

Abstract
Purpose
To describe the genotype-phenotype correlation and serial observations in a five-generation Czech family with X-linked retinitis pigmentosa (XLRP) associated with severe visual impairment in women.
Methods
Comprehensive ophthalmological examination including spectral domain optical coherence tomography (SD-OCT) was performed. Based on the pedigree structure and women being severely affected, autosomal dominant inheritance was suspected, and screening for known mutations by genotyping microarray was performed. Subsequently, direct sequencing of ORF15 RPGR was undertaken.
Results
Eighteen family members (nine women and nine men) were examined. A pathogenic variant, c.2543del in ORF15 of RPGR, was found to segregate with disease. The oldest woman and her two sisters had no perception of light in their sixth decade. Four women and five men had signs and symptoms of typical XLRP, including moderate to high myopia. Three other women also had moderate to high myopia and myopic astigmatism but without the presence of bone spicule-like formation. Severe disruption of macular architecture on SD-OCT was equally common in both sexes. Only one 32-year-old female carrier had clinically normal findings. Subfoveal choroidal thickness was decreased in all affected men and in all female carriers, except the only carrier with a normal fundus examination.
Conclusions
The c.2543del mutation in ORF15 of RPGR is associated with a severe phenotype in the women in this family. The presence of a significant myopic refractive error, in the absence of male-to-male transmission, may be indicative of X-linked inheritance. Measurements of choroidal thickness may help in clinically identifying carrier status.
Introduction
Retinitis pigmentosa (RP) is a diverse group of disorders characterized by progressive retinal dysfunction, and caused by mutations in at least 50 genes (RetNet accessed April 1, 2014). It has been estimated that about 40–50% of disease remains unaccounted for by screening of known genes, which suggests many more causative genes await identification [1,2]. Clinically, RP presents with night blindness and peripheral visual field constriction, with later central visual loss. At the cellular level, photoreceptor cell death occurs over time with the initial loss of rods, followed by cones at later stages of disease [2,3]. The phenotype, including onset and progression, shows considerable variability, even within families [4,5].
X-linked RP (XLRP) accounts for 5–20% of RP [2]. Three disease-causing genes have been identified to date: retinitis pigmentosa GTPase regulator (RPGR; OMIM 312610) [6], retinitis pigmentosa 2 (RP2; OMIM 312600) [7], and oral-facial-digital syndrome type 1 (OFD1; OMIM 300170) gene [8].
Mutations within RPGR cause approximately 80% of all XLRP, the highest rate of any RP locus identified to date [2,9]. The RPGR gene has 23 exons, including the alternatively spliced exons 9a, ORF15, 15a, and 15b [9,10]. Most mutations are located in ORF15, which is predominantly present in retinal transcripts. RPGR participates in intracellular protein trafficking through the connecting cilia of the photoreceptor cells [9-11].
The phenotype of XLRP associated with RPGR is considered one of the most severe, with an early onset in men, usually in the first or second decade, leading to significant visual impairment by the fourth decade. In female carriers, the disease is typically milder, presenting with a tapetal-like reflex or peripheral retinal pigmentary deposits, and can be asymptomatic [4,5,12].
Because of the anticipated introduction of gene therapy approaches, it is important to document specific genotype-phenotype correlations in detail. Herein we report the unusual severe phenotype associated with a c.2543del ORF15 RPGR mutation with profound visual loss in female carriers.
Methods
A large five-generation family of Caucasian Czech background with RP was studied. Graphical presentation of the family tree was done using HaploPainter software [13] (Haplopainter). The research followed the tenets of the Declaration of Helsinki, and informed consent approved by the local ethics committee was obtained from each participant before the study.
Molecular genetic analysis
Blood samples (6 ml) were taken from participating family members. DNA was extracted from peripheral blood leucocytes using the Nucleon Genomic DNA Extraction Kit BACC3 according to the manufacturer’s instructions (GE Healthcare, Little Chalfont, Buckinghamshire, UK). Genotyping microarray version 2.0 (Asper, Tartu, Estonia) testing was used for initial screening in the proband. This array simultaneously analyzes 370 known mutations and polymorphisms within 15 genes known to be implicated in autosomal dominant RP. Next, ORF15 screening of the RPGR gene (reference sequence NM_001034853.1) was performed at the National Genetic Reference Laboratory in Manchester, UK, as previously described [9].
Description of nucleotide and protein changes was as recommended by the Human Genome Variation Society [14,15] (HGVS).
