

Abstract
Type I interferons (IFNs) play central roles in innate immunity; however, overproduction of IFN can lead to immunopathology. Here, we demonstrate that adenosine deaminase acting on RNA 1 (ADAR1), an RNA-editing enzyme induced by interferon, is essential for cells to avoid inappropriate sensing of cytosolic RNA in an inducible knockout cell model – the primary mouse embryo fibroblast (MEF) derived from ADAR1 lox/lox & Cre-ER mice, as well as in HEK293 cells. ADAR1 suppresses viral and cellular RNA detection by RIG-I through its RNA binding rather than its RNA editing activity. DsRNA binds to both ADAR1 and RIG-I, but ADAR1 reduces RIG-I RNA binding. In the absence of ADAR1, cellular RNA stimulates type I IFN production without viral infection or exogenous RNA stimulation. Moreover, we showed in the ADAR1 inducible knockout mice that ADAR1 gene disruption results in a high level IFN production in neuronal tissues – the hallmark of Aicardi-Goutières Syndrome (AGS), a heritable autoimmune disease recently found to be associated with ADAR1 gene mutations. In summary, this study found that ADAR1 limits cytosolic RNA sensing by RIG-I through its RNA binding activity; therefore, ADAR1 suppresses type I IFN production stimulated by viral and cellular RNAs. These results explain why loss of ADARA1 causes IFN induction and also indicates a mechanism for the involvement of ADAR1 in autoimmune diseases such as AGS.
Keywords: adenosine deaminase acting on RNA 1 (ADAR1), interferon (IFN), Aicardi-Goutières Syndrome (AGS), retinoic acid-inducible gene 1 (RIG-I), RIG-I-like receptors (RLRs), inducible knockout mouse model
Introduction
While interferon (IFN) plays crucial roles in the defense against viral infection, excessive production may lead to pathogenesis of autoimmune diseases (1, 2). An example is Aicardi-Goutières syndrome (AGS), a hereditary neurodegenerative disorder characterized by early onset and progressive encephalopathy with a high level of IFN-α in the cerebrospinal fluid (3, 4). A subset of AGS was recently found to be associated with gene mutations in ADAR1 loci (5). ADAR1, an RNA editing enzyme, has two isoforms, P150 and P110, due to the alternative usage of exon 1a or 1b during RNA splicing (6, 7). P150 is located in the cytoplasm and its expression is induced by IFNs or upon infections, while P110 is mainly located in the nucleus and its expression is constitutive. Besides the dsRNA binding and catalytic deamination domains in ADAR1, which are required for RNA editing activity, P150 also has a Z-DNA binding domain in its N terminus. The function of the Z-DNA binding domain is not well understood but possibly binds to particular Z-DNA structures during gene transcription (8, 9). Both isoforms of ADAR1 convert adenosine to inosine on its double strand RNA substrates, potentially changing the coding information on mRNAs (10, 11). However, the majority of RNA editing occurs in noncoding regions with uncertain biological consequences (12) and the edited RNA molecules constitute the inosine-containing RNA population in the cells (13).
Besides its role in editing RNAs, ADAR1 exerts additional effects through binding to RNA (14, 15) or through interacting with other proteins (16, 17). Recent studies found that IFN signaling was highly activated in hematopoietic (18) and intestine stem cells (19) in the absence of ADAR1 in knockout animal models. These results point to a role for ADAR1 in the suppression of IFN production. ADAR1 is well-defined as an IFN Stimulated Gene (ISG) – an IFN-Stimulated Response Element (ISRE) sequence was identified in its promoter region, which is responsible for the elevated level of ADAR1 during infections (20, 21). It plays complicated roles in viral infection through RNA editing (22–24) and unknown mechanisms (25, 26), which often lead to viral proliferation (26–28). Suppression of the IFN signaling pathway could be a major role of ADAR1 in viral-infected cells and in normal cells to maintain homeostasis. Pathways governing IFN expression in viral-infected cells have been well-studied (29–31). After viruses enter host cells, viral RNA, either as a replicating genome or transcribed RNA, acts as a pathogen-associated molecular pattern (PAMP) that initiates IFN production signaling. The viral RNA usually forms long, double-stranded hairpin structures and may carry 5′ppp that can be detected by retinoic acid-inducible gene 1 (RIG-I)-like receptors (RLRs) (32). Upon binding to the viral RNAs, RLRs associate with the common adaptor MAVS after ubiquitin modifications, which lead to the activation of transcription factor IRF3 and/or IRF7. Binding of IRFs to ISRE in IFN promoters activates IFN expression. ADAR1 deficiency leads to increased expression of ISGs (18), but it is not known whether and how ADAR1 suppresses type I IFN expression. In this study, we took advantage of utilizing an inducible knockout cell model to clearly demonstrate that ADAR1 suppresses type I IFN production. Further, we found that ADAR1, via its RNA binding rather than its RNA editing activity, interacts with the cytosolic signaling pathway used for detection of viral RNA. Both ADAR1 and RIG-I bind to cellular dsRNA, but the former competes with the latter, therefore limiting RNA sensing by RIG-I. In the absence of ADAR1, type I IFN is produced without exogenous RNA stimulation. Deletion of the ADAR1 gene in an inducible knockout mouse model recapitulates the pathogenic change of excessive IFN production in neural tissues observed in AGS.
Materials and Methods
Mice and mouse embryonic fibroblasts
ADAR1 inducible knockout (iKO) mice were prepared by crossing floxed ADAR1 mice (33) and Cre-ER transgenic mice (34) as described previously (19). Mouse embryonic fibroblasts (MEFs) were prepared from each individual E14 embryo of ADAR1 lox/lox; Cre-ER(+) male and female breeding. Polymerase chain reaction (PCR) genotyping was used to identify the iKO and control MEFs prepared from embryos from the same litter. Primary MEFs were used in this study since ADAR1 gene deletion responding to tamoxifen (TM) induction was ineffective in immortalized cell lines. Procedures for all animal experiments were approved by the University of Pittsburgh Institutional Animal Care and Use Committee.
Plasmids
pCMV vector expressing wild type (WT) ADAR1 P150 and E/A mutation were gifts from Dr. Nishikura (35). ADAR1 P110, ADAR1 ΔR, and ADAR1 ΔC were sub-clones derived from a WT clone with deletions. Flag tag was added to all of the clones and the protein expressed from the transfected cells could be detected by M2 antibody on Western blot, except ADAR1 P110, which could be detected by anti-ADAR1 antibody.
