

Significance
The 22q11.2 deletion is the most common known genetic cause of congenital heart disease (CHD). The haploinsufficiency of TBX1 has been identified as the cause of CHD. Using mouse models of the disease, we found that reduced dosage of p53 suppresses the Tbx1 mutant phenotype. Tbx1 and p53 proteins coregulate the gene Gbx2, which is required for cardiovascular development. Gbx2 expression is positively regulated by Tbx1, whereas suppression of p53 results in reduced levels of the repressive chromatin mark H3K27me3 at a genetic element occupied by both Tbx1 and p53. These data illustrate a mechanism by which reduced p53, by genetic or pharmacological means, can counterbalance the consequences of reduced dosage of Tbx1.
Keywords: Tbx1 and second heart field, pharyngeal arch artery development
Abstract
T-box 1 (Tbx1), a gene encoding a T-box transcription factor, is required for embryonic development in humans and mice. Half dosage of this gene in humans causes most of the features of the DiGeorge or Velocardiofacial syndrome phenotypes, including aortic arch and cardiac outflow tract abnormalities. Here we found a strong genetic interaction between Tbx1 and transformation related protein 53 (Trp53). Indeed, genetic ablation of Trp53, or pharmacological inhibition of its protein product p53, rescues significantly the cardiovascular defects of Tbx1 heterozygous and hypomorphic mutants. We found that the Tbx1 and p53 proteins do not interact directly but both occupy a genetic element of Gbx2, which is required for aortic arch and cardiac outflow tract development, and is a known genetic interactor of Tbx1. We found that Gbx2 expression is down-regulated in Tbx1+/− embryos and is restored to normal levels in Tbx1+/−;Trp53+/− embryos. In addition, we found that the genetic element that binds both Tbx1 and p53 is highly enriched in H3K27 trimethylation, and upon p53 suppression H3K27me3 levels are reduced, along with Ezh2 enrichment. This finding suggests that the rescue of Gbx2 expression in Tbx1+/−;Trp53+/− embryos is due to reduction of repressive chromatin marks. Overall our data identify unexpected genetic interactions between Tbx1 and Trp53 and provide a proof of principle that developmental defects associated with reduced dosage of Tbx1 can be rescued pharmacologically.
The 22q11.2 deletion syndrome (22q11.2DS), also known as DiGeorge and Velocardiofacial syndromes are relatively frequent and are caused by a chromosomal deletion that includes TBX1 as well as other genes. However, heterozygous mutation of TBX1 alone recapitulates most of the disease phenotype (1–3). Many of the clinical findings are of clear embryonic origin, e.g., congenital heart disease and thymic hypo/aplasia. However, other symptoms are of a less obvious derivation, for example the complex adolescent/adult phenotype, such as the neurobehavioral phenotype (3), which could benefit from specific treatments. Therefore, we searched for strategies to rescue the T-box 1 (Tbx1) haploinsufficiency phenotype based upon our current understanding of disease pathogenesis. Tbx1 supports cell proliferation and inhibits cell differentiation in the second heart field (SHF), a population of cardiac progenitor cells that migrate and populate the outflow tract and right ventricle (4, 5). The gene plays an important, suppressive role in a switch from the progenitor state of SHF cells to the differentiated state as they enter the heart (6). Tbx1 loss of function leads to a reduced pool of cardiac progenitors in the SHF and their premature differentiation and, consequently, hypoplasia of the cardiac outflow tract (OFT). Tbx1 mutation is associated with reduced cell proliferation in several other tissues, for example the pharyngeal endoderm and the otic epithelium (7, 8). In addition, Tbx1 has also been implicated in the expansion of dental stem cells (9) and self-renewal of hair follicle stem cells (10). Based on this knowledge, we sought genetic strategies that could counteract the effects of reduced dosage of Tbx1. The transcription factor p53, which is encoded by the transformation related protein 53 (Trp53) gene, has prodifferentiation and antiproliferation activities in stem cells (11–13), and its loss of function facilitates reprogramming of differentiated cells (14, 15). Therefore, we asked whether Tbx1 and Trp53 interact during cardiovascular development. Results show that heterozygous deletion of Trp53 or pharmacological suppression of p53 partially rescues the cardiovascular phenotype associated with reduced dosage of Tbx1. Surprisingly, we found that Tbx1 and p53 cooccupy chromatin segments, suggesting that they have shared targets. Indeed, we found that Gbx2, a gene required during cardiovascular development and known to genetically interact with Tbx1 (16, 17), is targeted by both transcription factors. Specifically, it is down-regulated by Tbx1 deletion, but its expression is reestablished to wild-type levels in the presence of Trp53 deletion.
Our data reveal a previously unsuspected genetic interaction between Tbx1 and Trp53, provide novel insights into the mechanisms of Tbx1 function, and suggest a role of p53 in cardiac progenitors biology. Importantly, we have also established a proof of principle that the Tbx1 haploinsufficiency phenotype can be rescued pharmacologically.
Results
Epistatic Genetic Interaction Between Trp53 and Tbx1.