Clinical assessment
Clinical examination included best corrected visual acuity (BCVA) measurements using Early Treatment of Diabetic Retinopathy Study (ETDRS) charts, slit-lamp evaluation including funduscopy, Goldmann intraocular pressure measurements, the Lanthony 15-hue desaturated test, and static automatic perimetry (M-700, Medmont International, Vermont, Australia, and Peristat 433, G. Rodenstock Instruments GmbH, Ottobrunn-Riemerling, Germany). Spectral domain optical coherence tomography (SD-OCT) and peripapillary retinal nerve fiber layer (RNFL) thickness measurements were performed using two spectral-domain devices (Spectral OCT/SLO, OTI Ophthalmic Technologies Inc., Toronto, Canada, and Spectralis, Heidelberg Engineering GmbH, Heidelberg, Germany) allowing in vivo retinal cross sectioning with an axial resolution of 6 µm. RNFL thickness was acquired by circular scans 3.46 mm in diameter around the optic nerve in the temporal (316°–45°), superior (46°–135°), nasal (136°–225°), and inferior (226°–315°) quadrants. Choroidal thickness was measured manually on horizontal foveal SD-OCT scans.
The inner segment ellipsoid (ISe) band within the central 3.0 mm on the horizontal mid-line scan (gray-scale image) of the fovea was classified into three categories: absent; partially intact, i.e., a distinct line within the central 1.0 mm; and intact. Foveal minimum thickness, i.e., the distance between the signal transition at the vitreoretinal interface and the RPE layer in an area of the central 1.0 mm, was measured manually on horizontal SD-OCT scans. Family members refused to undergo electroretinography, since individual II:5 (Figure 1) was convinced that she had lost her residual light projection immediately after this examination in the past.
Figure 1.
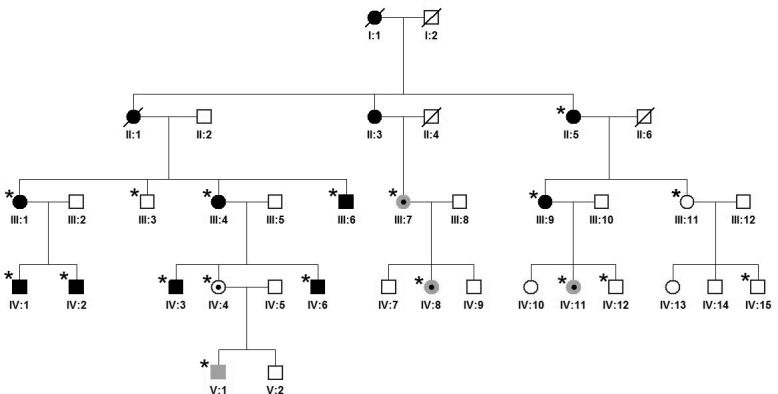
Pedigree of a family with retinitis pigmentosa associated with a c.2543del mutation in RPGR ORF15. Clinically examined individuals are indicated with an asterisk. All family members with bone spicules indicative of typical retinitis pigmentosa (RP) are shown shaded in black as affected. Individuals with myopia (−3.0 to −15.0 diopter sphere) but no pigmentary retinopathy are shown shaded in gray. Individuals III:4, III:9, IV:2, IV:4, and IV:11 were positive for the disease-causing mutation, whereas III:3 and IV:12 were negative. For individuals II:5, III:1, III:6, III:7, III:11, IV:1, IV:3, IV:6, IV:8, IV:15, and V:1, no DNA was available for laboratory investigation.
Results
The pedigree is shown in Figure 1. No pathogenic change was detected with genotyping microarray analysis of known mutations in 15 genes implicated in dominant RP. Next, an X-linked mode of inheritance was considered since there was no male-to-male transmission in the family (Figure 1) and several women with and without bone spicules had high myopia. Myopia has been reported in patients with RP2 and RPGR mutations, particularly in those who harbor ORF15 pathogenic changes [4,5,12,16].
Sequence analysis of the RPGR gene in the proband (III:4) identified a c.2543del; p.(Glu848Glyfs*241) mutation in ORF15 of RPGR. The same sequence variant was also present in two other family members with RP (III:9 and IV:2): one woman with high myopia and no pigmentary retinopathy (IV:11) and one woman who appeared clinically normal (IV:4). Two clinically unaffected first-degree relatives (III:3 and IV:12) did not harbor c.2543del in RPGR (Figure 1, Table 1). In summary, the mutation segregated with disease in the pedigree.
Table 1. Serial clinical observations in individuals from a family with retinitis pigmentosa caused by a c.2543del in RPGR ORF15.
