12 proteins were assayed for the Norrin-β-catenin signaling pathway with luciferase reporter assays.
Results
Three novel heterozygous mutations in TSPAN12 were identified: c.566G>A (p.C189Y), c.177delC (p.Y59fsX67), and c.C254T (p.T85M). All three mutations involved highly conserved residues and were not present in 200 normal individuals. Ocular phenotypes included increased ramification of the peripheral retinal vessels, a peripheral avascular zone, inferotemporal dragging of the optic disc and macula, and retinal folds. The probands showed relatively severe retinopathy, whereas the other family members were often asymptomatic. In SuperTopFlash (STF) cell line transfection studies, C189Y, Y59fsX67, and T85M mutants failed to induce luciferase reporter activity in response to Norrin.
Conclusions
We found three novel TSPAN12 mutations in Chinese patients with autosomal dominant FEVR, and suggest that TSPAN12 mutations cause FEVR. The phenotypes associated with the TSPAN12 mutations showed extensive variation in disease severity among members of the same family, which implied the complexity of FEVR mutations and phenotypes.
Introduction
Familial exudative vitreoretinopathy (FEVR, OMIM 133780, 305390, 605750, 601813, 613310) is a hereditary disorder with defects in the development of retinal vasculature [1]. A wide range of clinical manifestations have been found in patients with FEVR. Severely affected patients suffer from blindness during infancy, and manifest retinal folds or detachments. In contrast, mildly affected patients usually have no visual problems and do not need to undergo fluorescein angiography unless a severely affected family member is diagnosed.
In the past two decades or so, four genes that cause FEVR have been identified: NDP (OMIM 300658, X-linked), LRP5 (OMIM 603506, dominant and recessive), FZD4 (OMIM 604579, dominant) [2-4], and TSPAN12 (OMIM 613138, dominant and recessive) [5-10]. All the proteins encoded by the FEVR genes have been found to participate in the Norrin-β-catenin signaling pathway [4-7]. In addition, another locus for autosomal dominant FEVR (adFEVR), EVR3, has been mapped to chromosomal locus 11p12-p13 [11]. In this study, three novel TSPAN12 mutations in patients with FEVR were identified, and we demonstrated that these three mutants failed to induce luciferase reporter activity in HEK293 transfection studies.
Methods
Patients and clinical examinations
This study adhered to the ARVO statement on human subjects and was approved by the Institutional Review Board of the Xinhua Hospital affiliated with the Shanghai Jiao Tong University School of Medicine and the Institutional Review Board of the Sichuan Academy of Medical Sciences and Sichuan Provincial People’s Hospital, China. The study complied with the guidelines of the Declaration of Helsinki and the Guidance of Sample Collection of Human Genetic Disease of the Ministry of Public Health of China. Informed consent was obtained from all participants in the study. 85 outpatients diagnosed as FEVR were recruited: 47 were male and 38 were female. The age of all participants ranged from 2 months to 14 years old. Their parents were also recruited in this study. All participants were in good health when they were recruited, except for the eye. For minors, written consent was obtained from their parents. All participants underwent careful ophthalmology examinations, and were diagnosed by a clinical ophthalmologist and a geneticist based mainly on the presence of retinal abnormalities in fundus photographs deemed typical of FEVR, which included an avascular peripheral retina together with exudative and/or sequelae of retinal traction, such as macular ectopia, retinal folds, and retinal detachment. Fundus fluorescein angiography (FFA) was performed in selected cases to confirm further the diagnosis. In the 200 normal matched controls, all individuals underwent an eye examination and no signs of eye diseases were observed. Venous blood samples were obtained from all subjects in EDTA Vacutainers.
Detection of TSPAN12 mutations
Genomic DNA samples were extracted from the peripheral blood of the subject patients using a whole blood DNA extraction kit (BioTeke, Beijing, China). Venous blood in EDTA vautainers was stored in 4 ºC and processed within 24 h after blood drawn. Genomic DNA samples were extracted blood DNA extraction kit following manufacture’s instruction (Bioteke). All patients had been confirmed beforehand as carrying no mutations in FZD4, LRP5, or NDP. To identify mutations in the coding exons of the TSPAN12 gene, oligonucleotide primers flanking the coding regions and neighboring intronic sequences were designed (Table 1). The exons of the TSPAN12 gene were analyzed via direct sequencing of the PCR products. PCR working conditions are as following: initial denature temperature 95 ºC for 3 min, followed by 33 cycles of reaction: template denature at 95 ºC for 15 s, annealing for 15 s at 59 ºC and extension at 72 ºC for 20 s. A final step of 7 min reaction extension at 72 ºC was applied to fill in the gaps of PCR product. Amplified products were purified using the QIAquick Gel Extraction Kit (Qiagen, Valencia, CA), and were sequenced with forward and reverse primers by the BigDye Terminator v3.1 Cycle Sequencing Kit (ABI Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions. After sequence changes were detected in the probands, samples from the other family members were analyzed with direct sequencing.