SiRNA
siRNAs for ADAR1, RIG-I, and MDA-5 were purchased from Dhamacon. A set of four siRNAs with different sequences for each molecule were used to knock down the protein levels. For ADAR1, the sequences were CAUCAAAUGCCUCAAAUAA (Cat# D-008630-02), UAAAUGCUGUGCUAAUUGA (D-008630-03), GAAACCACCUGUUCAUUAC (D-008630-04), and ACUAAGGAGACAAGCGUCA (D-008630-17); For RIG-I, the sequences were CAGAAGAUCUUGAGGAUAA (Cat# D-012511-01); GCACAGAAGUGUAUAUUGG (Cat# D-012511-02); AGACAUGGGUAUAGAGUUA (Cat# D-012511-03) and CAACCGAUAUCAUUUCUGA (Cat# D-012511-04) ; For MDA-5, the sequences were GAGAAUAACUCAUCAGAAU (Cat# D-013041-01); GAAAUCAUCUGCAAAUGUG (Cat# D-013041-02); GGAAAUGAAUCAGGUGUAA (Cat# D-013041-03) and GAAUAACCCAUCACUAAUA (Cat# D-013041-04).
Inducible gene deletion in primary MEFs
Primary MEFs were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS) and penicillin-streptomycin at 37°C, 5% CO2. Once the cells were prepared, they were first expanded. For the IFN production experiments, 3.5 × 105 cells were seeded into each 6 cm dish on the first day. Control and iKO MEFs were split into two parallel groups: one for TM induction and another as a non-TM treatment control. 10 nM of 4-hydroxytamoxifen (4-OHT) was added to the cells on the second day. Gene deletion and ADAR1 protein depletion were monitored each day after TM addition. As ADAR1 protein completely disappeared at 48 hours, the cells were stimulated with virus or poly I:C at 48 hours after TM induction. For endogenous RNA stimulation, total RNA samples were harvested at this time point.
Cell culture and transfection
HEK293 cells were maintained in DMEM with 10% FBS and penicillin-streptomycin at 37°C, 5% CO2. When cells grew to −60% confluence, transfection was performed in 6-cm tissue culture dishes or 24-well plates (BD) using Lipofectamine 2000 according to the manufacturer’s protocol (Invitrogen, CA). Neon™ Transfection System (Invitrogen, Carlsbad, CA) was used for primary MEF transfection.
Viral infections
Sendai virus stock (SeV, Cantell Strain) purchased from Charles River Laboratories (Wilmington, MA). The titer was predetermined by the vendor and it was 2,000 HA units/ml upon arrival to the lab. The virus was aliquoted and kept at −80°C until use. For infections, cells were washed once with phosphate buffered saline, twice with viral infection medium, DMEM supplemented with 2% FBS, and then maintained in the above medium. SeV was added at a concentration of 80 hemagglutinating units/ml to the cell culture and incubated for the indicated time until the cells were harvested.
Luciferase reporter assays
The RL24 luciferase reporter cell line (36) was derived from HEK293 cells with stable expression of both IRF3-reporter gene and TLR3. This cell line is used for all the assays to monitor IRF3 activation leading to type I IFN production (Fig. 3A, 3B, 4A and 5C). For the reporter assay, the cells were seeded in 24-well plates at a density of 1 × 105 cells per milliliter. After 48 hours, the cells were lysed in passive lysis buffer (Promega) and reporter gene activity was measured by using a luminometer. The data were expressed as folds relative to the control levels. Each experiment was performed at least three times, and all statistical data are represented as means ± standard deviations (SD).
FIGURE 3. Suppression of IFN production by ADAR1 depends on its RNA binding rather than its RNA editing activity.

Total cellular RNA from wild type MEFs (RNA edited by ADAR1) and RNA from ADAR1 knockout MEFs (without ADAR1 editing) were tested for their capacity to induce IFN production in the RL24 reporter cell line. (A) Neither wild type RNA nor ADAR1 KO RNA stimulated IFN production when they were added into the culture medium. (B) RNA from both wild type and knockout MEFs stimulated IFN production when they were transfected into the cytoplasm. However, the wild type and KO RNAs did not show a significant difference. Poly I:C was used as a positive control, which stimulated IFN production in both the medium and cytosol. (C) Wild type and RNA editing incompetent ADAR1 (ADAR1 with E/A mutation in its catalytic domain) were over expressed in HEK293 cells to test the RNA editing activity for IFN suppression. Both of them inhibited IFN-β expression to the same extent. (D) Scheme of serial vectors that were constructed to express ADAR1 proteins with mutations or deletions of Z-DNA binding domain, RNA binding domain, and catalytic domain. These vectors were used to map the functional domain for the inhibitory effect on IFN production. (E) Protein expression of the truncated ADAR1 was confirmed before the functional assays. Western blot was conducted with anti-flag antibody as a flag tag added to wild type, ΔC, and ΔR ADAR1 proteins. However, the N terminal truncated form p110 was detected by anti-ADAR1 monoclonal antibody, as flag tag was not expressed. (F) Expressions of all of these truncated proteins, except RNA binding domain deletion (ΔR), significantly inhibited IFN-β expression stimulated by SeV in HEK293 cells. Data shown in this figure is mean ± S.D. (n=3. Experiments repeated three times). *** p<0.001.
FIGURE 4. ADAR1 interacts with RLR pathways and limits RIG-I binding to dsRNA.

(A) RL-24 cells were used to test the signaling pathway regulated by ADAR1. Cells were transfected with control siRNA or ADAR1 siRNA for 48 hours. Then, poly I: C (1μg/ml) was either added to the culture medium or transfected into the cytosol for a further six hours and cells were harvested for luciferase reporter assay. *** p<0.001. (B) IFN-β expression was measured in HEK293 cells when ADAR1, RIG-I, or both were knocked down. The knock down of ADAR1 and RIG-I were verified by Western blotting (right panel). (C) ADAR1 monoclonal antibody was used to pull down protein from HEK293 cells with or without viral infection. RIG-I was co-precipitated with ADAR1, where RNA is preserved. The co-precipitation of RIG-I was significantly decreased if the protein sample was subject to RNase treatment before immunoprecipitation. (D) Adding dsRNA to the protein extract increased the quantity of RIG-I co-precipitated with ADAR1. (E) In an in vitro RNA-protein binding assay, RIG-I could be pulled down by poly I:C conjugated with agarose beads, but not by poly C. Protein extracts made from iKO MEFs with or without TM induction, ADAR1 protein absent or not, showed obvious differences in the quantities of RIG-I pulled down by poly I:C beads. (F) The effect of different quantities of ADAR1 on RIG-I RNA binding capacity. Increasing amounts of ADAR1 input gradually decreased RIG-I quantity pulled down by poly I:C. The input proteins were extracts from SeV infected HEK293 cells with a high level expression of RIG-I and ADAR1 plasmid transfected cells with over expressed ADAR1 protein.
FIGURE 5. ADAR1 deletion or down-regulation led to IFN expression without exogenous RNA stimulation.