| Individual/gender | Age (yrs) |
BCVA |
Subjective refraction (DS/DC) |
Pigmentary deposits |
Color Vision Deficiencies |
Mutation | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| RE | LE | RE | LE | RE | LE | RE | LE | |||
| II:5/F |
67 | LP | HM | −15.0 | −15.0 | Y | Y | Not tested – obligate carrier |
||
| 72 |
TB |
TB |
UA |
UA |
Y |
Y |
||||
| III:1/F |
58 | 0.63 | 0.63 | −6.0/-2.5 | −4.0/-1.5 | Y | Y | Not tested – obligate carrier |
||
| 64 |
0.2 |
0.64 |
−6.0/-2.5 |
−5.0/-1.0 |
Y |
Y |
Moderate tritan |
Mild protan |
||
| III:3/M |
52 |
1.0 |
1.0 |
UA |
UA |
N |
N |
Not found |
||
| III:4/F |
47 | 0.25 | 0.4 | −15.0/-0.25 | −14.0/-0.75 | Y | Y | Present | ||
| 54 | 0.2 | 0.32 | −11.5 | −11.5 | Y | Y | Mild tritan | Mild protan | ||
| 58 |
0.2 |
0.4 |
−11.5 |
−11.5 |
Y |
Y |
||||
| III:6/M |
48 |
0.16 |
0.16 |
−6.5/-1.5 |
−6.0/-1.5 |
Y |
Y |
Not tested |
||
| III:7/F |
53 |
0.5 |
0.63 |
−11.0/-1.25 |
−8.0/-2.0 |
N |
N |
Not tested |
||
| III:9/F |
44 | 0.04 | 1.0 | −11.0/-2.0 | −4.0/-2.0 | Y | N | Present | ||
| 50 |
0.03 |
1.0 |
−11.0/-2.0 |
−4.5/-2.0 |
Y |
Y |
Impossible to test due to visual impairment |
Mild tritan |
||
| III:11/F |
41 |
1.0 |
1.0 |
None |
None |
N |
N |
Not tested |
||
| IV:1/M |
42 |
0.25 |
0.25 |
−3.0/-2.0 |
−3.0/-2.0 |
Y |
Y |
Severe protan |
Severe protan |
Not tested |
| IV:2/M |
32 | 0.4 | 0.5 | −8.5/-3.5 | −9.0/-1.0 | Y | Y | Present | ||
| 38 |
0.32 |
0.4 |
−9.5/-3.0 |
−9.5/-3.5 |
Y |
Y |
Moderate tritan |
Mild protan |
||
| IV:3/M |
23 | 0.5 | 0.5 | −11.0 | −11.0/-1.0 | Y | Y | Not tested | ||
| 27 | 0.4 | 0.4 | −11.0 | −11.0 | Y | Y | ||||
| 30 |
0.16 |
0.32 |
−11.0 |
−11.0 |
Y |
Y |
Moderate protan |
Severe protan |
||
| IV:4/F |
25 | 1.0 | 1.0 | None | None | N | N | Present | ||
| 29 | 1.0 | 1.0 | None | None | N | N | None | None | ||
| 32 |
1.0 |
1.0 |
None |
None |
N |
N |
||||
| IV:6/M |
14 | 0.8 | 0.8 | −3.25/-1.75 | −2.75/-1.5 | N | N | Not tested | ||
| 19 |
0.8 |
0.8 |
−5.5/-1.0 |
−5.0 |
Y |
Y |
None |
None |
||
| IV:8/F |
22 |
0.32 |
0.32 |
−10.5/-3.5 |
−13.5/-1.75 |
N |
N |
Not tested |
||
| IV:11/F |
25 |
0.63 |
0.5 |
−4.5/-3.0 |
−10.0/-2.5 |
N |
N |
Present |
||
| IV:12/M |
37 |
1.0 |
1.0 |
UA |
UA |
N |
N |
Not found |
||
| IV:15/M |
11 |
1.0 |
1.0 |
None |
None |
N |
N |
Not tested |
||
| V:1/M |
4 | UA | UA | −2.5/-2.5 | −2.5/-2.5 | N | N | Not tested | ||
| 6.5 | 1.0 | 0.5 | −2.5/-2.5 | −2.5/-2.5 | N | N | ||||
| 10 | 0.5 | 0.5 | −3.0/-2.5 | −3.0/-3.0 | N | N | ||||
BCVA: best corrected visual acuity, DS: diopter sphere, DC: diopter cylinder, F: female, HM: hand movement, M: male, LE: left eye, RE: right eye, N: No, Y: yes, yrs: years, TB: total blindness, UA: unavailable data. Color vision was tested using Lanthony 15-Hue Desaturated Test.
Eighteen family members (nine women and nine men) were clinically examined. In some subjects, serial observations were performed over a period of up to 11 years. A summary of their clinical findings is provided in Table 1. No systemic or sensory disorder was found to cosegregate with RP in the family.
Female patients I:1, II:1, II:3, and II:5 were reported to become “totally blind” in their 60s and 70s. Bilateral no perception of light was confirmed in patient II:5. There was a variable onset of nyctalopia. Female patients II:5 and III:9 noticed difficulties in dim light in childhood, III:1 at the age of 30 years, and III:4 had attended a school for visually handicapped children and had reported impaired orientation during the day and night since childhood. With one exception (IV:4), all women had reduced BCVA, high myopia, and astigmatism in at least one eye. Signs of RP (pigmentary retinopathy, attenuated vessels, optic disc pallor, and posterior subcapsular cataracts) in at least one eye were found in female individuals II:5, III:1, III:4, and III:9. Macular atrophy was also common (Table 1, Figure 2, Figure 3). White dots were noted in the fundus periphery of the right eye of individual III:9; her left eye could not be adequately examined since the patient refused dilation (Figure 2C). None of the examined women had evidence of a tapetal reflex.