Table 1. Primer sequences of TSPAN12.
| Exon | Forward primer (5′>3′) | Reverse primer (5′>3′) | Amplicon length (bp) |
|---|---|---|---|
| 2 |
GGTGAGATGTCCCGTGTTCT |
TCAAAGGCATTTTAAGAAGGTCA |
340 |
| 3 |
AATCCTGCAGTGAATGTTACG |
AGGCGCACCTTAAGGAGAAT |
314 |
| 4 |
TGCTATGTCTTGGGTGCATT |
AAACGAAAGCGTCCCTTCTT |
331 |
| 5 |
TGCCTCTGTTTTCTTGGTCA |
TTCACCTTCTGCCATGATTG |
367 |
| 6 |
CGAGTATGCGTGTGTACGTG |
GAAGAAAAGCAGGCCATGAA |
393 |
| 7 |
TTTGTGGTTTCTGAGGCTGA |
TTTCTTCTGCTTCTCCCCATA |
333 |
| 8 | ACAGATTGTTTGCTTTCAT | GCTTAGGTGTTATTTTATGGC | 505 |
Primer used for amplification and sequence analysis of human TSPAN12.
Construction of expression plasmids
The cDNA encoding wild-type TSPAN12 (Origene, Rockville, MD) was subcloned in-frame into the pCMV-entryMyc-Flag vector (Origene) using SgfI and MluI sites. All mutations were introduced into the wild-type TSPAN12 cDNA by site-directed mutagenesis using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA). LRP5, FZD4, and Norrin expression vectors (generously provided by Dr. Jeremy Nathans of Johns Hopkins University) have been described previously [12]. The recombinant plasmids containing TSPAN12-Myc-Flag fusion constructs were first verified with DNA sequencing, and then prepared for transfection using Qiagen plasmid Maxi preparation kit (Qiagen).
Luciferase assays
The SuperTopFlash (STF) reporter, in which firefly luciferase was driven by seven lymphoid enhancer factor/T-cell factor (LEF/TCF) consensus binding sites, was a kind gift from Dr. Jeremy Nathans (John Hopkins University, Baltimore, MD). This reporter plasmid was stably transfected into HEK 293 cells as reported previously to generate the STF cell line. In 24-well plates, 160 K STF cells/well were transfected with 800 ng DNA and 1.5 µl Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA). The DNA mix contained 200 ng of Norrin, 200 ng of FZD4, 200 ng of LRP5, 100 ng of pSV-β-galactosidase control vector and 100 ng of TSPAN12 plasmid (wild-type or mutation). Forty-eight hours after transfection, the cells were harvested and washed twice with PBS (1X; 120 mM NaCl, 20 mM KCl 10 mM NaPO4, 5 mM KPO4, pH 7.4), and the luciferase activities were measured with a Dual-Luciferase Assay Kit (Promega) according to the manufacturer’s instructions. Reporter activity was normalized to the coexpressed β-galactosidase activity in each well. Each test was performed in triplicate. The reporter assay was repeated three times, and a representative result was obtained.
Expression of TSPAN12 in Cos 7 cells
Cos 7 cells (American Type Culture Collection [ATCC], Manassas, VA) were cultured in DMEM (ATCC, Manassas, VA) with high glucose (HyClone) supplemented with 10% fetal bovine serum and 1% (vol/vol) penicillin/streptomycin at 37 °C in a 5% CO2 atmosphere. Cells were seeded in six-well plates (Corning Inc., Corning, NY) and transfected at 50% confluency with 1 μg of wild-type and mutant pCMV6-entry-TSPAN12 or empty vector using Lipofectimine 2000 (Invitrogen) according to the manufacturer’s instructions. After 48 h, the cells were washed with PBS, and fixed with 4% paraformaldehyde (PFA) for 15 min. Anti-Flag antibody was used to detect TSPAN12 expression using standard immunostaining method.
