

Abstract
Clindamycin is commonly prescribed to treat children with skin and skin structure infections (including those caused by community-acquired methicillin-resistant Staphylococcus aureus [CA-MRSA]), yet little is known about the pharmacokinetics (PK) across pediatric age groups. A population PK analysis was performed in NONMEM using samples collected in an opportunistic study from children receiving intravenous clindamycin per standard of care. The final model was used to optimize pediatric dosing to match adult exposure proven effective against CA-MRSA. A total of 194 plasma PK samples collected from 125 children were included in the analysis. Median age (range) was 3.3 years (0–20). Median dosing was 9.9 mg/kg/dose (3.8–15.1). A 1-compartment model described the data well. The final model included body weight and a sigmoidal maturation relationship between postmenstrual age (PMA) and clearance (CL): CL (L/h)=13.7*(weight/70)0.75*(PMA3.1/(43.63.1+PMA3.1)); V (L)=61.8*(weight/70). Maturation reached 50% adult CL values at ~44 weeks PMA. Our findings support age-based dosing.
Keywords: MRSA, population pharmacokinetics, clindamycin, pediatrics
Clindamycin is a lincosamide antibiotic commonly prescribed to treat children with skin and skin structure infections (including those caused by community-acquired methicillin-resistant Staphylococcus aureus [CA-MRSA]), pneumonia, osteomyelitis, and anaerobic infections.1 Due to a rise in the incidence of CA-MRSA infections,2 the use of clindamycin has increased significantly, and it is now the antibiotic most widely prescribed for this indication in hospitalized pediatric patients.3
Following intravenous administration, clindamycin distributes extensively into tissues (with the exception of cerebrospinal fluid) and, in healthy adults, has a volume of distribution at steady state of 0.79 L/kg.4 Clindamycin is metabolized predominantly by cytochrome P450 (CYP) 3A4 to clindamycin sulfoxide and N-demethyl clindamycin.5 When measured in vivo, metabolite concentrations, although quantifiable in bile and urine,6,7 are negligible in plasma.8,9 In healthy adults, clindamycin’s clearance and half-life are ~0.3–0.4 L/h/kg and 2.1 hours, respectively.4,9 In spite of its widespread use, pharmacokinetic (PK) studies to inform clindamycin dosing in children are limited. This is due in part to this drug being marketed before studies were required in children, but also because of the many challenges associated with conducting clinical trials in children, including low consent rates, limited access to the population of interest, and the frequent need for research blood draws to obtain PK samples.
To overcome these latter challenges, opportunistic study designs capitalizing on standard medical care procedures have been implemented successfully in children.10–12 In this study design, patients prescribed a drug of interest by the treating physician are identified and consented for PK sample collection, optimally, at the time of routine laboratory draws.13 By not administering a drug solely for research purposes and timing blood draws with standard-of-care procedures, there is minimal study-related risk to the patient, and higher consent rates are obtained. However, opportunistic study designs have not been broadly implemented in children because of a lack of consistently successful applications demonstrating their value.11,12
Clindamycin is an off-patent, widely used, yet understudied pediatric therapeutic, the dosing for which could be informed by such an efficient and cost-effective opportunistic design. Furthermore, given the biotransformation pathway involved, it is likely that important developmental changes during childhood will affect clindamycin drug disposition, and the PK data collected will inform pediatric dosing across age groups. An opportunistic study design facilitates collection of PK data across the pediatric age continuum, allowing for differences in clindamycin elimination and distribution to be assessed. A final rationale for reassessing clindamycin PK is that historical studies in children used bioassays incapable of separating clindamycin from its bioactive metabolites, perhaps over-estimating clindamycin exposure in children.14,15
The objectives of this study were to characterize clindamycin drug disposition across the age continuum from premature infants to adolescents using an opportunistic clinical trial design and to optimize clindamycin dosing in this population.
RESULTS
Patient characteristics
A total of 194 plasma PK samples collected from 125 subjects receiving intravenous clindamycin were included in the analysis; 2 additional samples below the limit of quantification were excluded. Sixty five percent of samples (127/194) were collected for research purposes. The five most common indications for clindamycin administration were Staphylococcus and skin and skin structure infections (N=41/125), prophylaxis (21/125), other infections (21/125), intra-abdominal infection (10/125), and pneumonia (7/125). The median (range) number of samples per subject, dose, and daily dosage were 1 sample (1–6), 9.9 mg/kg/dose (3.8–15.1), and 29.7 mg/kg/day (7.6–52.2), respectively. Patient demographic variables are summarized in Table 1. The median (range) postnatal age (PNA) and weight were 3.3 years (1 day–20 years) and 17.3 kg (0.5–139.8), respectively. Fifty six percent (70/125) of subjects were male; 76% (95/125) were white. Twenty five percent of subjects (31/125) were obese (body mass index ≥ 95th percentile). A total of 35 subjects were ≤120 days of PNA; of these 57% (20/35) were ≤32 weeks gestational age (GA) at birth and were studied at median (range) 9.5 days PNA (2–61) and 28.8 weeks postmenstrual age (PMA) (23.6–37.6).
Table 1.
Clinical data
| Na | 0–28 days (N=21) | Na | >28 days – 2 years (N=34) | Na | >2–12 years (N=34) | Na | >12 years (N=36) | |
|---|---|---|---|---|---|---|---|---|
| Gestational age (weeks) | 21 | 28.1 (22.9–40) | 14 | 35.9 (24.6–42) | - | - | - | - |
| Postnatal age (years) | 21 | 0 (0–0.1) | 34 | 0.5 (0.1–1.9) | 34 | 6.2 (2–11.5) | 36 | 16.3 (12.4–20) |
| Postmenstrual age (weeks) | 21 | 29 (23.6–42) | 34 | 65 (28.9–139.7) | 34 | 363.3 (145.4–639) | 36 | 889.6 (685.7–1081) |
| Weight (kg) | 21 | 1 (0.5–4.4) | 34 | 6.8 (0.9–12.2) | 34 | 24.3 (10.3–58.1) | 36 | 75.6 (28.6–139.8) |
| Serum creatinine (mg/dL) | 16 | 0.6 (0.3–1.5) | 15 | 0.3 (0.1–0.8) | 13 | 0.4 (0.2–1.6) | 17 | 0.6 (0.2–3.4) |
| AST (U/L) | 3 | 21 (18–28) | 3 | 28 (21–193) | 9 | 49 (8–389) | 6 | 39.5 (19–102) |
| ALT (U/L) | 3 | 11 (8–55) | 3 | 32 (10–78) | 9 | 29 (13–266) | 6 | 57.5 (29–162) |
| Total bilirubin (mg/dL) | 8 | 3.7 (0.5–8.5) | 2 | 1.1 (1–1.2) | 9 | 0.4 (0–11) | 6 | 1.9 (0.5–4.3) |
| Direct bilirubin (mg/dL) | 4 | 0.3 (0.1–0.7) | 2 | 0.4 (0.3–0.5) | 3 | 0.5 (0.2–7.9) | 2 | 0.7 (0–1.3) |
| Albumin (g/dL) | 4 | 2.6 (1.9–3.7) | 6 | 3.7 (2–4.5) | 11 | 3.5 (2.2–4.2) | 8 | 3.6 (2.3–4.3) |
| Male sex | 21 | 12 (57) | 34 | 23 (68) | 34 | 13 (38) | 36 | 22 (61) |
| White race | 21 | 18 (86) | 34 | 29 (85) | 34 | 23 (68) | 36 | 25 (69) |
| Hispanic ethnicity | 21 | 4 (19) | 34 | 11 (32) | 34 | 12 (35) | 36 | 9 (25) |
| Obeseb | - | - | - | - | 31 | 13 (38) | 36 | 18 (50) |
Values are medians (range) for continuous variables and counts (%) for categorical variables calculated based on values at the time of first recorded dose. For categorical variables, percentages were calculated as a function of the number of subjects in each respective postnatal age cohort.