We crossed Trp53+/− and Tbx1+/− mice and harvested embryos at embryonic day (E) 10.5 to test whether there is a genetic interaction between Tbx1 and Trp53, and test its effects on the typical haploinsufficiency phenotype, i.e., hypoplasia or aplasia of the fourth pharyngeal arch arteries (fourth PAAs) (18–20). We analyzed a total of 56 embryos; the results are summarized in Table 1. WT and Trp53+/− embryos were all normal (n = 32), whereas Tbx1+/− were mostly abnormal (12 of 13 embryos or 92%). Double heterozygous Trp53+/−;Tbx1+/− embryos, however, were mostly normal (10 out of 11 embryos, or 91%, P = 0.00004). Examples of ink injection data are shown on Fig. 1. We measured the diameter of “rescued” fourth PAAs of Trp53+/−;Tbx1+/− embryos and found it very similar compared with that of WT embryos: 55.1 ± 16.1 µm (n = 22) for the WT and 61.3 ± 9.6 µm (n = 18) in rescued Trp53+/−;Tbx1+/− embryos. Therefore, Trp53 deletion almost completely rescues this particular phenotype. To exclude that rescue was due to a gene mutation linked to the mutant Trp53 allele or to genetic background, we repeated the experiment using pharmacological suppression of p53 with the drug Pifithrin-α (21). Timing of drug administration to pregnant females was based on our findings that Tbx1 is required for fourth PAA development as early as E7.5 but it is not longer required after E9.5 (22). Therefore, we injected Pifithrin-α into pregnant females at E7.5, E8.5, and E9.5 and harvested embryos at E10.5 to score the fourth PAA phenotype. Results (Table 2) showed again a significant suppression of the fourth PAA defect (P = 0.00016). Thus, genetic ablation as well as temporally restricted pharmacological suppression of p53 rescues the Tbx1 haploinsufficiency phenotype.
Table 1.
Rescue of fourth PAA defects by genetic or pharmacological suppression of p53: Genetic experiment
| Genotype (E10.5) | Normal | Fourth PAA defects |
| Tbx1+/+ | 16 | 0 |
| Tbx1+/− | 1 | 12 (92%) |
| Trp53+/− ; Tbx1+/− | 10 | 1 (9%)** |
| Trp53+/− | 16 | 0 |
“Normal” refers to the pattern of the PAA system. **P = 0.00004.
Fig. 1.

Trp53 deletion rescues the fourth PAA defects in Tbx1+/− embryos. Examples of ink injections assays for the visualization of the pharyngeal arch arteries in E10.5 embryos. (A) Ink-injected, WT E10.5 embryo showing the normal anatomy of the third, fourth, and sixth pharyngeal arch arteries on the left. (B) Ink-injected, Tbx1+/−; Trp53+/+ E10.5 embryo. Note the absence of the left fourth PAA. (C) Ink-injected, Tbx1+/−; Trp53+/− E10.5 embryo. Note the presence and normal anatomy of the fourth PAA. Arrowheads indicate the fourth PAA.
Table 2.
Rescue of fourth PAA defects by genetic or pharmacological suppression of p53: Treatment with Pifithrin-α
| Genotype (E10.5) | Pifithrin-α | Normal | Fourth PAA defects |
| Tbx1+/+ | Untreated | 10 | 0 |
| Treated | 7 | 0 | |
| Tbx1+/− | Untreated | 0 | 10 (100%) |
| Treated | 14 | 5 (36%)* |
“Normal” refers to the pattern of the PAA system. *P = 0.00016.
Trp53 Mutation Modifies the Hypomorphic but Not the Null Tbx1 Phenotype.
Reduced p53 dosage may suppress the Tbx1 mutant phenotype indirectly (e.g., through a compensatory mechanism that buffers the consequences of loss of Tbx1), or it may act more specifically by interacting with the normal transcriptional functions of Tbx1. Although these hypotheses are not mutually exclusive, if the second mechanism is significant, the Tbx1 protein must be present for the rescue to occur. Therefore, we first tested whether Trp53 mutation can modify the intracardiac phenotype of Tbx1−/− embryos. To this end, we crossed Tbx1+/−; Trp53+/− and Tbx1+/−; Trp53+/+ mice and harvested embryos at E18.5, when the cardiac phenotype can be fully assessed morphologically. Results, summarized in Table S1, revealed the rescue of the cardiovascular haploinsufficiency phenotype (at this stage it consists of anomalous remodeling of the great arteries and aortic arch), thus confirming that the fourth PAAs were not only rescued morphologically at E10.5, but they also underwent normal remodeling. However, Tbx1−/−;Trp53+/+ and Tbx1−/−; Trp53+/− embryos were phenotypically indistinguishable (Table S1). Specifically, we examined, by external inspection and by histology, Tbx1−/−;Trp53+/+ and Tbx1−/−; Trp53+/− hearts and scored the pattern of the aortic arch and connected arteries, type of VSD, the alignment of the truncus and ventricles, and the morphology of the outflow valves. We could not find any morphological difference between the two genotypes. Next, we used the hypomorphic allele Tbx1neo2, which expresses ∼15% of the Tbx1 mRNA compared with the WT allele (23). We crossed Tbx1+/−;Trp53+/− and Tbx1+/neo2;Trp53+/+ mice and harvested embryos at E18.5. A tabulated summary of cardiac phenotyping data is shown in Fig. 2A. The external appearance of Tbx1-/neo2;Trp53+/+ and Tbx1-/neo2;Trp53+/− embryos was indistinguishable and embryo dissection showed that both genotypes had absence or severely hypoplastic thymus. However, cardiac phenotyping revealed some important differences. For practical purposes, we assigned a phenotypic score (from 1 to 7, where 1 is morphologically normal and 7 is the most severe) to individual samples. The most remarkable result was the finding of 3 of the 18 Tbx1-/neo2;Trp53+/− embryos examined (17%) showed a normally septated heart (Fig. 2 B–C', score 1), whereas all of the 17 Tbx1-/neo2;Trp53+/+ embryos examined had a complex septation defect (see, for example, Fig. 3 A, B, E, and F, scores from 4 to 7). In addition, 3 Tbx1−/neo2;Trp53+/− embryos presented with a milder form (score 3) of double outlet right ventricle (DORV) or incomplete truncus arteriosus (TA). In these cases, the aorta or the truncus arteriosus were overriding the ventricular septum, whereas in Tbx1−/neo2;Trp53+/+ embryos they originated from the right ventricle (Fig. 3). This finding indicates that Trp53 mutation improves the alignment between the outflow tract and the ventricles. Furthermore, we found a single case of isolated ventricular septal defect (VSD, score 2) among the Tbx1−/neo2;Trp53+/− embryos (Fig. 3 F–H). In contrast, all of the Tbx1−/neo2;Trp53+/+ embryos examined presented with VSD associated with conotruncal septation defects or DORV (Fig. 2A).