Results
Identification of novel TSPAN12 mutations and phenotypes
We identified three proband patients with FEVR in our clinic. To identify causative genes for FEVR, we sequenced all the exons and the flanking intonic sequences for four known FEVR genes: FZD4, LRP5, Norrin, and TSPAN12. Three novel mutations in the coding sequence of the TSPAN12 gene were found in these families, including c.566G>A (p.C189Y), c.177delC (Y59fsX67), and c.C254T (p.T85M; Figure 1).
Figure 1.

Chromatograms and pedigrees of three families with familial exudative vitreoretinopathy. Three novel mutations were identified in TSPAN12. A: In Family A, the affected mother and son had the c.566G>A (p.C189Y) mutation. C: In Family B, the patient and her affected mother had the c.177delC (p.Y59fsX67) mutation. E: In Family C, the affected mother and son had the c.C254T (p.T85M) mutation. The columns from left to right display the pedigree and the sequence chromatograms of these patients (A, C, E) and the normal controls (B, D, F). Arrows indicate the positions of the altered nucleotides.
In Family A, we identified a missense mutation in exon 7, c.566G>A, segregating with the disease. This mutation led to the replacement of a cysteine by a tyrosine at codon 189 (p.C189Y; Figure 1), which involved a highly evolutionarily conserved residue (Figure 2A) from Homo sapiens to Danio rerio. The proband of the family was a 1-year-old boy with bilateral retinal folds affecting the macula (Figure 3). His mutation-carrying mother had normal vision but with areas of avascular zone and abnormal vessels in the peripheral retina (Figure 3).
Figure 2.

Protein sequence alignment of human TSPAN12 with its orthologs. The conserved amino acid residues are shaded. The orthologs are from the following species: Homo sapiens (NP_036470), Pan troglodytes (XP_001142754), Musmusculus (NP_766595), Rattus norvegicus (NP_001015026), Bos taurus (NP_001039977), Equus caballus (XP_001502093), Canis lupus familiaris (XP_855095), Monodelphis domestica (XP_001364876), Gallus gallus (NP_001007850), Taeniopygia guttata (XP_002192381), Ornithorhynchus anatinus (XP_001516347), and Danio rerio (NP_957446). A: The residue of the missense mutation p.C189Y is highly conserved. B: The residue of the missense mutation p.T85M is also highly conserved.