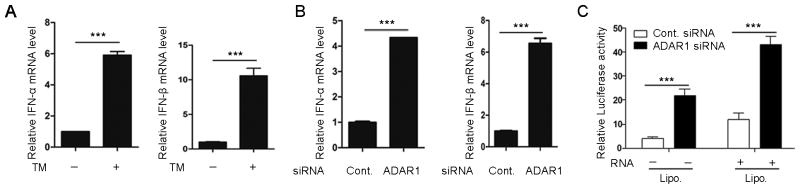
(A) IFN-α and IFN-β mRNA levels in iKO-MEFs were quantified by real time PCR without exogenous RNA stimulation. (B) Knockdown of ADAR1 with siRNA in HEK293 cells increased IFN-α and IFN-β expression without exogenous RNA stimulation. (C) Luciferase activity in RL24 cells was measured after knocking down ADAR1. RL24 cells were transfected with ADAR1 siRNA for 48 hours. The total cellular RNA from the same cells was added to the culture medium or transfected to the cells with Lipofectamine 2000 for six hours. Then, cells were harvested to measure luciferase activity. Data shown in this figure is mean ± S.D. (n=3). *** p<0.001.
RNA isolation and Real-Time PCR
Total RNA was isolated cultured cell or mouse tissues using TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. RNA (1 μg) was reverse transcribed using SuperScript reverse transcriptase (Bio-Rad) to generate first-strand cDNA. Real-time PCR experiments were performed using a Stratagene Mx3005P real-time thermocycler (Stratagene, La Jolla, CA). iTaq™ Universal SYBR® Green Supermix (Bio-Rad) was used for all reactions with the specific primers. Primers used are listed here: Human IFN-α Sense, TTGGCTGTGAAGAAATACTTCCG, antisense, ACCAGATGTTATTCCTTCCTCCTT; human IFN-β sense, GCACTGGCTGGAATGAGACT, antisense, TGCTCATGAGTTTTCCCCTGG; human IFN-γ sense, GGCTTTTCAGCTCTGCATCG, antisense, AGTTCCATTATCCGCTACATCTGA; mouse IFN-α sense, CTACTGGCCAACCTGCTCTC, antisense, AGACAGCCTTGCAGGTCATT; mouse IFN-β sense, TGACGGAGAAGATGCAGAAG, antisense, ACCCAGTGCTGGAGAAATTG; mouse IFN- γsense, CACGGCACAGTCATTGAAAG, antisense, CATCCTTTTGCCAGTTCCTC.
Protein extract and Western blot
Protein extract from cells or tissues was prepared in RIPA Lysis buffer (sc24948, Santa Cruz Biotechnology, Inc.) supplied with PMSF, protease inhibitor cocktail, sodium orthovanadate, or RNase inhibitor. Lysates proteins were quantified using a Bio-Rad protein assay kit. Forty micrograms of proteins were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto nitrocellulose membranes and blocked with 5% nonfat milk in TBS-T (20 mM Tris, 500 mM NaCl, and 0.1% Tween 20) at room temperature for one hour with rocking. The membranes were probed with primary antibodies overnight at 4°C. After washing with TBS-T, the membranes were incubated with horseradish-peroxidase-conjugated secondary antibodies (Thermo scientific) in 5% nonfat milk/TBS-T at room temperature for one hour. The protein–antibody complex was detected by Western blotting luminol reagent (ECL substrate,# 32106, Thermo scientific, Rockford, IL). Primary antibodies used were mouse anti-ADAR1(Cat# sc-73408, Santa Cruz ), rabbit anti-RIG-I (Cat# 3743, Cell Signaling), rabbit anti-MDA5 ( Cat# PA5-20337, Thermo Scientific ), rabbit anti-IRF3 (sc9082, Santa Cruz), rabbit anti-phospho-IRF3 (cat # 4947, Cell Signaling), rabbit anti-IkB-α (cat# 4812, Cell Signaling), rabbit anti-phospho IkB-α (Cat#2859, Cell Signaling), mouse anti-flag ( Cat# F3165, Sigma), mouse anti-β-actin(Cat # sc-47778, Santa Cruz).
Coimmunoprecipitation and protein detection
Protein extracts from HEK293 cells or primary MEF cells were incubated with antibody and 20μl of a 1:1 slurry of protein A/G PLUS-Agarose (Santa Cruz) at 4°C for four hours. Beads were centrifuged at 1,000g and then washed three times with 0.5 ml of lysate buffer. The precipitates were separated by SDS-PAGE and transferred to a PVDF membrane for the immunoblot assay.
In vitro RNA-protein binding assay (including dosage dependent assays)
The agarose beads (Sigma) conjugated to poly(C) were washed several times in 50 mM Tris (pH 7.0)–200 mM NaCl and then resuspended in 50 mM Tris (pH 7.0)–50 mM NaCl. To make poly(I-C)-coated agarose beads, poly(C)-coated beads were resuspended in two volumes of 2 mg of poly(I) (Sigma)/ml prepared in 50 mM Tris (pH 7.0)–150 mM NaCl. The mixture was then rocked gently overnight at 4°C, collected by centrifugation at 1,000g, washed with 50 mM Tris (pH 7.0)–150 mM NaCl, and resuspended in the same buffer as a 50% final slurry. For poly(C) and poly(I-C) pull-down assays, poly(C)- or poly(I-C)-coated beads were equilibrated in binding buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1% NP-40) as a 10% slurry and then combined with an equal volume of whole-cell extract that was prediluted to contain 300 ug of protein. The cell extracts from primary MEF cells or HEK293 cells were supplemented with protease and phosphatase inhibitors and 25 U of RNase inhibitor/ml. The mixture was incubated with gentle agitation for one hour at 4°C. Beads were centrifuged at 1,000g, rinsed three times with binding buffer, and then resuspended in three volumes of 1x SDS-PAGE sample buffer. Samples were incubated at 95°C for five minutes, centrifuged at 13,000g for 30 seconds, and loaded immediately onto SDS-PAGE gels and processed for immunoblot analysis.
In vivo ADAR1 gene deletion by tamoxifen induction
For in vivo ADAR1 gene deletion, 120 mg/kg tamoxifen (cat# T5648, Sigma, St. Louis, MO, USA) was given orally to three-day-old mice on day 1. Mice were sacrificed on day 5 to collect brain and spinal cord tissues for analysis of type I IFN transcription and protein levels.
ELISA
IFNα levels of brain or spinal cord tissues from ADAR1 WT and ADAR1 KO mice were measured by using a mouse IFNα ELISA kit ( Cat#:42120-1, R&D Systems), following the manufacturer’s protocol.
Statistical analysis
Statistical analysis was carried out using GraphPad Prism V software. Data are presented as mean ± S.D. Statistical significance was calculated with a Student’s t-test. P<0.05 was considered to be significant. The means ± S.D. are shown in the figures where applicable.
Results
ADAR1 suppresses type I IFN expression responding to viral RNA in inducible knockout cells
We developed a unique mouse primary cell model in which we could easily turn ADAR1 gene expression on or off. This is a primary mouse embryonic fibroblast (MEF) derived from an inducible knockout (iKO) mouse, referred to as iKO MEF, in which ADAR1 expression is intact until an inducer, tamoxifen (TM), is added to the culture medium. TM activates the Cre recombinase, leading to deletion of the floxed ADAR1 gene (Fig. 1A, 1B).
FIGURE 1. ADAR1 suppresses viral RNA-stimulated type I IFN expression in primary mouse cells.