Figure 2.

Spectrum of fundus finding in two women and one man with retinitis pigmentosa caused by a c.2543del RPGR ORF15 mutation. A: Composite fundus photography of the right eye of female III:4 at the age of 54 years. B: Composite fundus photography of the right eye of female III:9 at the age of 50 years; note the chorioretinal degeneration at the macula and in the periphery with intraretinal asymmetric involvement. C: Red-free imaging enhancing peripheral white dots in the upper nasal quadrant of the right eye of female III:9. D, E: Fundus photographs of male IV:6 aged 19; note the relatively normal appearing macula and pigment clumps in the mid-periphery.
Figure 3.
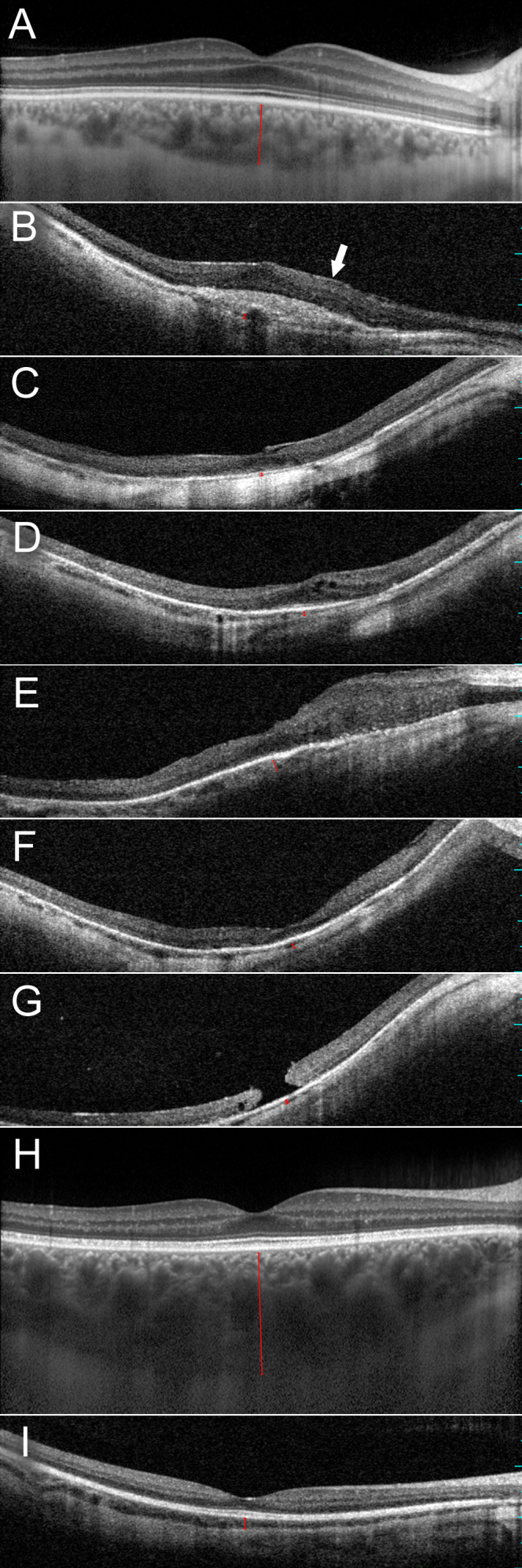
Horizontal optical coherence tomography images cross sectioning the fovea in the right eyes of affected men and women who carry a c.2543del mutation in RPGR ORF15. Choroidal thickness is indicated with a red bar. A: Normal retinal architecture in a 40-year-old control individual (enhanced depth imaging). B: Chorioretinal thinning, missing foveal depression, scar formation, and epiretinal membrane (arrow) in female III:1 at the age of 64 years. C: Chorioretinal thinning, missing foveal depression, and vitreomacular traction associated with foveal elevation in female III:4 at the age of 54 years. D: Chorioretinal thinning, myopic foveoschisis, and posterior staphyloma in female III:9 at the age of 50 years. E: Chorioretinal thinning and focal retinal thickening in male IV:1 at the age of 42 years. F: Chorioretinal thinning and myopic posterior staphyloma in male IV:2 at the age of 38 years. G: Chorioretinal thinning, macular hole, and myopic posterior staphyloma in male IV:3 at the age of 30 years. H: Normal retinal and choroidal architecture in female IV:4 at the age of 32 years (enhanced depth imaging). I: Choroidal thinning and normal retinal architecture in male IV:6 at the age of 19 years.
All affected male individuals, III:6, IV:1, IV:2, and IV:3 (except IV:6 aged 19 years who was asymptomatic), reported onset of nyctalopia between 10 and 20 years of age and refractive errors since early childhood. All had typical ocular features of RP (including IV:6; Figure 2). Mild to high myopia with astigmatism and macular atrophy was also present (Table 1 and Table 2, Figure 2, Figure 3). Serial examination with static perimetry demonstrated progressive visual field constriction (Figure 4). Disruption of the macular architecture including the absence of the ISe band together with retinal thinning was documented with SD-OCT in the majority of the subjects (Table 2, Figure 3).