Fig. 2.

Trp53 deletion ameliorates the outflow tract phenotype in Tbx1 hypomorphic mutants. (A) Tabulated summary of cardiovascular phenotype in hypomorphic mutants with and without Trp53 deletion. n, total number of embryos examined with the genotype indicated. “Normal” refers to heart phenotype. DORV, double outlet right ventricle; ovAo, overriding aorta; ovTA, overriding of (unseptated) truncus arteriosus; TA, Truncus arteriosus communis; VSD, ventricular septal defects. The number in parentheses is the phenotypic score, an arbitrary number that indicates the severity of the phenotype, 1 being the mildest and 7 being the most severe. Histological sections of E18.5 embryo hearts showing complete rescue of ventricular and conotruncal septation. (B and B') Two different sections of the same heart of a WT littermate of the embryo shown in C and C'. (C and C') Two different sections of the same heart of a Tbx1-/neo2;Trp53+/− embryo. Note the normal septation of the great arteries and ventricles, similar to WT embryos. A: Aorta; LV: left ventricle; P: Pulmonary trunk; RV: right ventricle.
Fig. 3.

Trp53 deletion ameliorates the outflow tract phenotype in Tbx1 hypomorphic mutants at E18.5. (A and A') Two sections of the same heart of a Tbx1-/neo2;Trp53+/+ embryo. Note that both the aorta and the pulmonary trunk originate from the right ventricle. This is an arrangement observed in all of the embryos with this genotype. (B, B', and C) Three sections of the same heart of a Tbx1-/neo2;Trp53+/− embryo. The pulmonary trunk communicates with the right ventricle, whereas the aorta communicates with both ventricles, indicating a better alignment between the outflow and the heart chambers, compared with the embryo shown in A and A'. In C, the section is shown at a lower magnification to fit the panel. (D and D') Two sections of the same heart of a Tbx1-/neo2;Trp53+/+ embryo. Note that the truncus arteriosus communis (T) is positioned directly above the RV. (E and E') Two sections of the same heart of a Tbx1-/neo2;Trp53+/− embryo. The truncus arteriosus communis communicates with both the RV and LV, above the interventricular septum. (F–H) Three sections of the same heart of a Tbx1-/neo2;Trp53+/− embryo. The pulmonary trunk is connected to the RV and the aorta to the LV. Although the latter is located above the septum, it does not directly communicate with the RV. In H, the section is shown at a lower magnification to fit the panel. A, aorta; DORV, double outlet right ventricle; Inc. TA, incomplete truncus arteriosus (there is truncal septation but not conal septation); LV, left ventricle; P, pulmonary trunk; RV, right ventricle; T, truncus arteriosus unseptated; VSD, ventricular septal defect. Arrowhead, interventricular communication (VSD).
Because the OFT phenotype of Tbx1 mutants is associated with reduced cell proliferation in the SHF, we tested whether Trp53 ablation increases cell proliferation in a Tbx1-/neo2 background, thus providing a possible explanation of the partial rescue of the OFT phenotype. To this end, we performed double immunofluorescence staining of histological sections of Tbx1-/neo2;Trp53+/+ and Tbx1-/neo2;Trp53+/− E9.5 embryos with anti-phosphorylated histone 3 (PH3, a mitotic marker) and anti-Isl1 (a cardiac progenitor marker) antibodies and evaluated the percentage of double positive Isl1+;PH3+ cells over the total of Isl1+ cells in the SHF area (operatively defined as the posterior pericardial wall excluding inflow and outflow proper) (Fig. S1). Results showed that 35.9% of the Isl1+ SHF cells were PH3+ in in Tbx1-/neo2;Trp53+/+ embryos and 62.7% in Tbx1-/neo2;Trp53+/−, significantly more than in Tbx1-/neo2;Trp53+/+ littermates (three embryos per genotype, P = 7.4 × 10−7). Thus, Trp53 ablation had a significant impact on Isl1+ SHF cell proliferation in a Tbx1 mutant background.
Tbx1 and p53 Proteins Cooccupy Chromatin Segments.