Mouse primary fibroblasts were prepared from embryos carrying floxed ADAR1 gene and inducible Cre-ER gene (iKO MEF) or littermates without Cre gene (cMEF). (A) The floxed ADAR1 gene allele is illustrated before and after tamoxifen (TM) induction in iKO MEF and cMEF. (B) ADAR1 protein was completely depleted in iKO MEFs between 24–48 hours after adding 10nm 4-OH TM to culture medium, but did not affect ADAR1 protein in cMEFs. (C) iKO MEFs were split into two groups in parallel for TM treatment and non-TM treatment. IFN production was compared in the exact same cells, except TM induction. cMEFs were included to exclude the potential effect of TM. (D, E) Sendai virus (SeV) was used to stimulate the cells for IFN production. SeV infection induced significant IFN-α and IFN-β expression in these primary cells, as shown by the mRNA levels measured by real-time PCR. TM had no effect on cMEFs, but it caused dramatic increases in IFN-α and IFN-β expression in iKO-MEFs. (F, G) Transfection of poly I:C into iKO MEFs caused the same responses with or without TM induction, as observed in SeV infection. IFN mRNA was measured after six hours of SeV infection or poly I:C transfection. Expression level of each sample was normalized by its beta-actin expression and the fold change was relative to the non-stimulated INF expression level. Data shown in this figure is mean ± S.D. (n=3. Experiments repeated five times with MEFs from different embryos). * indicates p<0.05; ** p<0.01; *** p<0.001.
Elevated levels of ISGs observed in ADAR1 knockout cells indicated a suppressive role of ADAR1 in IFN production; therefore, interaction between ADAR1 and a signaling pathway regulating IFN production might exist. Since the pathway for viral RNA sensing has been well-studied, the potential effect of ADAR1 on this pathway was examined. We used the Sendai virus (SeV) to stimulate iKO MEFs and measure the IFN expression levels. Before viral infection, TM was added to the medium to induce ADAR1 gene deletion. To exclude the non-specific effect of TM, control MEFs (cMEFs) derived from embryos carrying only floxed ADAR1 without the Cre gene were included (Fig. 1C). As expected, viral infection led to type I IFN gene expression. IFN levels were dramatically elevated in the TM-induced iKO MEFs compared to the non-TM induced iKO cells and TM-treated cMEFs (Fig. 1D, 1E). To confirm this effect of ADAR1 on the cellular RLR pathway, we also transfected poly I:C into the cells to stimulate IFN production. The results using poly I:C are consistent with the observation using SeV (Fig. 1F,1G). IFN-γ levels were minimally detected across all conditions in MEFs (data not shown). Cells made from embryos of independent litters yielded highly reproducible results. With very strict controls, we demonstrated in these experiments that ADAR1 interacts with the viral RNA sensing pathway to suppress type I IFN production.
ADAR1 suppresses type I IFN production in human cells
Next, we tested whether the suppressive effect of ADAR1 on IFN production also occurs in human cells. We choose to use HEK293 cells to assess the cytosol signaling pathway, as this cell line is known to lack toll-like receptors (TLRs) on the cell membrane that carry the extracellular ligand signals. To modify ADAR1 levels in HEK293 cells, ADAR1 siRNA and ADAR1-expressing plasmids were used to transfect the cells. The protein levels were confirmed before the IFN production assays (Fig. 2A). Consistent with the observation in iKO MEFs, knocking down ADAR1 led to an increase of type I IFN expression in HEK293 cells in response to viral infection or poly I:C exposure (Fig. 2B, 2C). Furthermore, we found that overexpression of ADAR1 inhibits type I IFN expression in response to SeV infection or poly I:C stimulation (Fig. 2D, 2E). Results from this human cell line further confirmed the suppressive effect of ADAR1 on type I IFN production and indicated that ADAR1 may interact with a molecule in the cytosol viral RNA signaling cascade.
FIGURE 2. ADAR1 suppresses viral RNA-stimulated type I IFN expression in human cells.

The suppressive effect of ADAR1 on type I IFN production was examined in HEK293 cells. (A) ADAR1 protein levels were monitored 48 hours after transfection of the cells with ADAR1 siRNA or ADAR1-expressing plasmid. (B, C) IFN-α and IFN-β mRNA expression levels were measured in the cells with ADAR1 siRNA knock down by real-time PCR as described in Fig. 1. Total RNA was isolated six hours after SeV infection or introducing poly I:C into the cytoplasm by transfection. Both SeV infection and poly I:C transfection stimulated IFN expression to a significantly higher level in ADAR1 siRNA transfected cells compare to those transfected with control siRNA. (D, E) In contrast to ADAR1 knock down, INF mRNA expression levels were decreased in the cells in which ADAR1 was over expressed. The conditions for viral and poly I:C stimulation and mRNA measurement were the same as those used for ADAR1 knock down experiments. Data shown in this figure is mean ± S.D. (n=3. Experiments repeated three times). * indicates p<0.05; ** p<0.01; *** p<0.001.
RNA binding activity is required for ADAR1’s inhibitory effect
Chemically synthesized dsRNA containing multiple inosine:uridine pairs (I:U pairs) is known to inhibit IFN production stimulated by poly I:C (37). ADAR1 catalyzes the potential formation of I:U pairs through the conversion of adenosine to inosine in cellular RNAs. Therefore, the increased IFN levels in ADAR1 knockout cells could be due to the decreased number of edited RNAs. To test this hypothesis, we compared the capacities of cellular RNAs edited in wild type and unedited in knockout MEFs to stimulate IFN production. Total RNAs isolated from wild type and knockout MEFs were applied to the RL24 luciferase reporter cell line (36). This cell line was derived from HEK293 cells with stable expression of both IRF3-reporter gene and TLR3. Luciferase activity was assessed to monitor the induction of type I IFNs. RNA from either wild type or knockout cells had little effect if the RNA was added to the medium (Fig. 3A). However, when RNA was introduced into the cytosol by transfection, both types of RNA stimulated IFN production, but no significant difference was observed between the effects of the RNAs from wild type and knockout cells (Fig. 3B). Next, we evaluated the role of RNA editing activity on IFN production by comparing wild type ADAR1 to editing-incompetent protein using mutant ADAR1 where a glutamic acid (E910) is replaced with alanine (A) at the catalytic domain, completely abolishing its RNA-editing capacity (35, 38). This mutation was found to be associated with a severe human neuronal autoimmune disease (see the following sections). Expression of the mutant and wild-type protein in HEK293 cells showed that editing-incompetent ADAR1 (ADAR1 E/A) inhibited IFN production to the same extent as did the wild type ADAR1 in the setting of viral infection (Fig. 3C), indicating that RNA editing activity is not required for its inhibitory effects on IFN production. In mammalian cells, especially human neurons, many potential RNA editing sites were identified on RNA transcripts. However, A-to-I RNA editing is still a rare event among the vast numbers of cellular RNA molecules (39, 40). Thus, it is unlikely that RNA editing catalyzed by ADAR1 yields a quantity of inosine containing RNA adequate to suppress IFN expression. We therefore determined which functional domain of ADAR1 is responsible for its role in IFN regulation through testing a series of truncated ADAR1 proteins (Fig. 3D, 3E). Results showed that all the truncated proteins tested retained their inhibitory effects, except for one, ADAR1 ΔR, in which the RNA binding domain was deleted (Fig. 3F). Therefore, these results support that ADAR1 inhibits IFN production through its RNA binding capacity, rather than its RNA editing activity.