AST, aspartate aminotransferase; ALT, alanine aminotransferase.
N signifies the number of subjects with data available for each respective variable and within each respective postnatal age cohort.
Only assessed in patients >2 years of age.
Population PK model development and evaluation
Clindamycin plasma concentrations versus time data are shown in Figure 1. A 1-compartment body model described the data well. Scaling of clearance (CL) and volume of distribution (V) parameters by weight using a fixed exponent allometric relationship (CL=0.75; V=1) resulted in a 341-point reduction in the -2 log likelihood objective function value (OFV). As a result, additional covariate relationships were evaluated after adjusting for size-based differences using total body weight. A sigmoidal EMAX maturation function accounting for PMA-based differences in drug CL resulted in a 66-point further reduction in the OFV and reduced the between-subject variability by 25.4% (Supplemental Table 1). When compared with a sigmoidal relationship, linear and power maturation functions using PMA and PNA had lesser reductions in the OFV. After accounting for size- and age-based differences in PK parameters, no other covariates (including obesity on V) reached statistical significance.
Figure 1.
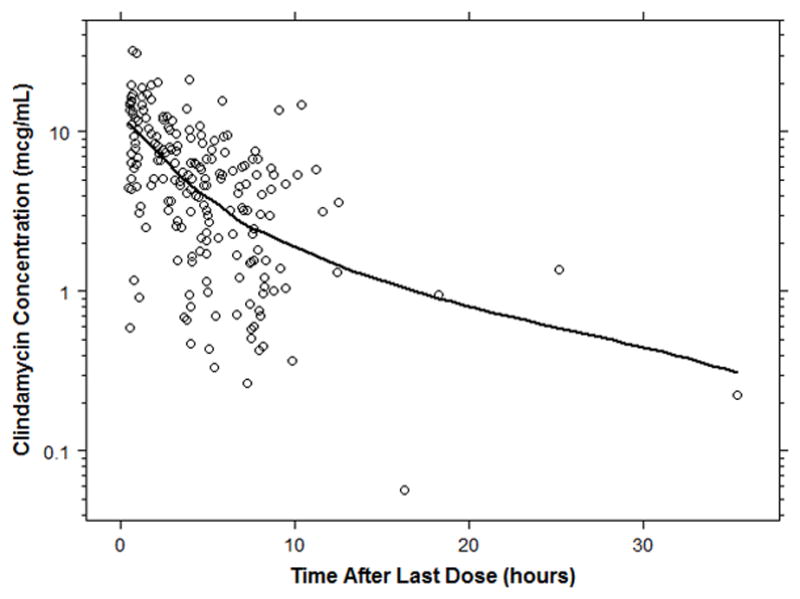
Clindamycin concentration versus time curve. The solid black line represents the loess curve.
The final population PK (PPK) model included body weight and a sigmoidal EMAX maturation relationship between PMA and CL (Table 2):
Table 2.
Parameter estimates and bootstrap analysis results
| Final Model | Bootstrap (n=1,000) | ||||
|---|---|---|---|---|---|
|
|
|||||
| Parameter | Estimate | RSE (%) | 2.5th Percentile | Median | 97.5th Percentile |
| CLstd (L/H/70 kg) | 13.7 | 8.5 | 11.8 | 13.9 | 17.6 |
| Vstd (L/70kg) | 61.8 | 8.7 | 53 | 61.9 | 74.5 |
| TM50 (weeks) | 43.6 | 15.7 | 36.1 | 44.2 | 83.0 |
| HILL | 3.1 | 37.1 | 1.2 | 3 | 5.1 |
| BSV (CL) | 57.1 | 16 | 46.7 | 56.3 | 66.5 |
| Proportional error (%) | 39.5 | 17.3 | 31.8 | 38.7 | 45.7 |
RSE, relative standard error; CLstd, population clearance estimate standardized to a 70 kg weight using an allometric model; Vstd population volume of distribution estimate standardized to a 70 kg weight using a linear model; TM50, maturation half-life calculated as a function of postmenstrual age (weeks); HILL, Hill coefficient in sigmoidal maturation function; BSV (CL), between-subject variability in drug clearance.
Maturation reached 50% adult CL (TM50) values at approximately 44 weeks PMA. Between-subject variability in CL was 57.1%. Between-subject random effect (η) shrinkage for CL was 9%, while epsilon (ε) shrinkage was 20.8%. Estimation of between-subject variability in V was not supported by the data; a high η-shrinkage (49%) was observed for this parameter. A proportional residual error model characterized unexplained residual variability well and was estimated at 39.5%. There were no obvious trends or model misspecification identified in the diagnostic plots for the final model (Figure 2). The visual predictive check (VPC) indicated that the final model described the observed data adequately; 11% of the observed concentrations fell outside the 90% simulated prediction interval (Supplemental Figure 1). Median bootstrap estimates were within approximately 3% of final model estimates.
Figure 2.
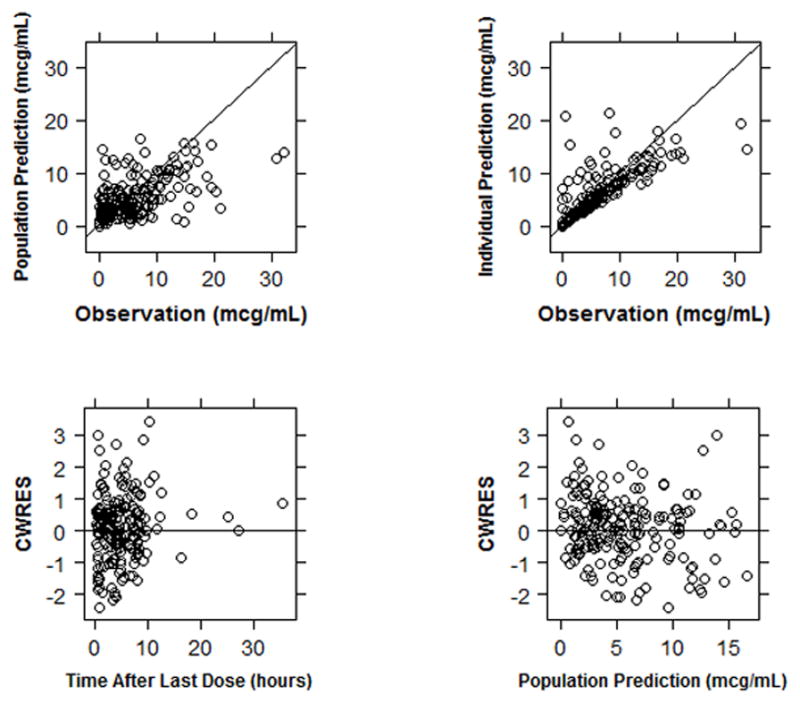
Diagnostic plots for the final model. CWRES, conditional weighted residuals.