To gain insights into the nature of the Tbx1–p53 interaction, we first excluded that Trp53 is a negative regulator of Tbx1 expression in mouse embryos (Fig. 4A). Next, we tested whether the two proteins interact directly. However, coimmunoprecipitation did not reveal any interaction (Fig. 4B). Next, we asked whether the two proteins, albeit not interacting directly, occupy the same chromatin regions. To this end, we used chromatin immunoprecipitation combined with Western blotting (ChIP–WB). Chromatin was extracted from cross-linked 5-Azacytidine/DMSO-induced P19Cl6 cells (48 h after induction), which express both Tbx1 and Trp53 genes (Fig. S2), fragmented to a range of 150–350 bp and immunoprecipitated using anti-Tbx1 or anti-p53 antibodies. Captured complexes were released and de-cross-linked, and proteins were subjected to Western blot analyses. Results showed that the p53 protein was present in the Tbx1-captured complexes (Fig. 4C) and vice versa (Fig. 4D). Thus, there are chromatin segments that are occupied by both Tbx1 and p53, suggesting that the two transcription factors coregulate shared targets.
Fig. 4.
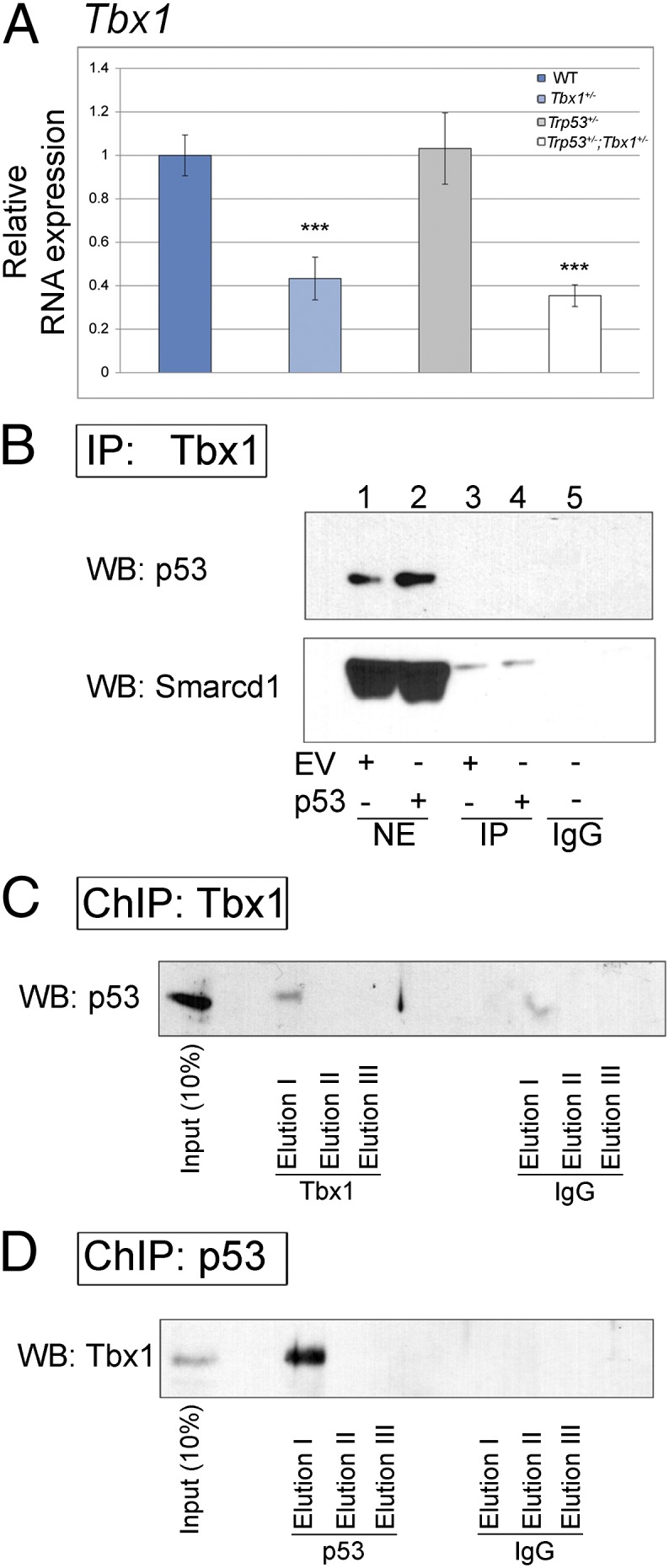
Molecular analyses of Trp53–Tbx1 interaction. (A) Quantitative real time PCR evaluation of Tbx1 expression in E8.5 embryos with the genotypes indicated. Tbx1 expression is associated with heterozygous deletion, as expected, but is not affected by the presence of Trp53 deletion. ***P = 0.001. Error bars indicate SEM of five independent experiments. (B) Immunoprecipitation experiment using nuclear extracts from differentiated (48 h) P19Cl6 cells and an antibody anti-Tbx1. In parallel, we used also cells transfected with a Trp53 expression vector (lanes 2 and 4). We carried out Western blotting using an antibody against p53 and Smarcd1/Baf60a (positive control). The p53 protein is not coimmunoprecipitated. (C) ChIP–Western blot experiment using an anti-Tbx1 antibody. The p53 protein is readily detected. (D) ChIP–Western blot experiment using an anti-p53 antibody. The Tbx1 protein is present in the eluate.