ADAR1 interacts with the cytosol RIG-I pathway
The above results indicate that ADAR1 binds with dsRNA to regulate the IFN inducing pathway. Besides cytosol RLRs, viral dsRNA also can be detected by TLRs as a PAMP, especially the TLR3 located on the endosome membrane (41, 42). Using the RL24 IRF3-reporter cell line, we tested whether signal transduction by either membrane bound TLR3, cytosolic RLRs, or both are affected by ADAR1. In addition to the cytosolic RLR pathways, RL24 cells also stably express TLR3 on its cell membrane (36), therefore, these cells could respond to poly I:C added to the medium through TLR3 and poly I:C transfected into the cytoplasm through RLRs. We knocked down ADAR1 expression before applying poly I:C to test the pathways. IFN activity was significantly increased in the ADAR1 siRNA transfected cells stimulated by cytosol poly I:C compared to the control siRNA transfected cells, whereas ADAR1 knockdown did not show a significant effect on stimulation from medium poly I:C (Fig. 4A). This data showed that ADAR1 predominantly affects the RLR pathway rather than TLR3-mediated IFN production.
Next, we tested whether ADAR1 regulation of IFN expression is RLR-dependent through knocking down the cellular RNA receptors. As shown in Fig. 4B and supplementary Fig. 1A, RIG-I siRNA attenuated the effect of ADAR1 on IFN production in non-stimulated and in virus infected HEK293 cells. Knocking down MDA-5 showed the same effect as RIG-I suppression (Supplemental Fig 1B and 1C). We also tested the signal relays downstream of RIG-I/MAVS activation in iKO MEFs in the absence of ADAR1. Phosphorylation of IRF3 and IkB-α were increased after poly I:C transfection or viral infections in TM-induced cells compared to cMEFs as shown in supplemental Fig 1D. This data further supports the presence of an interaction between ADAR1 and the RLR pathways. Next, we tested whether ADAR1 and RIG-I physically contact each other using co-immunoprecipitation assays. Interestingly, our results showed that ADAR1 forms complexes with RIG-I. RIG-I was detected in the precipitation pulled down by anti-ADAR1 antibody (Fig. 4C). However, the interaction was shown to be RNA-dependent, as RNase treatment dissociated ADAR1 and RIG-I (Fig. 4C), while adding dsRNA into the reaction increased the quantity of co-immunoprecipitated RIG-I (Fig. 4D). Because both ADAR1 and RIG-I are dsRNA-binding proteins, it is not unexpected that the interaction of ADAR1 and RIG-I is bridged by a dsRNA molecule. However, it raises the question as to whether ADAR1 prevents RNA sensing by interfering with RIG-I dsRNA recognition. This possibility was tested by measuring the quantity of RIG-I that bound to poly I:C in the presence or absence of ADAR1 in an in vitro RNA-protein binding assay (Fig. 4E). Protein extract was made from iKO MEFs either induced or not by TM, and then incubated with poly I:C conjugated to agarose-beads. The proteins bound to poly I:C were pulled down by the beads and analyzed by Western blot. The quantity of RIG-I bound to poly I:C was much greater in the lysates in which ADAR1 is absent. This was true in both viral-infected and non-infected cells, although the viral infection dramatically increased the quantity of RIG-I bound to poly I:C (Fig. 4E). In addition, we tested whether the level of ADAR1 correlates with its inhibitory effect. In the RNA-protein binding assay, we used protein extracts from HEK293 cells with over-expressed ADAR1 and gradually increased ADAR1 levels by controlling the protein input to the reactions (Fig. 4F). We observed that the inhibition of RIG-I-poly I:C binding was ADAR1 concentration-dependent and that RIG-I binding negatively correlated with the level of ADAR1 protein (Fig. 4F). The same result was observed with protein extracts from cells transfected with increasing amount plasmids (supplementary Fig. 2), while overexpression of ADAR1ΔR that lacks RNA binding domain did not affect RIG-I binding at any expression level (supplementary Fig. 3), consistent with its effect on IFN production (Fig. 3F).
ADAR1 absence causes IFN production without exogenous RNA stimulation
In the context of autoimmune disease, an excessive immune response is caused by endogenous substances. Such as in AGS, aberrant metabolism of nucleic acid is suspected to be involved in triggering IFN production (43). We examined if deletion of ADAR1 in iKO MEFs leads to IFN production without exogenous RNA stimulation. ADAR1 deletion induced by TM caused significant increases in IFN-α and –β expression without stimulation (Fig. 5A). Knocking down ADAR1 in HEK293 cells showed the same effect (Fig. 5B). These results indicate that when ADAR1 is suppressed, an endogenous component is capable of activating the IFN expression signaling pathway. Taken that ADAR1 inhibits cellular RNA binding to RIG-I, the above results hinted that removal of ADAR1 enables endogenous RNA to stimulate RIG-I and activates the signaling pathway. To examine if ADAR1 plays a role in preventing the immune recognition of endogenous RNA, we tested whether down-regulation of ADAR1 shows an effect on IFN signaling activity in response to cellular RNA. We isolated total cellular RNA from the RL24 cell line and used this cellular RNA to stimulate RL24 reporter cells. While knocking down ADAR1 in the reporter cells resulted in activation of the IFN pathway without transfection of the cellular RNA, it caused a much higher level of reporter activity when total RNA was transfected into the cells compared to the responses in control cells and non-RNA stimulated cells (Fig. 5C). These results indicated that ADAR1 prevents cellular RNA to stimulate IFN inducing pathways in the cells.
Inducing ADAR1 gene deletion causes excessive IFN production in mouse neural tissues
Multiple mutations in the ADAR1 gene loci were recently linked with a subcategory of AGS (5). The high level of IFN-α in the cerebrospinal fluid of AGS patients (up to 1,000 times higher in these patients) plays a critical role in the progressive encephalopathy (4). However, experimental evidence associating ADAR1 mutations with this immune disease is lacking. Support for this possibility would require data showing that loss of ADAR1 is sufficient for excessive IFN production. We tested whether the high level of IFN observed in AGS patients could be recapitulated in the ADAR1 inducible knockout mouse. Since the onset of AGS is seen at a very young age and even in newborns, we started to administer TM to iKO mice in the first week after birth (Fig. 6A). ADAR1 genotype (supplementary Fig. 4) and protein expression in the brain and spinal cord was confirmed by Western blot after five days of TM induction (Fig. 6B). Because cerebrospinal fluid is very limited in the newborn mouse, we chose to measure the IFN-α level in neural tissues by ELISA. Both the brains and spinal cords of ADAR1 knockout mice contained a much higher level of IFN-α than those of the control littermates (Fig. 6C). To verify that the high level of IFN-α was produced in the neural tissues, we monitored the gene expression of type I IFNs by measuring the mRNA levels in the tissues. The results showed that neural tissues expressed significantly higher type I IFN mRNA levels after ADAR1 was deleted (Fig. 6D, 6 E). These data provide direct evidence that loss of ADAR1 function in vivo leads to AGS-like pathogenic changes in terms of IFN gene expression.