Individual empirical Bayesian estimates were compared across age groups (Table 3). As described using the sigmoidal maturation function, CL increases from 2.8 L/h/70 kg at 28 weeks PMA to an adult value of 13.7 L/h/70 kg (Figure 3). A notable difference in median (range) weight-normalized CL was observed between infants ≤32 weeks PMA (0.11 L/h/kg [0.06–0.31]), >32–40 weeks PMA (0.16 L/h/kg [0.06–0.26]), and >40–60 weeks PMA (0.29 L/h/kg [0.08–0.75]). Because between-subject variability in V was not incorporated into the model, individual estimates of V did not vary across age groups.
Table 3.
Individual empirical Bayesian post-hoc parameter estimates stratified by age
| PMA Categories | ≤28 weeks PMA (N=7) | >28–32 weeks PMA (N=7) | >32–40 weeks PMA (N=8) | >40–60 weeks PMA (N=14) | >5 months– 1 year (N=11) | >1–6 years (N=25) | >6–12 years (N=17) | >12 years (N=36) |
|---|---|---|---|---|---|---|---|---|
| CL (L/h/kg) | 0.12 (0.07–0.19) | 0.11 (0.06–0.31) | 0.16 (0.06–0.26) | 0.29 (0.08–0.75) | 0.27 (0.1–0.52) | 0.25 (0.09–0.63) | 0.31 (0.11–0.70) | 0.2 (0.04–0.66) |
| CL (L/h/70 kg) | 2.3 (1.6–4.2) | 2.6 (1.5–7.0) | 4.4 (1.4–6.9) | 10.2 (2.3–29.1) | 11.0 (3.8–21.9) | 11.0 (3.9–27.9) | 17.5 (6.6–44.0) | 15.4 (3.2–38.4) |
| V (L/kg)a | 0.9 | 0.9 | 0.9 | 0.9 | 0.9 | 0.9 | 0.9 | 0.9 |
| Half-life (h) | 5.3 (3.3–8.8) | 5.4 (2.0–9.5) | 3.9 (2.4–10.9) | 2.1 (0.8–7.6) | 2.3 (1.2–6.31) | 2.5 (1.0–7.0) | 2.0 (0.9–5.6) | 3.1 (0.9–14.2) |
Median (range) calculated using age at time of first PK sample.
PMA, postmenstrual age; CL, clearance; V, volume of distribution; PNA, postnatal age.
No difference in individual estimates for weight-normalized V as between-subject variability (V) was not estimated.
Figure 3.
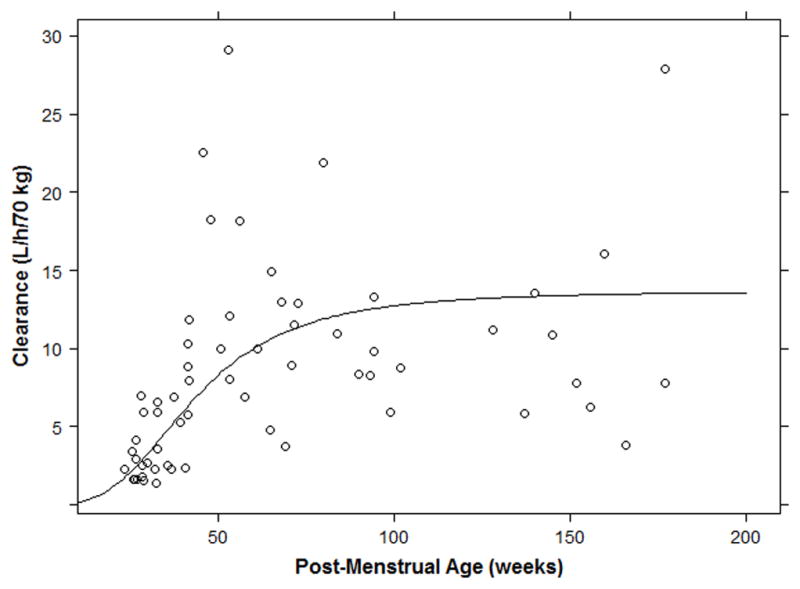
Clearance as a function of postmenstrual age. Individual clearance values for subjects with PMA≤200 weeks (~3 years age). The solid black line denotes the sigmoidal maturation function included in the final model.
Dose-exposure relationship
Dosing simulations were performed using the final PPK model. A virtual adult patient (70 kg weight) administered clindamycin 600 mg intravenously every 8 hours had a median (2.5th, 97.5th percentiles) area under the concentration versus time curve from 0–8 hours at steady state (AUCss,0–8) of 42.9 (14.2, 132) mcg*h/mL. Optimized PMA-based dosing resulted in simulated median AUCss,0–8 comparable to the virtual adult estimate (administered every 8 hours): 42.9 mcg*h/mL (5 mg/kg, PMA ≤32 weeks); 42.1 mcg*h/mL (7 mg/kg, PMA >32–40 weeks); 42.7 mcg*h/mL (9 mg/kg, PMA >40–60 weeks) (Figure 4). For older infants (≥5 months PNA) to adolescents, age-based dosing resulted in a median AUCss,0–8 similar to the adult prediction (administered every 8 hours): 42.2 mcg*h/mL (≥5 months–6 years, 12 mg/kg); 46.9 mcg*h/mL (>6–18 years, 10 mg/kg) (Figure 5). Median simulated maximal drug concentrations at steady state (Css,max) were also comparable to the predicted level for adults (within 25%) for all age groups except for PMA >28–32 weeks (~30% lower) (Supplemental Tables 2 and 3).
Figure 4.