Table 2. Spectral domain optical coherence tomography findings in individuals with a c.2543del mutation in RPGR ORF15.
| Individual/gender | Age (yrs) |
ISe |
Foveal thickness (µm) |
Choroidal thickness (µm) |
RNFL thickness (µm) |
||||
|---|---|---|---|---|---|---|---|---|---|
| RE | LE | RE | LE | RE | LE | RE | LE | ||
| III:1/F |
64 |
A |
I |
395 (epiretinal membrane) |
195 |
90 |
75 |
TQ 107 |
TQ 151 |
| III:4/F |
54 |
A |
P |
140 |
230 |
30 |
50 |
UA |
UA |
| III:9/F |
50 |
P |
P |
220 |
200 |
130 |
150 |
TQ 132 |
TQ 96 |
| IV:1/M |
42 |
P |
P |
190 (epiretinal membrane) |
150 |
120 |
110 |
TQ 162 |
TQ 172 |
| IV:2/M |
38 |
A |
A |
120 |
130 |
40 |
40 |
TQ 129 |
TQ 171 |
| IV:3/M |
30 |
A |
P |
Macular hole |
130 |
80 |
35 |
TQ 133 |
TQ 101 |
| IV:4/F |
32 |
I |
I |
170 |
165 |
600 |
590 |
Normal |
Normal |
| IV:6/M | 19 | I | I | 165 | 165 | 135 | 110 | Normal | Normal |
A: absent, F: female, I: intact, ISe: Inner segment ellipsoid band, LE: left eye, M: male, P: partially intact, RE: right eye, RNFL: retinal nerve fiber layer, TQ: temporal quadrant, UA: unavailable data. Minimal foveal thickness shown does not include pathological epiretinal membrane. Choroidal thickness was measured subfoveally. Exact values of RNFL thickness are shown for abnormal quadrants. Normal range of foveal thickness 261.31 ± 17.67 μm [33]. Normal range of choroidal thickness 336.60 ± 70.42 μm [27]. Normal range of RNFL thickness 79.5 ± 15.3 μm [34].
Figure 4.

Serial observations of disease progression on automated visual field for individuals with a c.2543del RPGR ORF15 mutation. Visual field scans (showing 50° nasal and temporal to fixation) of the right and left eyes of female III:4 at the age of 47 (A) and 58 (B) years; the right and left eyes of female III:1 at the age of 58 (C) and 64 (D) years; the right and left eyes of female III:9 at the age of 44 (E) and 50 (F) years; the right and left eyes of female IV:4 at the age of 25 (G) and 29 (H) years; the right and left eyes of male IV:3 at the age of 23 (I) and 30 (J) years; the right and left eyes of male IV:6 at the age of 14 (K) and 19 (L) years; the right and left eyes of male IV:2 at the age of 32 (M) and 38 (N) years; the right and left eyes of male IV:1 at the age of 42 (O) years. Static perimetry results are shown as 12-level gray scales of sensitivity loss. Black areas indicate no detection of stimuli. Physiologic blind spot is shown as a black circle at 12° in the temporal field.
All individuals with pigmentary retinopathy and family members who harbored the c.2543del mutation in RPGR, except female carrier IV:4, had abnormal subfoveal choroidal thickness (Table 2). In addition, a full-thickness macular hole (minimum diameter 460 µm, base diameter 890 µm) was present in the right eye of individual IV:3 (Figure 3G). In severely affected men, the RNFL thickness was increased in the temporal quadrants (Table 2).
Discussion
Reports of large families with XLRP mimicking autosomal dominant inheritance are uncommon [5,17-19]. Herein we describe a Czech family with RP with severe retinal degeneration in women carrying a c.2543del mutation (p.Glu848Glyfs*241) in RPGR ORF15. This sequence variant was previously found in one man from Central Europe, but no phenotypic description was provided [20]. RP is genetically heterogeneous. This is only the second report on the molecular genetic cause of RP from the Czech Republic [21].
It has been previously shown that in families with RP considered to be most likely autosomal dominant without male-to-male transmission and in severely affected sporadic men, X-linked inheritance should be considered [22,23]. Screening for mutations in RP2 and RPGR in a recent study revealed that 8.5% (22 out of 258) of such pedigrees were attributed to mutations in these genes and that they were in fact X-linked [22]. Initial evaluation of our pedigree with individuals affected in four consecutive generations including several severely affected women favored autosomal dominant inheritance. Failure to identify a disease-causing mutation using a microarray for detecting known mutations implicated in autosomal dominant RP and significant myopic refractive errors previously reported in men and women from families with XLRP [4,5,12,16] led us to screen RPGR.