Figure 3.
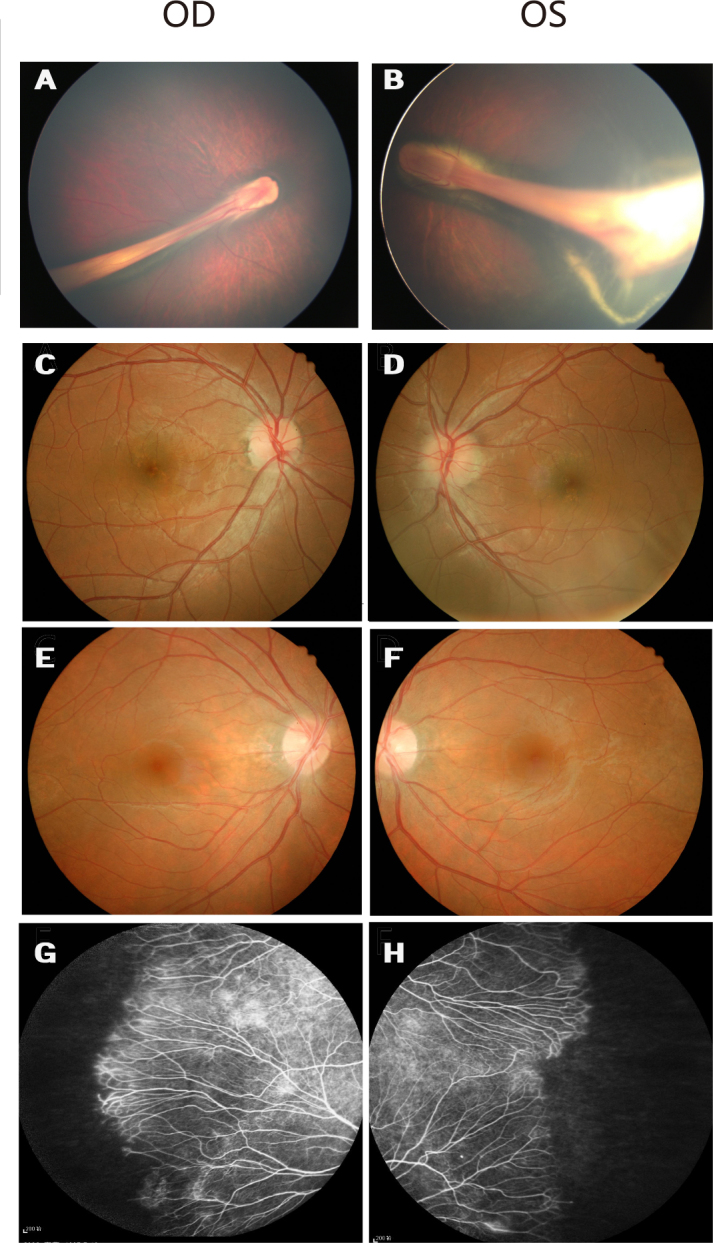
Fundus photographs of Family A with familial exudative vitreoretinopathy. A and B: Fundus photographs of the proband from Family A (individual II:1 in Figure 1), showing a retinal fold and a dragged macula. C and D: The unaffected father without the mutation has normal fundi. E and F: Fundus photographs of the asymptomatic mother with the c.566G>A mutation show normal posterior fundi. G and H: The mother has areas of avascularity and abnormal vessels in the peripheral retina.
We identified one base pair deletion in exon 4, c.177delC, in the proband of Family B and her asymptomatic mother. This mutation resulted in a frameshift followed by premature termination at codon 67 (p.Tyr59fsX67; Figure 1), which also involved highly evolutionarily conserved residues (Figure 2C) from Homo sapiens to Danio rerio. The proband was a 2-year-old girl with a complaint of esotropia. Fundus examination showed typical falciform retinal folds across the fovea in both eyes (Figure 4). Her father had normal fundi, but her mother had typical fundus changes in FFA examination, including increased ramification of the peripheral retinal vessels and an avascular zone in the peripheral retina, though she had normal visual acuity of 1.0 in both eyes and had no vision complaints (Figure 4).
Figure 4.
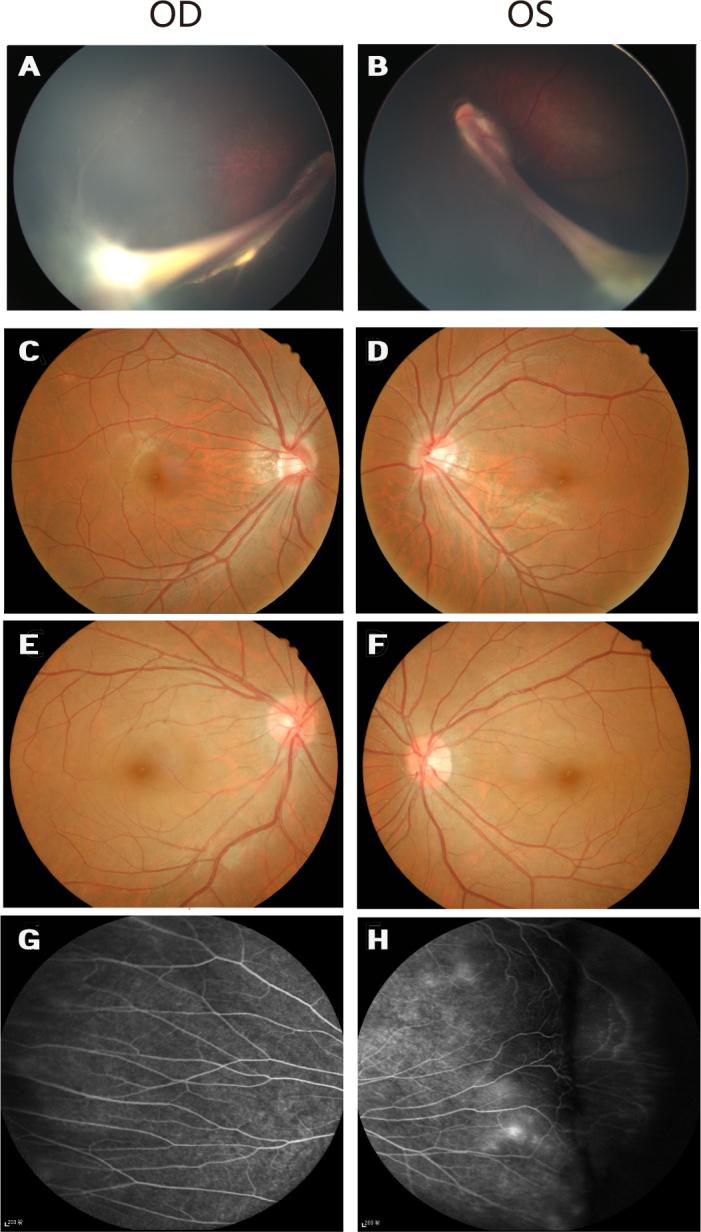
Fundus photographs of Family B with familial exudative vitreoretinopathy. A and B: Fundus photographs of the proband from Family B (individual II:1 in Figure 1), showing the retinal vessels drawn up in a retinal fold that is obscuring the macula. C and D: The unaffected father has normal fundi. E and F: Fundus photographs of the asymptomatic mother with the c.177delC mutation show normal posterior fundi. G and H: The mother has increased vessel branching in the equatorial area and an avascular zone on the peripheral retina.