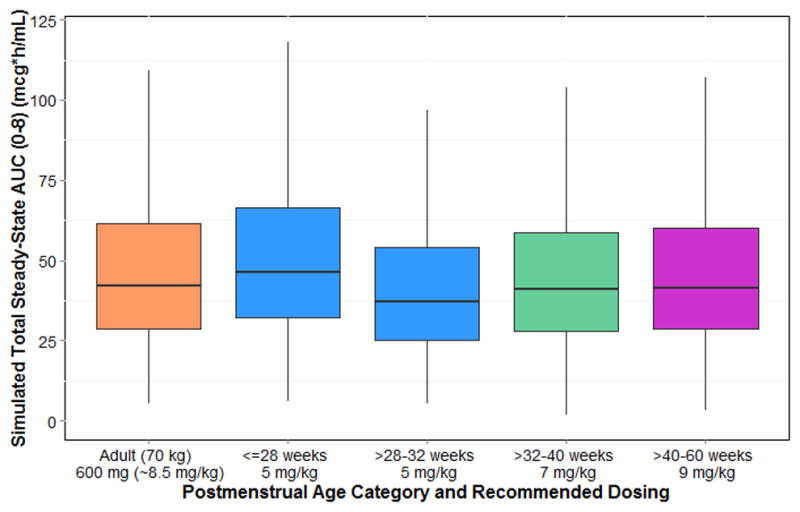
Optimized clindamycin dosing for infants of postmenstrual age ≤60 weeks (~5 months age). AUC, area under the curve at steady-state.
Figure 5.
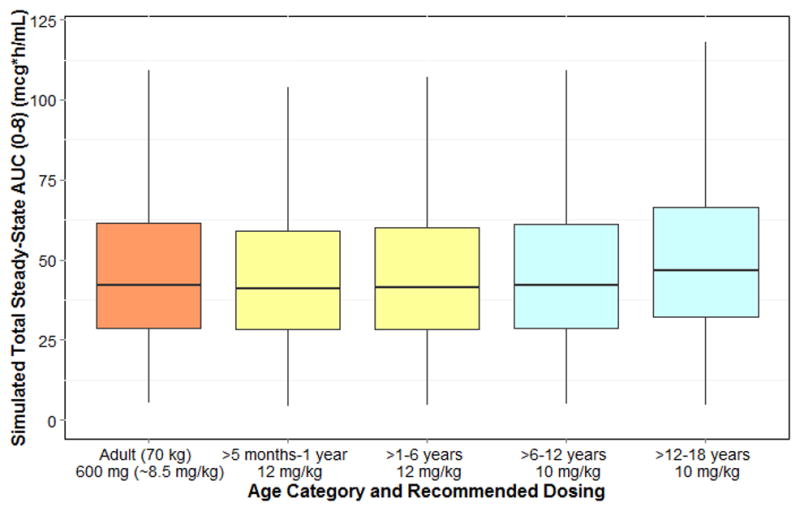
Optimized clindamycin dosing for infants (>5 months postnatal age) to adolescents. AUC, area under the curve at steady-state.
DISCUSSION
Clindamycin is prescribed frequently to children with common infections. Despite its widespread use, limited PK studies have been performed to characterize clindamycin disposition in the pediatric (i.e., neonatal to adolescent) population. Similar to a published clindamycin PPK model for adults,16 a 1-compartment model described the pediatric data well, and population estimates for CL and V, standardized to a 70 kg adult (13.7 L/h and 61.8 L, respectively), were comparable to values previously reported for adults (15.2 L/h and 66.2 L).16
After accounting for size-based (i.e., body weight) differences in PK parameters using a fixed exponent allometric relationship (CL=0.75; V=1), a sigmoidal EMAX maturation model successfully characterized developmental changes in CL across the age continuum from preterm infants to adolescents. This finding is consistent with prior evaluations of drug disposition in children, including dexmedetomidine, lamivudine, and vancomycin.17–19 The maturation half-life and slope factor for the maturation function were 43.5 weeks and 3.1, respectively. These estimates are similar to values reported previously for other CYP3A substrates: 39.7 weeks and 1 (slope factor not estimated) for lopinavir20 and 35.7 weeks and 3.87 for levobupivacaine.21,22
The estimate of maturation half-life observed in this study also agrees with available data describing the ontogeny of CYP3A expression in the human liver.23–25 Following birth, CYP3A7, the predominant isoform in the fetal liver, is known to decrease with PNA, though significant expression has been noted up to 6 months PNA.25 In contrast, CYP3A4 expression is low at birth, and functional enzymatic activity is rapidly acquired.23–25 One analysis pooled available hepatic CYP3A expression data and estimated a maturation half-life of 0.31 years (~16 weeks PNA).26 Given that the median GA was 31.1 weeks in our study, a maturation half-life of 47 weeks (PMA) corresponds well with the estimate observed in our PPK analysis.
Maturation in drug CL as a function of age is reflected in individual parameter estimates observed in this study. In infants, prematurity had a significant impact on CL; when stratified based on GA (≤32 weeks vs. >32 weeks), a notably lower CL was observed for infants ≤32 weeks. However, CL estimates are at least 2-fold higher than those published previously for infants, perhaps due to an older PNA in our cohort. One study that evaluated clindamycin PK following intravenous administration (17.9–44 mg/kg/day) in 12 newborn infants (GA 26–39 weeks; PNA 1–24 days; weight 0.8–2.6 kg) reported a mean (range) CL of 0.06 (0.02–0.13) L/h/kg.15 In a separate study of 40 infants (GA 28–40 weeks; PNA 2–27 days; weight 1–9.6 kg) who received intravenous clindamycin (15–20 mg/kg/day), CL was significantly higher in infants >4 weeks age (1.59 L/h) when compared with premature (0.29 L/h) or term (0.68 L/h) infants at birth (body weights were not reported for each respective age group).
Dosing simulations showed that the median simulated AUCss,0–8 following a 600 mg dose in a 70 kg adult was comparable to previously published values for adults.27 Simulations support age-based dosing (all regimens every 8 hours) with the lowest weight-normalized dose administered to premature infants ≤32 weeks PMA. Higher doses for infants 32–40 weeks and 40–60 weeks PMA are supported by a greater than 30% and 100% higher (relative to premature infants <32 weeks PMA) weight-normalized clearance, respectively. Older infants and children (>5 months–18 years) require higher weight-normalized doses relative to adults to match adult exposure. This finding is consistent with allometric scaling of CL across the spectrum of age using body weight.28
The clindamycin adult dose selected to match pediatric exposure was 600 mg every 8 hours, as is recommended in clinical practice guidelines for CA-MRSA infections.1 This dose has been shown to be bactericidal both in vitro and in a murine thigh infection model for up to 72 hours against a CA-MRSA strain lacking inducible clindamycin resistance.29 In children, current treatment guidelines recommend use of clindamycin 10–13 mg/kg/dose every 6–8 hours (not to exceed 40 mg/kg/day) for skin and soft tissue infections, pneumonia, and osteomyelitis, if clindamycin resistance rate is low (<10%) and CA-MRSA is the likely pathogen.1 Clinical studies evaluating clindamycin use for osteomyelitis and invasive infections caused by CA-MRSA have described use of clindamycin intravenous doses of 20–40 mg/kg/day in the pediatric population.30–32
In one study, 48 pediatric patients, age 1 month to 14 years, received 20–30 mg/kg/day of clindamycin (administered in 3 divided doses) for treatment of osteomyelitis and septic arthritis. Serum clindamycin concentrations were 8- to 32-fold higher than the minimum inhibitory concentration (MIC) of organisms isolated, bone and synovial fluid concentrations were 60–85% those reported in serum, and a favorable response was observed in 88% (42/48) of children (delayed response in 5 patients).30 Similarly, another study described use of clindamycin (30 mg/kg/day in 3 divided doses) in children (mean ± SD age, 6.3 ± 4 years) for treatment of osteomyelitis due to S. aureus, and 92% (11/12) of children had a favorable response to treatment.31 For treatment of bacteremia and complicated pneumonia, a retrospective study described effective use of clindamycin 40 mg/kg/day in children.32 Doses up to 50 mg/kg/day have been studied in pediatric patients (5 weeks to 15 years age) with osteomyelitis.33 A favorable safety profile for clindamycin was observed across all studies.