The Gbx2 Gene Is Down-Regulated in Tbx1+/− Embryos, Rescued by Trp53 Mutation, and Occupied by both Transcription Factors.
To identify putative shared targets, we followed a candidate gene approach. We considered three Tbx1 target genes that are known to interact with Tbx1 during fourth PAA development: Gbx2, Smad7, and Wnt5a (17, 24, 25). We first tested the expression of these genes in WT, Tbx1+/−, Trp53+/−, and Tbx1+/−;Trp53+/− E8.5 embryos using quantitative reverse transcription PCR. Results showed Gbx2 to be the most interesting target because its expression was the most affected by heterozygous Tbx1 deletion (Fig. 5 A–C), it was not affected by Trp53 deletion, and, strikingly, expression returned to WT levels in Tbx1+/−;Trp53+/− embryos (Fig. 5C). Homozygous deletion of Gbx2 causes similar fourth PAA abnormalities to those in Tbx1+/− embryos, and Gbx2 heterozygous deletion strongly enhances the Tbx1 haploinsufficiency phenotype (17). Therefore, it is possible that at least part of the gene haploinsufficiency rescue in Tbx1+/−;Trp53+/− embryos is due to rescue of Gbx2 expression. We asked whether the Gbx2 gene may be occupied by both Tbx1 and p53 proteins. To this end, we computationally searched for T-box binding elements (TBEs) and p53 binding elements (p53BEs) and found four candidates for each of these transcription factors (Fig. 6A). To test whether these sites are occupied, we performed ChIP of 5-Azacytidine/DMSO-induced P19Cl6 cells (48 h after induction) because, at this stage, all three endogenous genes (Tbx1, Trp53, and Gbx2) are expressed (Fig. S2). Quantitative ChIP using an anti-Tbx1 antibody revealed the occupation of TBE4 (Fig. 6B, Left), whereas ChIP with an anti-p53 antibody revealed occupation of p53BE3 (Fig. 6B, Right), in four independent experiments per antibody.
Fig. 5.

Trp53 deletion rescues the Gbx2 expression down-regulation in Tbx1+/− E8.5 embryos. (A) Quantitative real-time PCR (qPCR) evaluation of the Smad7 gene expression in E8.5 embryos (two embryos per genotype). None of the variation observed are statistically significant. (B) qPCR evaluation of the Wnt5a gene expression in E8.5 embryos (two embryos per genotype). None of the variation observed are statistically significant. (C) qPCR evaluation of the Gbx2 gene expression in E8.5 embryos (five embryos per genotype). The expression is significantly reduced in Tbx1+/− embryos and significantly enhanced in double heterozygous embryos. There is not significant difference between WT and double heterozygous embryos. **P = 0.004. Error bars indicate SEM of five independent experiments.
Fig. 6.

p53 and Tbx1 occupy the Gbx2 gene. (A, Left) schematic representation of the Gbx2 gene showing the position of computationally detected T-box (blue) and p53 (red) binding sites. (A, Right) Summary of results of ChIP experiments. +, enriched; −, not enriched; NT, not tested. The genomic coordinates (mm10) of TBE4 and p53BE3 are chr1:89925759–89925824 and chr1:89927115–89927206, respectively. (B) Quantitative ChIP (qChIP) results for the two enriched sites. (Left) ChIP using an anti-Tbx1 antibody. (Right) ChIP using an anti-p53 antibody. *P = 0.04; **P = 0.009. Error bars indicate mean ± SD of four independent experiments. (C) qChIP with an anti-H3K27me3 antibody at the p53BE3 site. The experiment was carried out from P19CL6 transfected with a control siRNA (NT) or with an anti-p53 siRNA (p53KD). The immunoprecipitation was carried out with an anti–Trimethyl-Histone H3 (Lys27) antibody. Note that the level of H3K27me3 at the p53BE3 site is significantly lower in the p53 knock down sample. *P < 0.05. Error bars indicate mean ± SD of three biological replicates. (D) qChIP experiment using an antibody anti-histone methyltransferase Ezh2 on the same site as in C. Enrichment is reduced in the samples with p53 knock down. The histogram shows the mean ± SD of three biological replicates. *P < 0.05.
The gene expression data shown above suggest that p53 alone does not have a direct effect on Gbx2 expression, but its deletion buffers the effect of the Tbx1 deletion. Therefore, considering that p53 has been shown to interact with the Phf1 component of the Polycomb repressive complex 2 (PRC2) (26), we asked whether p53 suppression may alter the chromatin state of the Gbx2 genetic element occupied by p53 and Tbx1. We performed qChIP with an anti-H3K27me3 antibody and found that the DNA segment is highly enriched for this histone modification, but after p53 suppression by siRNA, the levels of H3K27me3 are significantly reduced (Fig. 6C). Next, we asked whether H3K27me3 enrichment correlates with local enrichment of Ezh2, the histone methyltransferase that provides enzymatic activity to the PRC2 complex. To this end, we performed qChIP analysis with an anti-Ezh2 antibody and in cells with or without p53. Results revealed that this protein occupies the p53BE3 site, but its enrichment is significantly reduced following p53 knock down (Fig. 6D). However, Ezh2 does not coimmunoprecipitate with p53 (Fig. S3). Overall, these data suggest that p53 indirectly recruits Ezh2 to p53BE3 site, thus regulating its H3K27 methylation status.