FIGURE 6. Inducing ADAR1 deletion in mice increased type I interferon expression in the brain and spinal cord.
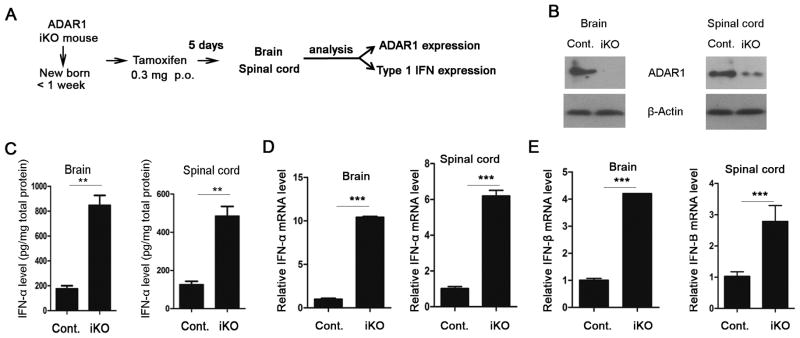
The inducible ADAR1 knockout mouse was used to test the in vivo effect of ADAR1 on type I IFN production in neural tissues. (A) The strategy for inducing ADAR1 gene deletion and neural tissue analysis. (B) The protein levels of ADAR1 in the brain and spinal cord after TM induction were monitored by Western blot. (C) Level of IFN-α protein in the neural tissues was measured using an ELISA kit and is shown in the pictogram per mg total protein. (D) IFN-α and (E) IFN-β mRNA levels were measured by real time PCR as described in Fig. 1. Experiments were done with three wild type and three knockout mouse newborns. Data shown is the mean ± S.D. ** p<0.01; *** p<0.001.
Discussion
In this study, we found that ADAR1 interacts with the cytosol RNA-sensing pathway to suppress IFN production responding to exogenous and endogenous RNAs. As demonstrated with RIG-I, ADAR1, through its RNA binding activity, limits the access of dsRNA to the RLRs and suppresses the signaling pathway used for viral RNA detection. In the absence of ADAR1, endogenous RNA stimulates type I IFN production without exogenous stimulation. Disruption of the ADAR1 gene in mice recapitulates the pathogenic change of AGS in terms of high-level IFN production in neural tissues.
We developed an inducible gene knockout model for identification of the direct effect of ADAR1 on IFN production. Adding 10 nM TM to the culture medium effectively eliminates ADAR1 protein in the cells, which enabled us to compare IFN production and its signaling pathway in the same cell. The inducible gene deletion did not work well in the immortalized cell lines; therefore, we used the primary cells for our studies, as it would more closely reassemble the in vivo mechanism than a cell line. Critical data led us to find the interaction of ADAR1 and RLR pathway that was originally obtained from this cell model, such as that ADAR1 deletion increases type I IFN expression (Fig. 1) and ADAR1 competes with RIG-I to bind dsRNAs (Fig. 4).
Signaling pathways for detecting viral RNA by RLRs that trigger type I interferon production have been very well-studied (29–31, 44, 45). Each component of this pathway is well-defined with protein crystal structure information. Besides RLRs, the invaded RNA also encounters other RNA-binding proteins (46, 47). Less knowledge has been obtained on whether these proteins interact with RLRs in RNA sensing. We demonstrated here that ADAR1 acts as an RNA-binding protein to suppress IFN production via interfering with RNA sensing by RLRs. Both RIG-I and MDA-5 detect viral RNA and analogs. It is well-known that RIG-I mediates Sendai virus-stimulated IFN production and has a high-affinity to 5′ppp-RNA and a lower-affinity to longer dsRNAs (>200bp), including poly I:C (48). MDA-5 prefers to bind poly I:C and picornavirus RNAs (44). RIG-I and MDA-5 share the same pathway to induce IFN production via MAVS (44). Through poly I:C beads pull-down we demonstrated that ADAR1 limits RIG-I binding to dsRNA therefore inhibits cytosolic RNA sensing. While poly I:C can be detected by both RIG-I and MDA-5, 5′-triphosphate RNA is the specific ligand of RIG-I. The poly I:C pull-down experiment did not prove that ADAR1 interferes with binding of RIG-I with its specific ligand 5′-triphosphate RNA. On the other hand, MDA-5 preferentially binds to long dsRNA and induces type I IFN production. It is conceivable that ADAR1 also affect MDA-5 RNA sensing pathway. Indeed, when we used siRNA to knock down MDA-5 in HEK293 cells an attenuation of the effect of ADAR1 suppression was observed (Supplementary Fig 1B and 1C). Thus, ADAR1 plays a general role in suppressing RNA sensing by RLRs for IFN production. ADAR1, stimulated by IFN, formulates a negative feedback loop through RLRs. It may act to prevent the overreaction during viral infection. This notion is consistent with the observation that overexpression of ADAR1 increases viral replications (27).
Although ADAR1 is an RNA-editing enzyme, emerging evidence shows it also exerts RNA editing independent functions (14–17, 49). A recent study found that ADAR1 forms a heterodimer with dicer, increases the maximum rate of pre-microRNA cleavage, and facilitates miRNA loading onto RNA-induced silencing complexes (RISC) (17). In contrast to heterodimer formation, our data showed that ADAR1 complexes with RIG-I is bridged by dsRNA. However, we could not completely rule out the possibility that direct binding of ADAR1 and RIG-I may also occur, since a weak signal of RIG-I remained in the pulled down proteins by ADAR1 antibody after RNase treatment (Fig. 4C). The major form of ADAR1 and RIG-I complex was still dependent on the presence of dsRNA (Fig. 4D).
ADAR1 gene mutation is often found in patients with a mild skin disorder, dyschromatosis symmetrica hereditaria (DSH) (50). However, AGS, a severe hereditary neurodegenerative disorder characterized by early onset and progressive encephalopathy, has recently been found to be associated with ADAR1 gene mutations (5). Single nucleotide mutation was predominantly found in the DSH family, but no neural phenotype likes AGS was associated with the mutations. In contrast, multiple mutations concur in all AGS patients and both alleles were affected. The pedigree information and the mutation frequency in normal population did not support that these mutations are gain-function mutations although particular exception could not be excluded.