Use of an opportunistic study design was an efficient approach that resulted in development of a PPK model that characterizes clindamycin drug disposition in children. The model was then used to assess optimal dosing for treatment of CA-MRSA infections in children. However, study limitations include: 1) the timing and number of samples varied significantly across patients, which may have prevented precise estimation of the between-subject variability in V and determination of the influence of obesity on PK parameter estimates; 2) despite accounting for a size and age effect on CL, between-subject variability was 57%, suggesting that additional sources of variability need to be characterized; 3) protein binding was not assessed, potentially limiting our ability to characterize differences in unbound drug concentrations in infants versus older populations; and 4) laboratory values (e.g., serum creatinine) were not available for the majority of patients enrolled in this study and may have limited our ability to evaluate the influence of these covariates on PK parameter estimates. For adults, however, product labeling does not recommend dosage adjustments in the presence of mild or moderate renal or hepatic impairment, and it is unlikely that these covariates would play an important role in the absence of severe disease.34
METHODS
Patient population
PK samples used to develop the PPK model were collected from the Pharmacokinetics of Understudied Drugs Administered to Children per Standard of Care (POPS) trial (clinicaltrials.gov #NCT01431326; protocol NICHD-2011-POP01), a multi-center (N=24), prospective, PK and safety study in children (<21 years of age). Children who received one of the targeted drugs of interest (including clindamycin) per standard of care as administered by their treating caregiver were eligible for enrollment. Exclusion criteria included failure to obtain consent/assent or known pregnancy. PK samples were collected either at the time of clinical laboratory collections, or if the parent/patient consented, following a specific collection for study purposes. Because this was a standard-of-care study, dosing and PK sample collection times varied between subjects. Standard-of-care laboratory assessments (e.g., comprehensive metabolic panel) were recorded if collected within 24 hours of a study dose of the drug. GA and PNA were collected in infants ≤120 days PNA. For infants >120 days of age, a GA of 40 weeks was assumed. Subjects were enrolled in the study for up to 90 days. Each study protocol was reviewed and approved by the institutional review board of each participating institution.
Drug dosing and sample collection
Dosing information was collected for up to 8 doses prior to the sampling dose (last dose prior to plasma PK sample collection). PK samples were collected at the time of routine laboratory (i.e., standard-of-care) blood draws unless the parent/guardian of the participating child provided consent allowing for sample collection to be performed for research purposes (specifically for the study). In the latter case, PK blood collections were obtained without regard to alignment with timing of routine laboratory blood draws. The guidelines for PK sampling time points were designed according to the frequency of administration of the study drug (Supplemental Table 4). The same sample processing and storage procedures were followed for all PK samples (collected with standard-of-care blood draws and those collected for research purposes). A maximum of 10 blood samples were collected per study participant.
Analytical methods
Whole blood was collected (200 μL) in an EDTA-K2 microtainer and was processed immediately prior to freezing at the study sites. PK samples were sent to the Pediatric Trials Network central laboratory (OpAns, LLC, Durham, NC) for storage and analysis. Clindamycin concentrations were quantified using a validated liquid chromatography-tandem spectrometry (LC-MS/MS) assay (assay did not quantify metabolite concentrations). The chromatography system and mass spectrometer used for sample analysis were the Agilent 1200 series high-performance liquid chromatography (HPLC) and an Agilent 6400 series triple quadrupole system, respectively. The Pursuit XRS Ultra C18 column (50 mm length x 2 mm internal diameter, 2.8 μm particle size, Agilent) and a gradient mobile phase (water containing 0.5% (v/v) formic acid; methanol containing 0.1% (v/v) formic acid) were used. The validation range for the assay was 50–50,000 ng/mL. The lower limit of quantification was 50 ng/mL. Accuracy and precision were within the FDA bioanalytical assay validation criteria (e.g., ±15%).
Structural model development
Clindamycin plasma PK data collected following intravenous administration were analyzed with a nonlinear mixed effects modeling approach using the software NONMEM (version 7.2, Icon Solutions, Ellicott City, MD). The first-order conditional estimation method with interaction (FOCE-I) was used for all model runs. Run management was performed using Pirana (version 2.8.1).35 Visual predictive checks and bootstrap methods were performed with Perl-speaks-NONMEM (PsN, version 3.6.2).36 Data manipulation and visualization were performed using R (version 3.0.2, R Foundation for Statistical Computing, Vienna, Austria) and RStudio (version 0.97.551, RStudio, Boston, MA), with the packages lattice, Xpose and ggplot2 used for the latter.37–39
Based on visual inspection of the PK data and a review of the primary literature, 1- and 2-compartment PK models were evaluated using the ADVAN1 TRANS2 and ADVAN3 TRANS4 subroutines, respectively, in NONMEM. Between-subject variability was assessed for PK model parameters using an exponential relationship (Equation 1).
| (1) |
Where Pij denotes the estimate of parameter j in the ith individual; θPop,j is the population value for parameter j; and ηij denotes the deviation from the average population value for parameter j in the ith individual. The random variable η is assumed to be normally distributed with a mean zero and variance ω2. The covariances between η parameters (e.g., CL and V) were estimated. Proportional, additive, and combined (proportional plus additive) residual error models were evaluated.
Covariate analysis
The potential effects of clinical covariates on PK parameters were evaluated if a relationship was suggested by visual inspection of scatter and box plots (continuous and categorical variables, respectively) of the individual deviations from the population-typical value for PK parameters (ETAs) against covariates. The following covariates were explored: PNA, PMA, GA (≤32 weeks vs. >32 weeks), aspartate aminotransferase, alanine aminotransferase, serum creatinine, bilirubin (total [TBIL] and direct [DBIL]), serum albumin, obese status (body mass index ≥95th percentile), ethnicity, and sex. PMA is defined as the sum of the GA (weeks) plus PNA in weeks (days/7). A 32-week GA cutoff was selected to evaluate the impact of prematurity on drug disposition.40
Body weight (WT) was assumed to be a significant covariate for CL and V and was included in the base model prior to assessment of other covariates. The relationship between WT and PK parameters was characterized using a fixed exponent allometric relationship for CL parameters and a linear-scale for V parameters (Equations 2 and 3).21 Scaling was based on a 70 kg standardized adult weight as shown below.
| (2) |
| (3) |
Where CLstd and Vstd represent population estimates of CL and V in a 70 kg subject and WTi denotes body weight for the ith subject.