The disease severity varied among the affected women in our family. In addition, we observed inter-ocular asymmetry, in contrast to the symmetry observed in the men. Myopia and astigmatism were present in all but one carrier and in all affected men. Refractive errors were also documented in three other women without signs of typical RP. One was found to be a carrier for the c.2543del mutation in RPGR ORF15, while the other two were not available for molecular genetic analysis.
Although the structure of the pedigree presented here and known phenotypic variability in XLRP expression [4,5,12,16,24] make it difficult to compare the phenotype between men and women of the same age, comparison of three siblings in generations III and IV corroborates that women show later onset and less severe loss of visual field when compared to similarly aged men [17]. Nevertheless, women in generation II had no perception of light bilaterally, which is unusually severe. The mechanism(s) underlying variable disease expression in women as well as the factors that contribute to inter-individual phenotypic heterogeneity remain elusive, but include skewed inactivation in women and other genetic modifiers [5,12,25].
To aid the clinical characterization of the severe phenotype in women, we performed detailed SD-OCT. A range of findings from normally appearing retinal architecture to gross abnormalities was observed. Marked structural disruption precluded reliable distinction between retinal layers. In one eye of a 30-year-old man, we documented a macular hole. Macular hole is an uncommon finding in RP. Hagiwara et al. (2011) found only three patients out of 323 (three eyes out of 622) with this pathology on OCT imaging [26]. Recently, several studies indicated that changes in choroidal morphology and thinning represent a distinct feature in RP [27,28]. Our study not only confirms these findings but is also the first one that provides correlation with the genotype. Since we also observed choroidal thinning in female carriers with no bone spicule-like formation or tapetal reflex, choroidal evaluation may be a useful non-invasive, readily available assessment for carrier status. However, since decreased choroidal thickness can also be observed in myopic eyes [29] and myopia was also present in the women examined in our study, the broad relevance and resulting implications of this parameter in RP diagnosis and evaluation of progression warrant further investigation.
Increased thickness, as well as decreased thickness of the RNFL in RP, has been reported previously [30-32], however, not in molecularly confirmed RP. It has been suggested that increased RNFL thickness may be associated with cone system abnormality [30,31]. Clinical findings in our patients support this observation. Nine out of 10 eyes observed to have increased RNFL thickness also had abnormalities of the foveal architecture, including an absent or markedly disrupted ISe band; in the remaining eye with an intact ISe band, the 15-hue test revealed a mild color vision defect.
In conclusion, in pedigrees where male-to-male transmission is absent, X-linked inheritance should be considered despite the presence of severe signs of retinal degeneration in women. Moderate and high myopia in affected individuals of both sexes, as well as ocular asymmetry in women, may also be suggestive of X-linked inheritance. Evaluation of choroidal thickness may prove to be a valuable readily available method for clinically identifying female carriers.
Acknowledgments
This work was supported by UNCE 204011 and PRVOUK-P24/LF1/3 programs of the Charles University in Prague and by a short visit grant to PL by the European Science Foundation. BK is supported by SVV-2014-260022, TF, NH, AG by RP Fighting Blindness (UK Registered Charity No. GR570) and Fight For Sight (Registered Charity No. 1111438), MM by a Foundation Fighting Blindness Career Development Award, CC by The US Foundation Fighting Blindness, and MM and AJH are supported by the National Institute for Health Research Biomedical Research Centre at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology.
References
- 1.Daiger SP, Sullivan LS, Bowne SJ. Genes and mutations causing retinitis pigmentosa. Clin Genet. 2013;84:132–41. doi: 10.1111/cge.12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berger W, Kloeckener-Gruissem B, Neidhardt J. The molecular basis of human retinal and vitreoretinal diseases. Prog Retin Eye Res. 2010;29:335–75. doi: 10.1016/j.preteyeres.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 3.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 4.Koenekoop RK, Loyer M, Hand CK, Al Mahdi H, Dembinska O, Beneish R, Racine J, Rouleau GA. Novel RPGR mutations with distinct retinitis pigmentosa phenotypes in French-Canadian families. Am J Ophthalmol. 2003;136:678–87. doi: 10.1016/s0002-9394(03)00331-3. [DOI] [PubMed] [Google Scholar]