Mutations on the other five genes (TREX1, RNASEH2B, RNASEH2C, RNASEH2A, and SAMHD1) were identified, showing that each of them is responsible for a subcategory of AGS. Interactions of these molecules with the IFN pathway were studied and documented (43). ADAR1 gene mutation, however, was just found very recently to be associated with AGS. Whether and how mutations on the ADAR1 gene cause excessive IFN production was not elucidated. Our present study using an iKO animal model suggested a mechanism by which ADAR1 regulates IFN production. It may help us to understand the pathogenesis of AGS caused by ADAR1 mutations. Given the advantage of our inducible ADAR1 gene knockout model, we provided direct evidence that disruption of the ADAR1 gene can result in excessive production of type I IFN in neural tissues. Discovering how ADAR1 regulates IFN production will likely accelerate the finding of more autoimmune diseases with type I interferon signatures as just found in bilateral striatal necrosis (51).
Although our data demonstrated that ADAR1 limits cellular RNA sensing by RLRs through RNA binding activity, competitive binding of the endogenous RNA by ADAR1 might not be the only mechanism that regulates type I IFN induction. ADAR1 was also found to be involved in cellular RNA distribution (52), degradation (49) and to interact with other proteins (16). Directly or indirectly changing the cellular RNA modifications and enhancing the degradation by ADAR1 could also prevent RLR activation and type I IFN induction by directly or indirectly enhancing the degradation or immune recognition of endogenous RNAs. Further studies of these possibilities will need to be conducted.
In conclusion, our study revealed a mechanism by which ADAR1 suppresses IFN production and also indicated potential involvement of ADAR1 in autoimmune diseases with an IFN signature, such as AGS.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants R21CA158650, GM050441, the University of Pittsburgh School of Medicine Dean’s Bridge fund, and a startup fund from the University of Pittsburgh, Department of Surgery.
We thank Christine Heiner (Scientific Writer, University of Pittsburgh Department of Surgery) for editing the manuscript.
Abbreviations used in this article
- ADAR1
adenosine deaminase acting on RNA 1
- IFN
interferon
- ISG
IFN Stimulated Gene
- iKO
inducible knockout
- ISRE
IFN-Stimulated Response Element
- MEFs
mouse embryonic fibroblasts
- PAMP
pathogen-associated molecular pattern
- RIG-I
retinoic acid-inducible gene 1
- RLRs
RIG-I like receptors
Footnotes
Disclosures
The authors declare no financial conflicts of interest.
References
- 1.Hall JC, Rosen A. Type I interferons: crucial participants in disease amplification in autoimmunity. Nat Rev Rheumatol. 2010;6:40–49. doi: 10.1038/nrrheum.2009.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baccala R, Kono DH, Theofilopoulos AN. Interferons as pathogenic effectors in autoimmunity. Immunol Rev. 2005;204:9–26. doi: 10.1111/j.0105-2896.2005.00252.x. [DOI] [PubMed] [Google Scholar]
- 3.Aicardi J, Goutieres F. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol. 1984;15:49–54. doi: 10.1002/ana.410150109. [DOI] [PubMed] [Google Scholar]
- 4.Chahwan C, Chahwan R. Aicardi-Goutieres syndrome: from patients to genes and beyond. Clin Genet. 2012;81:413–420. doi: 10.1111/j.1399-0004.2011.01825.x. [DOI] [PubMed] [Google Scholar]
- 5.Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet. 2012;44:1243–1248. doi: 10.1038/ng.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.George CX, Samuel CE. Human RNA-specific adenosine deaminase ADAR1 transcripts possess alternative exon 1 structures that initiate from different promoters, one constitutively active and the other interferon inducible. Proc Natl Acad Sci U S A. 1999;96:4621–4626. doi: 10.1073/pnas.96.8.4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.George CX, Samuel CE. Characterization of the 5′-flanking region of the human RNA-specific adenosine deaminase ADAR1 gene and identification of an interferon-inducible ADAR1 promoter. Gene. 1999;229:203–213. doi: 10.1016/s0378-1119(99)00017-7. [DOI] [PubMed] [Google Scholar]
- 8.Herbert A, Schade M, Lowenhaupt K, Alfken J, Schwartz T, Shlyakhtenko LS, Lyubchenko YL, Rich A. The Zalpha domain from human ADAR1 binds to the Z-DNA conformer of many different sequences. Nucleic Acids Res. 1998;26:3486–3493. doi: 10.1093/nar/26.15.3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwartz T, Rould MA, Lowenhaupt K, Herbert A, Rich A. Crystal structure of the Zalpha domain of the human editing enzyme ADAR1 bound to left-handed Z-DNA. Science. 1999;284:1841–1845. doi: 10.1126/science.284.5421.1841. [DOI] [PubMed] [Google Scholar]
- 10.Nishikura K. Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem. 2010;79:321–349. doi: 10.1146/annurev-biochem-060208-105251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosenthal JJ, Seeburg PH. A-to-I RNA editing: effects on proteins key to neural excitability. Neuron. 2012;74:432–439. doi: 10.1016/j.neuron.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, Fligelman ZY, Shoshan A, Pollock SR, Sztybel D. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat Biotechnol. 2004;22:1001–1005. doi: 10.1038/nbt996. [DOI] [PubMed] [Google Scholar]
- 13.Paul MS, Bass BL. Inosine exists in mRNA at tissue-specific levels and is most abundant in brain mRNA. Embo J. 1998;17:1120–1127. doi: 10.1093/emboj/17.4.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang W, Wang Q, Howell KL, Lee JT, Cho DS, Murray JM, Nishikura K. ADAR1 RNA deaminase limits short interfering RNA efficacy in mammalian cells. J Biol Chem. 2005;280:3946–3953. doi: 10.1074/jbc.M407876200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heale BS, Keegan LP, McGurk L, Michlewski G, Brindle J, Stanton CM, Caceres JF, O’Connell MA. Editing independent effects of ADARs on the miRNA/siRNA pathways. Embo J. 2009;28:3145–3156. doi: 10.1038/emboj.2009.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nie Y, Ding L, Kao PN, Braun R, Yang JH. ADAR1 interacts with NF90 through double-stranded RNA and regulates NF90-mediated gene expression independently of RNA editing. Mol Cell Biol. 2005;25:6956–6963. doi: 10.1128/MCB.25.16.6956-6963.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ota H, Sakurai M, Gupta R, Valente L, Wulff BE, Ariyoshi K, Iizasa H, Davuluri RV, Nishikura K. ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell. 2013;153:575–589. doi: 10.1016/j.cell.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hartner JC, Walkley CR, Lu J, Orkin SH. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol. 2009;10:109–115. doi: 10.1038/ni.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qiu W, Wang X, Buchanan M, He K, Sharma R, Zhang L, Wang Q, Yu J. ADAR1 is essential for intestinal homeostasis and stem cell maintenance. Cell Death Dis. 2013;4:e599. doi: 10.1038/cddis.2013.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.George CX, Samuel CE. Characterization of the 5′-flanking region of the human RNA-specific adenosine deaminase ADAR1 gene and identification of an interferon-inducible ADAR1 promoter. Gene. 1999;229:203–213. doi: 10.1016/s0378-1119(99)00017-7. [DOI] [PubMed] [Google Scholar]