With the exception of WT and age, continuous covariates were normalized to the population median value as described in Equation 4, whereas for categorical covariates such as obese status, a relationship as shown in Equation 5 was used.
| (4) |
| (5) |
Where covi denotes the individual covariate value; covm is the population median covariate value; θcov is a parameter which represents the covariate effect; and OBESE is a categorical variable which takes on a value of unity when obese and zero when non-obese or in cases where obese status was not assessed (<2 years age) or unavailable.
The relationship between age (e.g., PMA, PNA) and CL was explored using a sigmoidal EMAX maturation function as shown in Equation 6.41
| (6) |
Where FPMA denotes the fraction of the adult CL value; TM50 represents the value of PMA (weeks) when 50% adult clearance is reached; and HILL is a slope parameter for the sigmoidal maturation model. Power and linear functions using PMA and PNA were also evaluated.
A forward inclusion (p<0.05 and ΔOFV > 3.8) and backward elimination (p<0.001 and ΔOFV >10.8) approach was used to assess the statistical significance of relevant covariates. Missing covariate values were imputed using the median value for the sample.
Model evaluation
During the PPK model-building process, successful minimization, diagnostic plots, plausibility and precision of parameter estimates, as well as objective function and shrinkage values, were used to assess model appropriateness.42 Diagnostic plots generated included: individual predictions (IPRED) and population predictions (PRED) vs. observations; conditional weighted residuals (CWRES) vs. PRED and time; and individual weighted residuals (IWRES) vs. IPRED.
Parameter precision for the final PPK was evaluated using non-parametric bootstrapping (1000 replicates) to generate 95% confidence intervals (CI) for parameter estimates using the percentile method. Visual predictive checks were performed whereby the base and final models were used to generate 1000 Monte Carlo simulation replicates per time point of clindamycin exposure, and simulated results were compared with those observed in the study. The dosing and covariate values used to generate the simulations in the visual predictive checks were the same as those used in the study population.
Dose-exposure relationship
Clindamycin has a prolonged post-antibiotic effect, and AUC is an important predictor of drug efficacy.43 Pediatric dosing regimens were optimized to match (within 20%) median adult (70 kg WT) clindamycin exposure, specifically AUCss,0–8, following intravenous administration of 600 mg every 8 hours—the dose recommended for CA-MRSA pneumonia, osteomyelitis, and skin and soft tissue infections in adults.1 Css,max and minimum steady-state concentrations (Css,min) were also computed using the intermittent infusion equation.
Covariate values used in the simulations were the same as those in the study population used for model development. Using PK parameter estimates from the final population model, 1000 concentrations versus time profiles were simulated for each virtual patient. A maximum absolute dose of 900 mg every 8 hours and infusion duration of 0.5 hours was used in all dosing simulations.
CONCLUSION
A PPK model was developed with data collected from premature infants to adolescents using an opportunistic study design. A 1-compartment model, accounting for size-based differences with body weight and maturational factors and using a sigmoidal function, characterized drug disposition across pediatric age groups. The final model was applied to simulate drug exposure. Age-based dosing is recommended to match dosing recommended in adults.
Supplementary Material
Supplemental Figure 1. Visual predictive check (n=1000 simulations).
Supplemental Table 1. Summary of covariate model building process
Supplemental Table 2. Simulated parameter estimates and drug exposure following optimized drug dosing in infants ≤ 60 weeks postmenstrual age
Supplemental Table 3. Simulated parameter estimates and drug exposure following optimized drug dosing in infants to adolescents 5 months to 18 years of age
Supplemental Table 4. Optimal sample collection scheme for clindamycin PK samples collected for research purposes only
STUDY HIGHLIGHTS.
What is the current knowledge on the topic?
Limited published data suggest that there are important age-dependent differences in clindamycin disposition between children and adults.
What question does this study address?
This study sought to characterize clindamycin drug disposition across the age continuum from premature infants to adolescents using an opportunistic clinical trial design and to optimize dosing against CA-MRSA infections in this population.
What does this study adds to our knowledge?
Based on Monte Carlo simulations, age-based dosing, which matches adult exposure proven effective against CA-MRSA, is recommended: PMA≤32 weeks, 5 mg/kg; PMA >32–40 weeks, 7 mg/kg; PMA >40–60 weeks, 9 mg/kg; 5 months – 6 years, 12 mg/kg; >6 – 18 years of age, 10 mg/kg.
How this might change clinical pharmacology and therapeutics?
This study showed that an opportunistic study design can be used to efficiently characterize drug disposition for an off-patent therapeutic in the pediatric population and can serve as an example for future investigations.
Acknowledgments
This work was funded under National Institute of Child Health and Human Development (NICHD) contract HHSN201000003I for the Pediatric Trials Network (PI: Benjamin) and HHSN27500006 (PI: Melloni, Cohen-Wolkowiez) for the Pharmacokinetics of Understudied Drugs Administered to Children per Standard of Care Study (POPS; protocol NICHD-2011-POP01). Research reported in this publication was also supported by the National Center for Advancing Translational Sciences of the National Institutes of Health (NIH) under award number UL1TR001117.
The Pediatric Trials Network Administrative Core Committee
Katherine Y. Berezny, Duke Clinical Research Institute, Durham, NC; Jeffrey Barrett, Sanofi, Bridgewater, NJ; Gregory L. Kearns, Children’s Mercy Hospital, Kansas City, MO; Matthew Laughon, University of North Carolina at Chapel Hill, Chapel Hill, NC; Andre Muelenaer, Virginia Tech Carilion School of Medicine, Roanoke, VA; T. Michael O’Shea, Wake Forest Baptist Medical Center, Winston Salem, NC; Ian M. Paul, Penn State College of Medicine, Hershey, PA; P. Brian Smith, Duke Clinical Research Institute Durham, NC; John van den Anker, George Washington University School of Medicine and Health, Washington, DC; Kelly Wade, Children’s Hospital of Philadelphia, Philadelphia, PA; Thomas J. Walsh, MD, Weill Cornell Medical College of Cornell University, New York, NY.
The Eunice Kennedy Shriver National Institute of Child Health and Human Development: David Siegel, Perdita Taylor-Zapata, Anne Zajicek, Zhaoxia Ren, Alice Pagan.
The EMMES Corporation (Data Coordinating Center): Ravinder Anand, Traci Clemons, Gina Simone.
Pediatric Trials Network Study Team, Principal Investigators, and Study Coordinators
Andrew Lewandowski, PhD (statistician) and Barrie Harper, MT (ASCP), PMP (project leader), Duke Clinical Research Institute, Durham, NC; Ram Yogev, MD (PI) and Laura Fern, RN (study coordinator [SC]), Ann & Robert H. Lurie Children’s Hospital, Chicago, IL; Brenda Poindexter, MD (PI) and Lucy Smiley, BA (SC), Riley Hospital for Children, Indianapolis, IN; Susan Mendley, MD (PI) and Donna Cannonier, LPN, MS, MBA (SC), University of Maryland Hospital, Baltimore, MD; Paula Delmore, MSN (PI & SC), Wesley Medical Center, Wichita, KS; Janice Sullivan, MD, (PI) and Tressa Bratton, MSN, CPN (SC), Kosair Children’s Hospital, Louisville, KY.