In Family C, a missense mutation c.C254T (p.T85M) segregating with the disease was identified (Figure 1). This mutation led to the change in a threonine residue to a methionine at codon 85 (p.T85M), which is extremely conserved from Homo sapiens to Danio rerio. (Figure 1, Figure 2B). The proband was a 7-year-old boy with dragged disc in his left eye (Figure 5). There was pigment deposit in his right retina. His mutation-carrying mother had normal vision but had areas of avascular peripheral retina. All three mutations cosegregated with the disease phenotype of the respective families, and were not detected in 200 ethnically matched control individuals.
Figure 5.
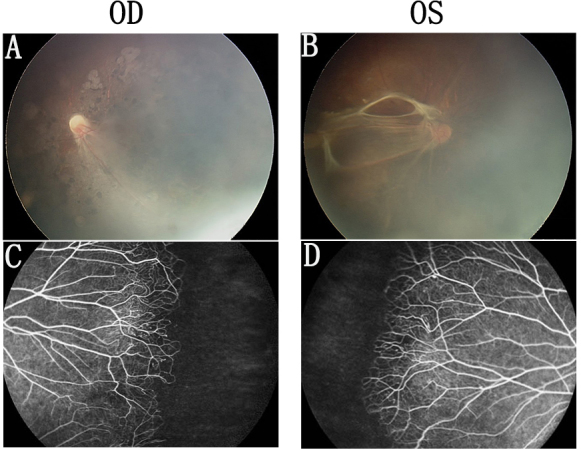
Fundus photographs of Family C with familial exudative vitreoretinopathy. A and B: Fundus photographs of the proband from Family C (individual II:1 in Figure 1), showing a retinal fold and a dragged macula. C and D: The mother has areas of avascularity and abnormal vessels in the peripheral retina.
Defective Norrin signaling mediated by mutant TSPAN12 proteins in vitro
To determine the effect of the TSPAN12 mutants on its biologic activity in Norrin/β-catenin signaling, we performed luciferase assays in STF cells using wild-type and three mutant TSPAN12 cDNAs. c.177delC, c.566G>A, and c.C254T were introduced into TSPAN12 with the site-direct mutagenesis method. Our in vitro analyses suggested that compared to wild-type TSPAN12, all three TSPAN12 mutants failed to induce luciferase reporter activity in STF cells in response to Norrin (Figure 6). Western blot analysis indicated that the T85M and Y59fsX67 mutant proteins were not stable (Figure 7). The C189Y mutant expression level was compatible to that of the wild-type (Figure 7 and Figure 8).The results suggest that the three TSPAN12 mutations identified in our study are pathogenic, and are consistent with the notion that defective Norrin/TSPAN12 signaling underlies FEVR.
Figure 6.