- 21.George CX, Wagner MV, Samuel CE. Expression of interferon-inducible RNA adenosine deaminase ADAR1 during pathogen infection and mouse embryo development involves tissue-selective promoter utilization and alternative splicing. J Biol Chem. 2005;280:15020–15028. doi: 10.1074/jbc.M500476200. [DOI] [PubMed] [Google Scholar]
- 22.Casey JL. Control of ADAR1 editing of hepatitis delta virus RNAs. Curr Top Microbiol Immunol. 2012;353:123–143. doi: 10.1007/82_2011_146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biswas N, Wang T, Ding M, Tumne A, Chen Y, Wang Q, Gupta P. ADAR1 is a novel multi targeted anti-HIV-1 cellular protein. Virology. 2012;422:265–277. doi: 10.1016/j.virol.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Samuel CE. Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology. 2011;411:180–193. doi: 10.1016/j.virol.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Samuel CE. Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology. 2011;411:180–193. doi: 10.1016/j.virol.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Z, Okonskiand CE, Samuel KM. Adenosine deaminase acting on RNA 1 (ADAR1) suppresses the induction of interferon by measles virus. J Virol. 2012;86:3787–3794. doi: 10.1128/JVI.06307-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Chassey B, Aublin-Gex A, Ruggieri A, Meyniel-Schicklin L, Pradezynski F, Davoust N, Chantier T, Tafforeau L, Mangeot PE, Ciancia C. The interactomes of influenza virus NS1 and NS2 proteins identify new host factors and provide insights for ADAR1 playing a supportive role in virus replication. PLoS Pathog. 2013;9:e1003440. doi: 10.1371/journal.ppat.1003440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 30.Pichlmair A, Sousa CRE. Innate recognition of viruses. Immunity. 2007;27:370–383. doi: 10.1016/j.immuni.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 31.Sun L, Liu S, Chen ZJ. SnapShot: pathways of antiviral innate immunity. Cell. 2010;140:436–436 e432. doi: 10.1016/j.cell.2010.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M. 5 ′-triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 33.Wang Q, Miyakoda M, Yang W, Khillan J, Stachura DL, Weiss MJ, Nishikura K. Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J Biol Chem. 2004;279:4952–4961. doi: 10.1074/jbc.M310162200. [DOI] [PubMed] [Google Scholar]
- 34.Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244:305–318. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- 35.Lai F, Drakas R, Nishikura K. Mutagenic analysis of double-stranded RNA adenosine deaminase, a candidate enzyme for RNA editing of glutamate-gated ion channel transcripts. J Biol Chem. 1995;270:17098–17105. doi: 10.1074/jbc.270.29.17098. [DOI] [PubMed] [Google Scholar]
- 36.Zhu J, Smith K, Hsieh PN, Mburu YK, Chattopadhyay S, Sen GC, Sarkar SN. High-throughput screening for TLR3-IFN regulatory factor 3 signaling pathway modulators identifies several antipsychotic drugs as TLR inhibitors. J Immunol. 2010;184:5768–5776. doi: 10.4049/jimmunol.0903559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vitali P, Scadden AD. Double-stranded RNAs containing multiple IU pairs are sufficient to suppress interferon induction and apoptosis. Nat Struct Mol Biol. 2010;17:1043–1050. doi: 10.1038/nsmb.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Y, Samuel CE. Mechanism of interferon action: functionally distinct RNA-binding and catalytic domains in the interferon-inducible, double-stranded RNA-specific adenosine deaminase. J Virol. 1996;70:1961–1968. doi: 10.1128/jvi.70.3.1961-1968.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kleinman CL, Adoue V, Majewski J. RNA editing of protein sequences: a rare event in human transcriptomes. Rna. 2012;18:1586–1596. doi: 10.1261/rna.033233.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eisenberg E, Nemzer S, Kinar Y, Sorek R, Rechavi G, Levanon EY. Is abundant A-toI RNA editing primate-specific? Trends Genet. 2005;21:77–81. doi: 10.1016/j.tig.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 41.Gowen BB, Wong MH, Jung KH, Sanders AB, Mitchell WM, Alexopoulou L, Flavell RA, Sidwell RW. TLR3 is essential for the induction of protective immunity against Punta Toro Virus infection by the double-stranded RNA (dsRNA), poly(I:C12U), but not Poly(I:C): differential recognition of synthetic dsRNA molecules. J Immunol. 2007;178:5200–5208. doi: 10.4049/jimmunol.178.8.5200. [DOI] [PubMed] [Google Scholar]
- 42.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 43.Rabe B. Aicardi-Goutieres syndrome: clues from the RNase H2 knock-out mouse. J Mol Med (Berl) 2013;91(11):1235–40. doi: 10.1007/s00109-013-1061-x. [DOI] [PubMed] [Google Scholar]
- 44.Takeuchi O, Akira S. MDA5/RIG-I and virus recognition. Curr Opin Immunol. 2008;20:17–22. doi: 10.1016/j.coi.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 45.Wilkins C, Jr, Gale M. Recognition of viruses by cytoplasmic sensors. Curr Opin Immunol. 2010;22:41–47. doi: 10.1016/j.coi.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Doria M, Neri F, Gallo A, Farace MG, Michienzi A. Editing of HIV-1 RNA by the double-stranded RNA deaminase ADAR1 stimulates viral infection. Nucleic Acids Res. 2009;37:5848–5858. doi: 10.1093/nar/gkp604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Polson AG, Bassand JL, Casey BL. RNA editing of hepatitis delta virus antigenome by dsRNA-adenosine deaminase. Nature. 1996;380:454–456. doi: 10.1038/380454a0. [DOI] [PubMed] [Google Scholar]
- 48.Kolakofsky D, Kowalinski E, Cusack S. A structure-based model of RIG-I activation. RNA. 2012;18:2118–2127. doi: 10.1261/rna.035949.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Agranat L, Raitskin O, Sperling J, Sperling R. The editing enzyme ADAR1 and the mRNA surveillance protein hUpf1 interact in the cell nucleus. Proc Natl Acad Sci U S A. 2008;105:5028–5033. doi: 10.1073/pnas.0710576105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suzuki N, Suzuki T, Inagaki K, Ito S, Kono M, Horikawa T, Fujiwara S, Ishiko A, Matsunaga K, Aoyama Y. Ten novel mutations of the ADAR1 gene in Japanese patients with dyschromatosis symmetrica hereditaria. J Invest Dermatol. 2007;127:309–311. doi: 10.1038/sj.jid.5700528. [DOI] [PubMed] [Google Scholar]
- 51.Livingston JH, Lin JP, Dale RC, Gill D, Brogan P, Munnich A, Kurian MA, Gonzalez-Martinez V, De Goede CG, Falconer A. A type I interferon signature identifies bilateral striatal necrosis due to mutations in ADAR1. J Med Genet. 2014;51(2):76–82. doi: 10.1136/jmedgenet-2013-102038. [DOI] [PubMed] [Google Scholar]
- 52.Chen LL, Carmichael GG. Altered nuclear retention of mRNAs containing inverted repeats in human embryonic stem cells: functional role of a nuclear noncoding RNA. Mol Cell. 2009;35:467–478. doi: 10.1016/j.molcel.2009.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.