Footnotes
See Acknowledgments for listing of committee members
Author Contributions
D.G. and M.C-W. wrote the manuscript. D.G., K.M.W., and M.C-W analyzed the data. D.G., C.M., K.M.W., D.K.B., and M.C-W. designed the research. D.G., C.M., R.Y., B.B.P., S.M., P.D., J.S., J.A., A.L., B.H., K.M.W., K.C.L, E.V.C., D.K.B., and M.C-W performed the research.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The assay measuring clindamycin concentrations was performed at OpAns Laboratory (Durham, NC, USA) by Lisa St. John-Williams, Christine Grosse, and Randall Armour.
Conflict-of-Interest Disclosures
D.G. is funded by training grant T32GM086330 from the National Institute of General Medical Sciences (NIGMS). K.M.W. receives support from NIGMS (1T32GM086330-01A1) and the Thrasher Research Fund (www.thrasherresearch.org) for his work in pediatric clinical pharmacology. E.V.C. receives salary support from the United States government (U54 HD071600-01) and research support from Trius, Cerexa Pharmaceuticals, Abbott, and Theravance. D.K.B. Jr. receives support from the United States government for his work in pediatric and neonatal clinical pharmacology (1R01HD057956-05, 1K24HD058735-05, UL1TR001117, and NICHD contract HHSN275201000003I) and the nonprofit organization Thrasher Research Fund for his work in neonatal candidiasis (www.thrasherresearch.org); he also receives research support from industry for neonatal and pediatric drug development (www.dcri.duke.edu/research/coi.jsp). M.C.W. receives support for research from the NIH (1K23HD064814), the National Center for Advancing Translational Sciences of the NIH (UL1TR001117), the Food and Drug Administration (1U01FD004858-01), the Biomedical Advanced Research and Development Authority (BARDA) (HHSO100201300009C), the nonprofit organization Thrasher Research Fund (www.thrasherresearch.org), and from industry for drug development in adults and children (www.dcri.duke.edu/research/coi.jsp). The remaining authors have no funding to disclose.
References
- 1.Liu C, et al. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis. 2011;52:e18–55. doi: 10.1093/cid/ciq146. [DOI] [PubMed] [Google Scholar]
- 2.Gerber JS, Coffin SE, Smathers SA, Zaoutis TE. Trends in the incidence of methicillin-resistant Staphylococcus aureus infection in children’s hospitals in the United States. Clin Infect Dis. 2009;49:65–71. doi: 10.1086/599348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herigon JC, Hersh AL, Gerber JS, Zaoutis TE, Newland JG. Antibiotic management of Staphylococcus aureus infections in US children’s hospitals, 1999–2008. Pediatrics. 2010;125:e1294–300. doi: 10.1542/peds.2009-2867. [DOI] [PubMed] [Google Scholar]
- 4.Gatti G, et al. Comparative study of bioavailabilities and pharmacokinetics of clindamycin in healthy volunteers and patients with AIDS. Antimicrob Agents Chemother. 1993;37:1137–1143. doi: 10.1128/aac.37.5.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wynalda M, Hutzler J, Koets M. In vitro metabolism of clindamycin in human liver and intestinal microsomes. Drug Metab Dispos. 2003;31:878–887. doi: 10.1124/dmd.31.7.878. [DOI] [PubMed] [Google Scholar]
- 6.DeHaan RM, Metzler CM, Schellenberg D, Vandenbosch WD. Pharmacokinetic studies of clindamycin phosphate. J Clin Pharmacol. 1973;13:190–209. doi: 10.1002/j.1552-4604.1973.tb00208.x. [DOI] [PubMed] [Google Scholar]
- 7.DeHaan R, Metzler C, Schellenberg D, VandenBosch W, Masson E. Pharmacokinetic studies of clindamycin hydrochloride in humans. Int J Clin Pharmacol. 1972;6:105–119. [PubMed] [Google Scholar]
- 8.Gatti G, et al. Penetration of clindamycin and its metabolite N-demethylclindamycin into cerebrospinal fluid following intravenous infusion of clindamycin phosphate in patients with AIDS. Antimicrob Agents Chemother. 1998;42:3014–3017. doi: 10.1128/aac.42.11.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flaherty J, et al. Comparative pharmacokinetics and serum inhibitory activity of clindamycin in different dosing regimens. Antimicrob Agents Chemother. 1988;32:1825–1829. doi: 10.1128/aac.32.12.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wade KC, et al. Population pharmacokinetics of fluconazole in young infants. Antimicrob Agents Chemother. 2008;52:4043–4049. doi: 10.1128/AAC.00569-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cohen-Wolkowiez M, et al. Population pharmacokinetics of piperacillin using scavenged samples from preterm infants. Ther Drug Monit. 2012;34:312–319. doi: 10.1097/FTD.0b013e3182587665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen-Wolkowiez M, et al. Population pharmacokinetics of metronidazole evaluated using scavenged samples from preterm infants. Antimicrob Agents Chemother. 2012;56:1828–1837. doi: 10.1128/AAC.06071-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laughon MM, et al. Innovative clinical trial design for pediatric therapeutics. Expert Rev Clin Pharmacol. 2011;4:643–652. doi: 10.1586/ecp.11.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bell MJ, Shackelford P, Smith R, Schroeder K. Pharmacokinetics of clindamycin phosphate in the first year of life. J Pediatr. 1984;105:482–486. doi: 10.1016/s0022-3476(84)80033-5. [DOI] [PubMed] [Google Scholar]
- 15.Koren G, Zarfin Y, Maresky D. Pharmacokinetics of intravenous clindamycin in newborn infants. Pediatr Pharmacol. 1985;5:287–292. [PubMed] [Google Scholar]
- 16.Bouazza N, et al. Population pharmacokinetics of clindamycin orally and intravenously administered in patients with osteomyelitis. Br J Clin Pharmacol. 2012;74:971–977. doi: 10.1111/j.1365-2125.2012.04292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Potts AL, et al. Dexmedetomidine pharmacokinetics in pediatric intensive care—a pooled analysis. Paediatr Anaesth. 2009;19:1119–1129. doi: 10.1111/j.1460-9592.2009.03133.x. [DOI] [PubMed] [Google Scholar]