- 5.Banin E, Mizrahi-Meissonnier L, Neis R, Silverstein S, Magyar I, Abeliovich D, Roepman R, Berger W, Rosenberg T, Sharon D. A non-ancestral RPGR missense mutation in families with either recessive or semi-dominant X-linked retinitis pigmentosa. Am J Med Genet A. 2007;143A:1150–8. doi: 10.1002/ajmg.a.31642. [DOI] [PubMed] [Google Scholar]
- 6.Meindl A, Dry K, Herrmann K, Manson F, Ciccodicola A, Edgar A, Carvalho MR, Achatz H, Hellebrand H, Lennon A, Migliaccio C, Porter K, Zrenner E, Bird A, Jay M, Lorenz B, Wittwer B, D’Urso M, Meitinger T, Wright A. A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X-linked retinitis pigmentosa (RP3). Nat Genet. 1996;13:35–42. doi: 10.1038/ng0596-35. [DOI] [PubMed] [Google Scholar]
- 7.Schwahn U, Lenzner S, Dong J, Feil S, Hinzmann B, van Duijnhoven G, Kirschner R, Hemberger M, Bergen AA, Rosenberg T, Pinckers AJ, Fundele R, Rosenthal A, Cremers FP, Ropers HH, Berger W. Positional cloning of the gene for X-linked retinitis pigmentosa 2. Nat Genet. 1998;19:327–32. doi: 10.1038/1214. [DOI] [PubMed] [Google Scholar]
- 8.Webb TR, Parfitt DA, Gardner JC, Martinez A, Bevilacqua D, Davidson AE, Zito I, Thiselton DL, Ressa JH, Apergi M, Schwarz N, Kanuga N, Michaelides M, Cheetham ME, Gorin MB, Hardcastle AJ. Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23). Hum Mol Genet. 2012;21:3647–54. doi: 10.1093/hmg/dds194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shu X, Black GC, Rice JM, Hart-Holden N, Jones A, O'Grady A, Ramsden S, Wright AF. RPGR mutation analysis and disease: an update. Hum Mutat. 2007;28:322–8. doi: 10.1002/humu.20461. [DOI] [PubMed] [Google Scholar]
- 10.Neidhardt J, Glaus E, Barthelmes D, Zeitz C, Fleischhauer J, Berger W. Identification and characterization of a novel RPGR isoform in human retina. Hum Mutat. 2007;28:797–807. doi: 10.1002/humu.20521. [DOI] [PubMed] [Google Scholar]
- 11.Wright RN, Hong DH, Perkins B. RpgrORF15 connects to the Usher protein network through direct interactions with multiple whirlin isoforms. Invest Ophthalmol Vis Sci. 2012;53:1519–29. doi: 10.1167/iovs.11-8845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pelletier V, Jambou M, Delphin N, Zinovieva E, Stum M, Gigarel N, Dollfus H, Hamel C, Toutain A, Dufier JL, Roche O, Munnich A, Bonnefont JP, Kaplan J, Rozet JM. Comprehensive survey of mutations in RP2 and RPGR in patients affected with distinct retinal dystrophies: genotype–phenotype correlations and impact on genetic counseling. Hum Mutat. 2007;28:81–91. doi: 10.1002/humu.20417. [DOI] [PubMed] [Google Scholar]
- 13.Thiele H, Nürnberg P. HaploPainter: a tool for drawing pedigrees with complex haplotypes. Bioinformatics. 2005;21:1730–2. doi: 10.1093/bioinformatics/bth488. [DOI] [PubMed] [Google Scholar]
- 14.den Dunnen JT, Antonarakis SE. Nomenclature for the description of human sequence variations. Hum Genet. 2001;109:121–4. doi: 10.1007/s004390100505. [DOI] [PubMed] [Google Scholar]
- 15.Antonarakis SE. Recommendations for a nomenclature system for human gene mutations. Nomenclature Working Group. Hum Mutat. 1998;11:1–3. doi: 10.1002/(SICI)1098-1004(1998)11:1<1::AID-HUMU1>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 16.Sharon D, Sandberg MA, Rabe VW, Stillberger M, Dryja TP, Berson EL. RP2 and RPGR mutations and clinical correlations in patients with X-linked retinitis pigmentosa. Am J Hum Genet. 2003;73:1131–46. doi: 10.1086/379379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu DM, Khanna H, Atmaca-Sonmez P, Sieving PA, Branham K, Othman M, Swaroop A, Daiger SP, Heckenlively JR. Long-term follow-up of a family with dominant X-linked retinitis pigmentosa. Eye (Lond) 2010;24:764–74. doi: 10.1038/eye.2009.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y, Dong B, Hu AL, Cui TT, Zheng YY. A novel RPGR gene mutation in a Chinese family with X-linked dominant retinitis pigmentosa. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2005;22:396–8. [PubMed] [Google Scholar]
- 19.Rozet JM, Perrault I, Gigarel N, Souied E, Ghazi I, Gerber S, Dufier JL, Munnich A, Kaplan J. Dominant X linked retinitis pigmentosa is frequently accounted for by truncating mutations in exon ORF15 of the RPGR gene. J Med Genet. 2002;39:284–5. doi: 10.1136/jmg.39.4.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pusch CM, Broghammer M, Jurklies B, Besch D, Jacobi FK. Ten novel ORF15 mutations confirm mutational hot spot in the RPGR gene in European patients with X-linked retinitis pigmentosa. Hum Mutat. 2002;20:405. doi: 10.1002/humu.9072. [DOI] [PubMed] [Google Scholar]