Overall, these results indicate that Gbx2 is regulated by both transcription factors.
Discussion
Gene haploinsufficiency is often a cause of genetic disease and indicates absence or insufficiency of genetic buffering system(s) to compensate for the loss of one copy of the gene. Tbx1 is haploinsufficient in humans and mice and its deletion affects a number of morphogenetic and developmental processes. Phenotypic anomalies also extend into adult life, including neuropsychiatric disease, for which there is no specific treatment. Thus, discovery of strategies to buffer gene haploinsufficiency is not only important from a biological point of view, but it is also important for the development of future treatments.
In this work, we followed a rationale that was based on the contrasting effects of Tbx1 and p53 on cell differentiation and proliferation to devise a genetic and pharmacological rescue strategy. We found that Trp53 heterozygous deletion was effective in rescuing the Tbx1 haploinsufficient phenotype in mice, partially effective in rescuing the cardiac outflow tract phenotype in hypomorphic mutants, and ineffective in null mutants. We found an interaction of p53 and Tbx1 in regulating the gene Gbx2. Both Tbx1 and p53 occupy a genetic element of the Gbx2 gene, the expression of which is reduced by Tbx1 heterozygosity and rescued in compound Tbx1+/−;Trp53+/− mutant embryos. Therefore, Gbx2 is a shared target, and the rescue of its expression in double mutants could explain phenotypic rescue. Gbx2 and Tbx1 are coexpressed in the pharyngeal surface ectoderm and in the pharyngeal endoderm (17), tissues that have been linked to the fourth PAA phenotype (17, 27, 28). In addition, Gbx2 is required for PAA and OFT development (16). We found that p53 regulates positively the enrichment of Ezh2 and H3K27me3 at the genetic element occupied by Tbx1 and p53. These data support the hypothesis that suppression of p53 reduces repressive marks on the Gbx2 gene and thereby “facilitates” its regulation by Tbx1. This rescue mechanism requires the presence of the Tbx1 protein, thus explaining why p53 suppression has no effect on the null Tbx1 phenotype.
The enhancement of cell proliferation in the SHF of Tbx1-/neo2;Trp53+/− embryos, compared with Tbx1-/neo2 embryos, could contribute to the improvement of the OFT phenotype. Indeed, reduced cell proliferation in the SHF has been associated with OFT abnormalities (5, 8, 29). In contrast, it is unlikely that rescue may be due to the antiapoptotic effects of loss of p53 because abnormal apoptosis is not part of the Tbx1 mutant cardiovascular phenotype (17, 30) (Fig. S1 C and D).
In view of the complexities of p53 functions in the context of embryonic development and cell differentiation (31–34), it is possible that Gbx2 is not the only player in phenotypic rescue. Nevertheless, our data about the Gbx2 gene illustrate an intriguing mechanism of interaction that might function also on other target genes yet to be identified. In addition, recent human genetics data have implicated the p53 pathway in a population of patients affected by Tetralogy of Fallot, a cardiac outflow tract abnormality (35), thus further suggesting that this pathway has relevance for cardiovascular development. Last but not least, our data provide a proof of concept that the Tbx1 mutant phenotype can be significantly ameliorated using pharmacological approaches, thus encouraging further studies into the functional mechanisms and interactions of Tbx1 with the aim of identifying potential drug targets.
Materials and Methods
Details can be found in SI Materials and Methods.
Mouse Mutant Lines.
We have used lines carrying the alleles Tbx1lacZ (here referred to as Tbx1−) (18), Tbx1neo2 (36), and Trp53− (37). All animal handling and experimentations were performed in accordance with the regulations of the Italian Ministry of Health, in a specific pathogen free (SPF) environment. All mouse lines were maintained in a mixed C57Bl6/129SvEv background.
Quantitative Real-Time PCR, RNA Extraction.
Total RNA was isolated using TRIzol (Invitrogen), and RNA from cells or E8.5 embryos was transcribed into cDNA using the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems). Quantitative real-time PCR was performed with FastStart Universal SYBR Green (Roche). Primer sequences are listed in Table S2.
Ink Injection and Histology.
Intracardiac ink injection was performed on E10.5 embryos as described (38). For histology and immunohistochemistry, embryos or hearts were embedded in paraffin and cut into 7 or 10 μm sections, respectively. For histology, sections were counter stained with eosin. Immunohistochemistry was carried out using an anti–phospho-Histone H3 (Ser10) antibody and an anti-Isl1 antibody (Developmental Studies Hybridoma Bank).
Coimmunoprecipitation.
Coimmunoprecipitation experiments were performed using 5-Azacytidine-DMSO-induced P19CL6 cells after 48 h of induction. Nuclear extracts were immunoprecipitated with an anti-TBX1 antibody (Abcam), with an anti-Ezh2 antibody (BD Biosciences) or with rabbit IgG (Santa Cruz Biotechnology). After immunoprecipitation, samples were extensively washed, resuspended in SDS sample buffer and analyzed by Western blot with an anti-p53 antibody (Santa Cruz Biotechnology) or with an anti-Smarcd1 antibody (BD Bioscience).
Chromatin Immunoprecipitation (ChIP) and Western Blotting.