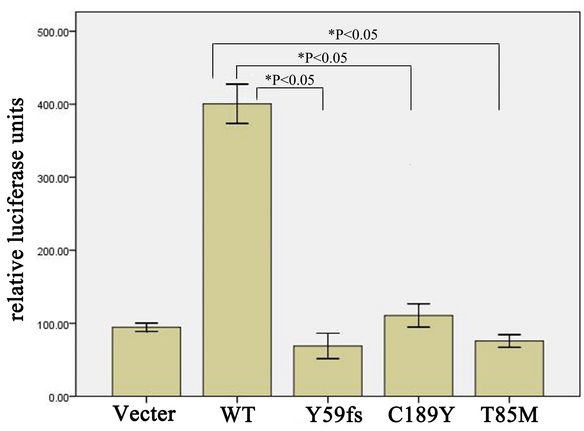
Luciferase assays with the SuperTopFlash cell line transfected with the indicated plasmids. SuperTopFlash (STF) cells/well were transfected with 800 ng DNA (200 ng of Norrin, 200 ng of FZD4, 200 ng of LRP5, 100 ng of pSV-β-galactosidase control vector, and 100 ng of TSPAN12 plasmid [wild-type or mutation]) and 1.5 µl Lipofectamine 2000 transfection reagent. Forty-eight hours after transfection, the cells were harvested and washed twice with PBS. Luciferase activities were measured with a dual-luciferase assay kit. Reporter activity was normalized to the coexpressed β-galactosidase activity in each well. Each test was performed in triplicate. The reporter assay was repeated three times, and a representative result was obtained.
Figure 7.
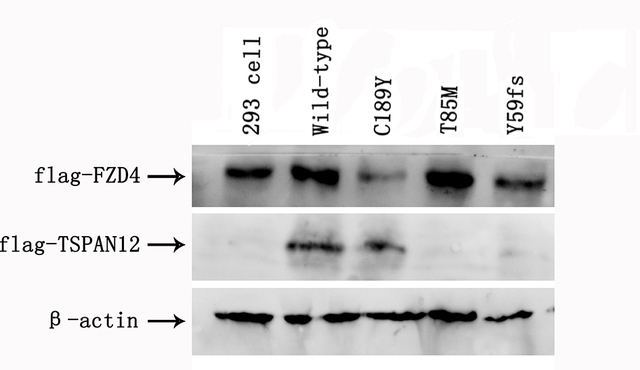
Western blot analysis by SDS-PAGE of the TSPAN12 mutants. Total protein (10 μg) isolated from cell lysates from luciferase assays was mixed with sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) loading buffer and subjected to SDS-PAGE and western blot analysis using anti-Flag antibody to detect TSPAN12 and FZD4 expression. Beta-actin was used as the loading control. The expression level of TSPAN12 C189Y was compatible with that of the wild-type. However, the T85M and Y59fs mutant proteins were not stable.
Figure 8.

Immunofluorescence staining of the TSPAN12 C189Y mutant. Cos 7 cells were transfected either with human wild-type or mutant TSPAN12 cloned into the pCMV6-entry vector, or empty vector. Cells were washed with PBS after 48 h and fixed with 4% PFA for 15 min. Mouse monoclonal anti-Flag antibody and Alexa Fluor 594 goat anti-mouse immunoglobulin (IgG) secondary antibody were used to detect TSPAN12 expression with the standard immunostaining method. Red channel, TSPAN12; blue channel, 4',6-diamidino-2-phenylindole (DAPI) for nuclei staining.
Discussion
In our study, three novel TSPAN12 mutations were detected in three families with FEVR, but not in 200 normal individuals. The clinical signs and symptoms showed variations among the patients with TSPAN12 mutations, from mild avascular peripheral retina with retinal degeneration to severe bilateral retinal folds. Our data suggest variable expressivity of FEVR: The probands manifested relatively severe retinopathy, whereas the other family members had normal vision and were often asymptomatic, as had been reported in cases carrying mutations in FZD4 or LRP5 [4,13-15]. Our results are consistent with the data reported by Kashani et al. [16]. These family members might develop advanced FEVR later on, which could deteriorate to vision loss. Therefore, it is necessary to screen immediate family members of the patients with FEVR with retina angiography and clinical treatment.