- 18.Bouazza N, et al. Developmental pharmacokinetics of lamivudine in 580 pediatric patients ranging from neonates to adolescents. Antimicrob Agents Chemother. 2011;55:3498–3504. doi: 10.1128/AAC.01622-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson BJ, Allegaert K, Van den Anker JN, Cossey V, Holford NHG. Vancomycin pharmacokinetics in preterm neonates and the prediction of adult clearance. Br J Clin Pharmacol. 2007;63:75–84. doi: 10.1111/j.1365-2125.2006.02725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Urien S, et al. Lopinavir/ritonavir population pharmacokinetics in neonates and infants. Br J Clin Pharmacol. 2011;71:956–960. doi: 10.1111/j.1365-2125.2011.03926.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holford N, Heo Y, Anderson B. A pharmacokinetic standard for babies and adults. J Pharm Sci. 2013;102:2941–2952. doi: 10.1002/jps.23574. [DOI] [PubMed] [Google Scholar]
- 22.Chalkiadis G, Anderson BJ. Age and size are the major covariates for prediction of levobupivacaine clearance in children. Paediatr Anaesth. 2006;16:275–282. doi: 10.1111/j.1460-9592.2005.01778.x. [DOI] [PubMed] [Google Scholar]
- 23.Lacroix D, Sonnier M, Moncion A, Cheron G, Cresteil T. Expression of CYP3A in the human liver—evidence that the shift between CYP3A7 and CYP3A4 occurs immediately after birth. Eur J Biochem. 1997;247:625–634. doi: 10.1111/j.1432-1033.1997.00625.x. [DOI] [PubMed] [Google Scholar]
- 24.Treluyer J, et al. Oxidative metabolism of amprenavir in the human liver. Effect of the CYP3A maturation. Drug Metab Dispos. 2003;31:275–281. doi: 10.1124/dmd.31.3.275. [DOI] [PubMed] [Google Scholar]
- 25.Stevens J, Hines R, Gu C. Developmental expression of the major human hepatic CYP3A enzymes. J Pharmacol Exp Ther. 2003;307:573–582. doi: 10.1124/jpet.103.054841. [DOI] [PubMed] [Google Scholar]
- 26.Johnson TN, Rostami-Hodjegan A, Tucker GT. Prediction of the clearance of eleven drugs and associated variability in neonates, infants, and children. Clin Pharmacokinet. 2006;45:931–956. doi: 10.2165/00003088-200645090-00005. [DOI] [PubMed] [Google Scholar]
- 27.Gatti G, Flaherty J, Bubp J. Comparative study of bioavailabilities and pharmacokinetics of clindamycin in healthy volunteers and patients with AIDS. Antimicrob Agents Chemother. 1993;37:1137–1143. doi: 10.1128/aac.37.5.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anderson BJ, Holford NHG. Understanding dosing: children are small adults, neonates are immature children. Arch Dis Child. 2013;98:737–744. doi: 10.1136/archdischild-2013-303720. [DOI] [PubMed] [Google Scholar]
- 29.LaPlante KL, Leonard SN, Andes DR, Craig W, Rybak MJ. Activities of clindamycin, daptomycin, doxycycline, linezolid, trimethoprim-sulfamethoxazole, and vancomycin against community-associated methicillin-resistant Staphylococcus aureus with inducible clindamycin resistance in murine thigh infection and in vitro pharmacodynamic models. Antimicrob Agents Chemother. 2008;52:2156–2162. doi: 10.1128/AAC.01046-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feigin R, Pickering L, Anderson D. Clindamycin treatment of osteomyelitis and septic arthritis in children. Pediatrics. 1975;55:213–223. [PubMed] [Google Scholar]
- 31.Kaplan S, Mason E, Feigin R. Clindamycin versus nafcillin or methicillin in the treatment of Staphylococcus aureus osteomyelitis in children. South Med J. 1982;75:138–142. doi: 10.1097/00007611-198202000-00005. [DOI] [PubMed] [Google Scholar]
- 32.Martínez-Aguilar G, Hammerman W, Mason EO, Kaplan SL. Clindamycin treatment of invasive infections caused by community-acquired, methicillin-resistant and methicillin-susceptible Staphylococcus aureus in children. Pediatr Infect Dis J. 2003;22:593–598. doi: 10.1097/01.inf.0000073163.37519.ee. [DOI] [PubMed] [Google Scholar]
- 33.Rodriguez W, Ross S, Khan W, McKay D, Moskowitz P. Clindamycin in the treatment of osteomyelitis in children: a report of 29 cases. Am J Dis Child. 1977;131:1088–1093. doi: 10.1001/archpedi.1977.02120230034005. [DOI] [PubMed] [Google Scholar]
- 34.Bedford Laboratories. [Accessed on February 17, 2014];Clindamycin phosphate injection. Available at: http://dailymed.nlm.nih.gov.
- 35.Keizer RJ, van Benten M, Beijnen JH, Schellens JHM, Huitema ADR. Pirana and PCluster: a modeling environment and cluster infrastructure for NONMEM. Comput Methods Programs Biomed. 2011;101:72–79. doi: 10.1016/j.cmpb.2010.04.018. [DOI] [PubMed] [Google Scholar]
- 36.Lindbom L, Pihlgren P, Jonsson EN, Jonsson N. PsN-Toolkit—a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed. 2005;79:241–257. doi: 10.1016/j.cmpb.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 37.Jonsson EN, Karlsson MO. Xpose—an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed. 1999;58:51–64. doi: 10.1016/s0169-2607(98)00067-4. [DOI] [PubMed] [Google Scholar]
- 38.Wickham H. ggplot2: Elegant Graphics for Data Analysis. Springer; New York: 2009. [Google Scholar]
- 39.Sarkar D. Lattice: Multivariate Data Visualization with R. Springer; New York: 2008. [Google Scholar]
- 40.Engle WA. American Academy of Pediatrics Committee on Fetus and Newborn. Age terminology during the perinatal period. Pediatrics. 2004;114:1362–1364. doi: 10.1542/peds.2004-1915. [DOI] [PubMed] [Google Scholar]
- 41.Anderson BJ, Holford NHG. Mechanism-based concepts of size and maturity in pharmacokinetics. Annu Rev Pharmacol Toxicol. 2008;48:303–332. doi: 10.1146/annurev.pharmtox.48.113006.094708. [DOI] [PubMed] [Google Scholar]
- 42.Karlsson M, Savic R. Diagnosing model diagnostics. Clin Pharmacol Ther. 2007;82:17–20. doi: 10.1038/sj.clpt.6100241. [DOI] [PubMed] [Google Scholar]
- 43.Craig W, Kiem S, Andes D. Free drug 24-hour AUC/MIC is the PK/PD target that correlates with in vivo efficacy of macrolides, azilides, ketolides and clindamycin. Presented at: 42nd Interscience Conference on Antimicrobial Agents and Chemotherapy; San Diego, CA. 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Visual predictive check (n=1000 simulations).
Supplemental Table 1. Summary of covariate model building process
Supplemental Table 2. Simulated parameter estimates and drug exposure following optimized drug dosing in infants ≤ 60 weeks postmenstrual age
Supplemental Table 3. Simulated parameter estimates and drug exposure following optimized drug dosing in infants to adolescents 5 months to 18 years of age
Supplemental Table 4. Optimal sample collection scheme for clindamycin PK samples collected for research purposes only