5-Azacytidine and DMSO induction of differentiation of P19CL6 cells was performed as described (39). Cells were used after 48 h of differentiation. At this stage, cells express Tbx1, Trp53, and Gbx2 but do not express cardiac differentiation markers (Fig. S2). For ChIP assay, cells were fixed with 1% formaldehyde Buffer A (Transcription Factor ChIP kit reagent, Diagenode) at room temperature for 10 min. The cross-linking reaction was stopped using 0.125 mol/L glycine at room temperature. Cells were lysed and chromatin was sonicated using S2 Covaris System. Sonicated chromatin (6 μg) was immunoprecipitated with an anti-TBX1 antibody (Abcam), an anti-p53 antibody (Santa Cruz Biotechnology), an anti-H3K27me3 (Millipore), an anti-Ezh2 antibody (BD Biosciences), or Rabbit Control IgG (Abcam) and then incubated at 4 °C overnight with Preblocked protein A/G coated beads. After incubation, immunoprecipitation, samples were extensively washed and reverse cross-linked. DNA was purified and subjected to quantitative PCR amplification.
For ChIP–Western blotting, sonicated chromatin (obtained as described above) was immunoprecipitated with an anti-TBX1 antibody, an anti-p53 antibody, or Control Rabbit IgG; all antibodies were conjugated with Dynabeads Protein G (Novex by Life Technologies). Samples were extensively washed, and DNA–protein complexes were eluted, reverse cross-linked, and subjected to Western blot analysis.
Supplementary Material
Acknowledgments
We thank Drs. F. Gabriella Fulcoli for help with biochemical experiments, Claudia Angelini for help with statistical analyses, and Elizabeth Illingworth for critical reading of the manuscript. We acknowledge the support of the Institute of Genetics and Biophysics core facilities Integrated Microscopy, Mouse Transgenics and Flow cytometry. This work was funded in part by Italian Telethon Foundation Grant GGP11029 and Italian Ministry of Research Grant PON01_02342, and FareBio (to A.B.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1401923111/-/DCSupplemental.
References
- 1.Papangeli I, Scambler P. The 22q11 deletion: DiGeorge and velocardiofacial syndromes and the role of TBX1. Wiley Interdiscip Rev Dev Biol. 2013;2(3):393–403. doi: 10.1002/wdev.75. [DOI] [PubMed] [Google Scholar]
- 2.Yagi H, et al. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362(9393):1366–1373. doi: 10.1016/s0140-6736(03)14632-6. [DOI] [PubMed] [Google Scholar]
- 3.Paylor R, et al. Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: Implications for 22q11 deletion syndrome. Proc Natl Acad Sci USA. 2006;103(20):7729–7734. doi: 10.1073/pnas.0600206103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen L, Fulcoli FG, Tang S, Baldini A. Tbx1 regulates proliferation and differentiation of multipotent heart progenitors. Circ Res. 2009;105(9):842–851. doi: 10.1161/CIRCRESAHA.109.200295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vincent SD, Buckingham ME. How to make a heart: The origin and regulation of cardiac progenitor cells. Curr Top Dev Biol. 2010;90:1–41. doi: 10.1016/S0070-2153(10)90001-X. [DOI] [PubMed] [Google Scholar]
- 6.Watanabe Y, et al. Fibroblast growth factor 10 gene regulation in the second heart field by Tbx1, Nkx2-5, and Islet1 reveals a genetic switch for down-regulation in the myocardium. Proc Natl Acad Sci USA. 2012;109(45):18273–18280. doi: 10.1073/pnas.1215360109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu H, et al. Tbx1 regulates population, proliferation and cell fate determination of otic epithelial cells. Dev Biol. 2007;302(2):670–682. doi: 10.1016/j.ydbio.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu H, et al. Tbx1 has a dual role in the morphogenesis of the cardiac outflow tract. Development. 2004;131(13):3217–3227. doi: 10.1242/dev.01174. [DOI] [PubMed] [Google Scholar]
- 9.Cao H, et al. Tbx1 regulates progenitor cell proliferation in the dental epithelium by modulating Pitx2 activation of p21. Dev Biol. 2010;347(2):289–300. doi: 10.1016/j.ydbio.2010.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen T, et al. An RNA interference screen uncovers a new molecule in stem cell self-renewal and long-term regeneration. Nature. 2012;485(7396):104–108. doi: 10.1038/nature10940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jain AK, et al. p53 regulates cell cycle and microRNAs to promote differentiation of human embryonic stem cells. PLoS Biol. 2012;10(2):e1001268. doi: 10.1371/journal.pbio.1001268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin T, et al. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol. 2005;7(2):165–171. doi: 10.1038/ncb1211. [DOI] [PubMed] [Google Scholar]
- 13.Meletis K, et al. p53 suppresses the self-renewal of adult neural stem cells. Development. 2006;133(2):363–369. doi: 10.1242/dev.02208. [DOI] [PubMed] [Google Scholar]
- 14.Tapia N, Schöler HR. p53 connects tumorigenesis and reprogramming to pluripotency. J Exp Med. 2010;207(10):2045–2048. doi: 10.1084/jem.20101866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Menendez S, Camus S, Izpisua Belmonte JC. p53: Guardian of reprogramming. Cell Cycle. 2010;9(19):3887–3891. doi: 10.4161/cc.9.19.13301. [DOI] [PubMed] [Google Scholar]