The location of three novel TSPAN12 mutations identified in our study reveals their pathogenic nature (Figure 9). The c.177delC mutation in exon 4 caused a frameshift with a premature termination at condon 67, resulting in truncated proteins that might not be synthesized because of nonsense-mediated decay of the messenger ribonucleic acid. The highly conserved cysteine residue at position 189 among tetraspanin family members plays a role in the formation of disulfide bridges, and is located in the large extracellular loop between the third and fourth transmembrane region. This extracellular loop is known to harbor most of the protein–protein interaction sites described for tetraspanins [17]. Thus, the substitution of a tyrosine residue for the cysteine at position 189 disrupts one of these interactions and, therefore, may have significant functional consequences, such as the interaction with the FZD4/LRP5/Norrin protein complex. Furthermore, the 189th cysteine is next to another TSPAN12 mutation G188R, a known FEVR mutation [6]. T85M is likely to bring about the change in the second transmembrane domain, as the other three known FEVR mutations (L101H, C105R, A237P) in the transmembrane domain do.
Figure 9.

Diagram showing 9 known TSPAN12 mutations and three novel mutations identified in this study. The novel mutations are red.
In addition, in our STF cell line transfection studies, the defective luciferase reporter activity mediated by the mutant TSPAN12 proteins revealed that Y59fsX67, C189Y, and T85M mutations in TSPAN12 led to a decrease in Norrin/FZD4/LRP5 signaling, which further suggested these three mutations were pathogenic. In conclusion, our study has identified three novel TSPAN12 mutations causing autosomal dominant FEVR. The findings provide additional evidence that mutations in TSPAN12 are causative in patients with FEVR, and TSPAN12 is crucial for the development of the retinal vessels.
Acknowledgments
We thank all of the participating FEVR patients and their relatives. This research was supported by the National Natural Science Foundation of China (81271045 (P. Z.), and 81025006, 81170883 (Z. Y.), 81271007 (X. Z.), 81300802 (L. H.)), Department of Science and Technology of Sichuan Province, China (SZ20120209, Z. Y.) and Shanghai Health Bureau (2012Y044, Y. X.). All authors have declared that they have no conflict of interest. Zhenglin Yang (zliny@yahoo.com) and Peiquan Zhao (zhaopeiquan@126.com) are co-corresponding authors of this paper.
References
- 1.Criswick VG, Schepens CL. Familial exudative vitreoretinopathy. Am J Ophthalmol. 1969;68:578–94. doi: 10.1016/0002-9394(69)91237-9. [DOI] [PubMed] [Google Scholar]
- 2.Chen ZY, Battinelli EM, Fielder A, Bundey S, Sims K, Breakefield XO, Craig IW. A mutation in the Norrie disease gene (NDP) associated with X-linked familial exudative vitreoretinopathy. Nat Genet. 1993;5:180–3. doi: 10.1038/ng1093-180. [DOI] [PubMed] [Google Scholar]
- 3.Robitaille J, MacDonald ML, Kaykas A, Sheldahl LC, Zeisler J, Dube MP, Zhang LH, Singaraja RR, Guernsey DL, Zheng B, Siebert LF, Hoskin-Mott A, Trese MT, Pimstone SN, Shastry BS, Moon RT, Hayden MR, Goldberg YP, Samuels ME. Mutant Frizzled-4 disrupts retinal angiogenesis in familial exudative vitreoretinopathy. Nat Genet. 2002;32:326–30. doi: 10.1038/ng957. [DOI] [PubMed] [Google Scholar]
- 4.Toomes C, Bottomley HM, Jackson RM, Towns KV, Scott S, Mackey DA, Craig JE, Jiang L, Yang Z, Trembath R, Woodruff G, Gregory-Evans CY, Gregory-Evans K, Parker MJ, Black GC, Downey LM, Zhang K, Inglehearn CF. Mutations in LRP5 or FZD4 underlie the common familial exudative vitreoretinopathy locus on chromosome 11q. Am J Hum Genet. 2004;74:721–30. doi: 10.1086/383202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Junge HJ, Yang S, Burton JB, Paes K, Shu X, French DM, Costa M, Rice DS, Ye W. TSPAN12 regulates retinal vascular development by promoting Norrin- but not Wnt- induced FZD4/beta-catenin signaling. Cell. 2009;139:299–311. doi: 10.1016/j.cell.2009.07.048. [DOI] [PubMed] [Google Scholar]