- 21.Liskova P, Colclough T, Hart-Holden N, Chakarova CF, O'Grady A, Kondrova L, Skalicka P, Diblik P, Hardcastle AJ. Molecular genetic cause of X-linked retinitis pigmentosa in a Czech family. Acta Ophthalmol (Copenh) 2011;89:e213–5. doi: 10.1111/j.1755-3768.2009.01802.x. [DOI] [PubMed] [Google Scholar]
- 22.Churchill JD, Bowne SJ, Sullivan LS, Lewis RA, Wheaton DK, Birch DG, Branham KE, Heckenlively JR, Daiger SP. Mutations in the X-linked retinitis pigmentosa genes RPGR and RP2 found in 8.5% of families with a provisional diagnosis of autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2013;54:1411–6. doi: 10.1167/iovs.12-11541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Breuer DK, Yashar BM, Filippova E, Hiriyanna S, Lyons RH, Mears AJ, Asaye B, Acar C, Vervoort R, Wright AF, Musarella MA, Wheeler P, MacDonald I, Iannaccone A, Birch D, Hoffman DR, Fishman GA, Heckenlively JR, Jacobson SG, Sieving PA, Swaroop A. A comprehensive mutation analysis of RP2 and RPGR in a North American cohort of families with X-linked retinitis pigmentosa. Am J Hum Genet. 2002;70:1545–54. doi: 10.1086/340848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zahid S, Khan N, Branham K, Othman M, Karoukis AJ, Sharma N, Moncrief A, Mahmood MN, Sieving PA, Swaroop A, Heckenlively JR, Jayasundera T. Phenotypic conservation in patients with X-linked retinitis pigmentosa caused by RPGR mutations. JAMA Ophthalmol. 2013;131:1016–25. doi: 10.1001/jamaophthalmol.2013.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fahim AT, Bowne SJ, Sullivan LS, Webb KD, Williams JT, Wheaton DK, Birch DG, Daiger SP. Allelic heterogeneity and genetic modifier loci contribute to clinical variation in males with X-linked retinitis pigmentosa due to RPGR mutations. PLoS ONE. 2011;6:e23021. doi: 10.1371/journal.pone.0023021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hagiwara A, Yamamoto S, Ogata K, Sugawara T, Hiramatsu A, Shibata M, Mitamura Y. Macular abnormalities in patients with retinitis pigmentosa: prevalence on OCT examination and outcomes of vitreoretinal surgery. Acta Ophthalmol (Copenh) 2011;89:e122–5. doi: 10.1111/j.1755-3768.2010.01866.x. [DOI] [PubMed] [Google Scholar]
- 27.Ayton LN, Guymer RH, Luu CD. Choroidal thickness profiles in retinitis pigmentosa. Clin Experiment Ophthalmol. 2013;41:396–403. doi: 10.1111/j.1442-9071.2012.02867.x. [DOI] [PubMed] [Google Scholar]
- 28.Dhoot DS, Huo S, Yuan A, Xu D, Srivistava S, Ehlers JP, Traboulsi E, Kaiser PK. Evaluation of choroidal thickness in retinitis pigmentosa using enhanced depth imaging optical coherence tomography. Br J Ophthalmol. 2013;97:66–9. doi: 10.1136/bjophthalmol-2012-301917. [DOI] [PubMed] [Google Scholar]
- 29.Chebil A, Ben Achour B, Chaker N, Jedidi L, Mghaieth F, El Matri L. Choroidal thickness assessment with SD-OCT in high myopia with dome-shaped macula. J Fr Ophtalmol. 2014;37:237–41. doi: 10.1016/j.jfo.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 30.Sobací G, Ozge G, Gundogan FC. Cone dysfunctions in retinitis pigmentosa with retinal nerve fiber layer thickening. Clin Ophthalmol. 2012;6:473–8. doi: 10.2147/OPTH.S28938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hwang YH, Kim SW, Kim YY, Na JH, Kim HK, Sohn YH. Optic nerve head, retinal nerve fiber layer, and macular thickness measurements in young patients with retinitis pigmentosa. Curr Eye Res. 2012;37:914–20. doi: 10.3109/02713683.2012.688163. [DOI] [PubMed] [Google Scholar]
- 32.Anastasakis A, Genead MA, McAnany JJ, Fishman GA. Evaluation of retinal nerve fiber layer thickness in patients with retinitis pigmentosa using spectral-domain optical coherence tomography. Retina. 2012;32:358–63. doi: 10.1097/IAE.0b013e31821a891a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Solé Gonzalez L, Abreu Gonzalez R, Alonso Plasencia M, Abreu Reyes P. Normal macular thickness and volume using spectral domain optical coherence tomography in a reference population. Arch Soc Esp Oftalmol. 2013;88:352–8. doi: 10.1016/j.oftal.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 34.Kampougeris G, Spyropoulos D, Mitropoulou A, Zografou A, Kosmides P. Peripapillary retinal nerve fibre layer thickness measurement with SD-OCT in normal and glaucomatous eyes: distribution and correlation with age. Int J Ophthalmol. 2013;6:662–5. doi: 10.3980/j.issn.2222-3959.2013.05.21. [DOI] [PMC free article] [PubMed] [Google Scholar]