- 16.Byrd NA, Meyers EN. Loss of Gbx2 results in neural crest cell patterning and pharyngeal arch artery defects in the mouse embryo. Dev Biol. 2005;284(1):233–245. doi: 10.1016/j.ydbio.2005.05.023. [DOI] [PubMed] [Google Scholar]
- 17.Calmont A, et al. Tbx1 controls cardiac neural crest cell migration during arch artery development by regulating Gbx2 expression in the pharyngeal ectoderm. Development. 2009;136(18):3173–3183. doi: 10.1242/dev.028902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lindsay EA, et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. 2001;410(6824):97–101. doi: 10.1038/35065105. [DOI] [PubMed] [Google Scholar]
- 19.Merscher S, et al. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell. 2001;104(4):619–629. doi: 10.1016/s0092-8674(01)00247-1. [DOI] [PubMed] [Google Scholar]
- 20.Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet. 2001;27(3):286–291. doi: 10.1038/85845. [DOI] [PubMed] [Google Scholar]
- 21.Komarov PG, et al. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. 1999;285(5434):1733–1737. doi: 10.1126/science.285.5434.1733. [DOI] [PubMed] [Google Scholar]
- 22.Xu H, Cerrato F, Baldini A. Timed mutation and cell-fate mapping reveal reiterated roles of Tbx1 during embryogenesis, and a crucial function during segmentation of the pharyngeal system via regulation of endoderm expansion. Development. 2005;132(19):4387–4395. doi: 10.1242/dev.02018. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Z, Baldini A. In vivo response to high-resolution variation of Tbx1 mRNA dosage. Hum Mol Genet. 2008;17(1):150–157. doi: 10.1093/hmg/ddm291. [DOI] [PubMed] [Google Scholar]
- 24.Papangeli I, Scambler PJ. Tbx1 genetically interacts with the transforming growth factor-β/bone morphogenetic protein inhibitor Smad7 during great vessel remodeling. Circ Res. 2013;112(1):90–102. doi: 10.1161/CIRCRESAHA.112.270223. [DOI] [PubMed] [Google Scholar]
- 25.Chen L, et al. Transcriptional control in cardiac progenitors: Tbx1 interacts with the BAF chromatin remodeling complex and regulates Wnt5a. PLoS Genet. 2012;8(3):e1002571. doi: 10.1371/journal.pgen.1002571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Y, et al. Polycomb group protein PHF1 regulates p53-dependent cell growth arrest and apoptosis. J Biol Chem. 2013;288(1):529–539. doi: 10.1074/jbc.M111.338996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Z, et al. Tbx1 expression in pharyngeal epithelia is necessary for pharyngeal arch artery development. Development. 2005;132(23):5307–5315. doi: 10.1242/dev.02086. [DOI] [PubMed] [Google Scholar]
- 28.Randall V, et al. Great vessel development requires biallelic expression of Chd7 and Tbx1 in pharyngeal ectoderm in mice. J Clin Invest. 2009;119(11):3301–3310. doi: 10.1172/JCI37561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prall OW, et al. An Nkx2-5/Bmp2/Smad1 negative feedback loop controls heart progenitor specification and proliferation. Cell. 2007;128(5):947–959. doi: 10.1016/j.cell.2007.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vitelli F, Morishima M, Taddei I, Lindsay EA, Baldini A. Tbx1 mutation causes multiple cardiovascular defects and disrupts neural crest and cranial nerve migratory pathways. Hum Mol Genet. 2002;11(8):915–922. doi: 10.1093/hmg/11.8.915. [DOI] [PubMed] [Google Scholar]
- 31.Rivlin N, Koifman G, Rotter V. p53 orchestrates between normal differentiation and cancer. Semin Cancer Biol. 2014 doi: 10.1016/j.semcancer.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 32.Danilova N, Sakamoto KM, Lin S. p53 family in development. Mech Dev. 2008;125(11-12):919–931. doi: 10.1016/j.mod.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 33.Armstrong JF, Kaufman MH, Harrison DJ, Clarke AR. High-frequency developmental abnormalities in p53-deficient mice. Curr Biol. 1995;5(8):931–936. doi: 10.1016/s0960-9822(95)00183-7. [DOI] [PubMed] [Google Scholar]
- 34.Sah VP, et al. A subset of p53-deficient embryos exhibit exencephaly. Nat Genet. 1995;10(2):175–180. doi: 10.1038/ng0695-175. [DOI] [PubMed] [Google Scholar]
- 35.Grunert M, et al. Rare and private variations in neural crest, apoptosis and sarcomere genes define the polygenic background of isolated Tetralogy of Fallot. Hum Mol Genet. 2014;23(12):3115–3128. doi: 10.1093/hmg/ddu021. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Z, Huynh T, Baldini A. Mesodermal expression of Tbx1 is necessary and sufficient for pharyngeal arch and cardiac outflow tract development. Development. 2006;133(18):3587–3595. doi: 10.1242/dev.02539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jacks T, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4(1):1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 38.Lindsay EA, et al. Congenital heart disease in mice deficient for the DiGeorge syndrome region. Nature. 1999;401(6751):379–383. doi: 10.1038/43900. [DOI] [PubMed] [Google Scholar]
- 39.Mueller I, Kobayashi R, Nakajima T, Ishii M, Ogawa K. Effective and steady differentiation of a clonal derivative of P19CL6 embryonal carcinoma cell line into beating cardiomyocytes. J Biomed Biotechnol. 2010;2010:380561. doi: 10.1155/2010/380561. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.