- 6.Nikopoulos K, Gilissen C, Hoischen A, van Nouhuys CE, Boonstra FN, Blokland EA, Arts P, Wieskamp N, Strom TM, Ayuso C, Tilanus MA, Bouwhuis S, Mukhopadhyay A, Scheffer H, Hoefsloot LH, Veltman JA, Cremers FP, Collin RW. Next-generation sequencing of a 40 Mb linkage interval reveals TSPAN12 mutations in patients with familial exudative vitreoretinopathy. Am J Hum Genet. 2010;86:240–7. doi: 10.1016/j.ajhg.2009.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poulter JA, Ali M, Gilmour DF, Rice A, Kondo H, Hayashi K, Mackey DA, Kearn LS, Ruddle JB, Craiq JE, Pierce EA, Downey LM, Mohamed MD, Markham AF, Inqlehearn CF, Toomes C. Mutations in TSPAN12 cause autosomal-dominant familial exudative vitreoretinopathy. Am J Hum Genet. 2010;86:248–53. doi: 10.1016/j.ajhg.2010.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kondo H, Kusaka S, Yoshinaga A, Uchio E, Tawara A, Hayashi K, Tahira T. Mutations in the TSPAN12 gene in Japanese patients with familial exudative vitreoretinopathy. Am J Ophthalmol. 2011;151:1095–100. doi: 10.1016/j.ajo.2010.11.026. [DOI] [PubMed] [Google Scholar]
- 9.Yang H, Xiao X, Li S, Mai G, Zhang Q. Novel TSPAN12 mutations in patients with familial exudative vitreoretinopathy and their associated phenotypes. Mol Vis. 2011;17:1128–35. [PMC free article] [PubMed] [Google Scholar]
- 10.Poulter JA, Davidson AE, Ali M, Gilmour DF, Parry DA, Mintz-hittner HA, Carr IM, Bottomley HM, Long VW, Downey LM, Sergouniotis PI, Wright GA, MacLaren RE, Moore AT, Webster AR, Inglehearn CF, Toomes C. Recessive mutations in TSPAN12 cause retinal dysplasia and severe familial exudative vitreoretinopathy (FEVR). Invest Ophthalmol Vis Sci. 2012;53:2873–9. doi: 10.1167/iovs.11-8629. [DOI] [PubMed] [Google Scholar]
- 11.Downey LM, Keen TJ, Roberts E, Mansfield DC, Bamashmus M, Inglehearn CF. A new locus for autosomal dominant familial exudative vitreoretinopathy maps to chromosome 11p12–13. Am J Hum Genet. 2001;68:778–81. doi: 10.1086/318790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Q, Wang Y, Dabdoub A, Smallwood PM, Williams J, Woods C, Kelley MW, Jiang L, Tasman W, Zhang K, Nathans J. Vascular development in the retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell. 2004;116:883–95. doi: 10.1016/s0092-8674(04)00216-8. [DOI] [PubMed] [Google Scholar]
- 13.Benson WE. Familial exudative vitreoretinopathy. Trans Am Ophthalmol Soc. 1995;93:473–521. [PMC free article] [PubMed] [Google Scholar]
- 14.Qin M, Hayashi H, Oshima K, Tahira T, Hayashi K, Kondo H. Complexity of the genotype-phenotype correlation in familial exudative vitreoretinopathywithmutations in the LRP5 and/or FZD4 genes. Hum Mutat. 2005;26:104–12. doi: 10.1002/humu.20191. [DOI] [PubMed] [Google Scholar]
- 15.Downey LM, Bottomley HM, Sheridan E, Ahmed M, Gilmour DF, Inglehearn CF, Reddy A, Agrawal A, Bradbury J, Toomes C. Reduced bone mineral density and hyaloid vasculature remnants in a consanguineous recessive FEVR family with a mutation in LRP5. Br J Ophthalmol. 2006;90:1163–7. doi: 10.1136/bjo.2006.092114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kashani AH, Learned D, Nudleman E, Drenser KA, Capone A, Trese MT. High prevalence of peripheral retinal vascular anomalies in family members of patients with familial exudative vitreoretinopathy. Ophthalmology. 2014;121:262–8. doi: 10.1016/j.ophtha.2013.08.010. [DOI] [PubMed] [Google Scholar]
- 17.Garcia-España A, Chung PJ, Sarkar IN, Stiner E, Sun TT, Desalle R. Appearance of new tetraspanin genes during vertebrate evolution. Genomics. 2008;91:326–34. doi: 10.1016/j.ygeno.2007.12.005. [DOI] [PubMed] [Google Scholar]