

Abstract
Severe bacterial sepsis leads to a pro-inflammatory condition that can manifest as septic shock, multiple organ failure, and death. Neutrophils are critical for the rapid elimination of bacteria, however, the role of neutrophil chemoattractant CXCL1 in bacterial clearance during sepsis remains elusive. To test the hypothesis that CXCL1 is critical to host defense during sepsis. We used CXCL1-deficient mice and bone marrow chimeras to demonstrate the importance of this molecule in sepsis. We demonstrate that CXCL1 plays a pivotal role in mediating host defense to polymicrobial sepsis following cecal ligation and puncture (CLP) in gene-deficient mice. CXCL1 appears to be essential for restricting bacterial outgrowth and death in mice. CXCL1 derived from both hematopoietic and resident cells contributed to bacterial clearance. Moreover, CXCL1 is essential for neutrophil migration, expression of pro-inflammatory mediators, activation of Nuclear-Factor-κ-B (NF-κB) and Mitogen-Activated Protein (MAP) kinases and upregulation of adhesion molecule Intercellular Adhesion Molecule-1 (ICAM-1). Recombinant interleukin 17 (IL-17) rescued impaired host defenses in cxcl1−/− mice. CXCL1 is important for IL-17A production via Th17 differentiation. CXCL1 is essential for Nicotinamide Adenine Dinucleotide Phosphate (NADPH) oxidase-mediated reactive oxygen species production and neutrophil extracellular trap (NET) formation. This study reveals a novel role for CXCL1 in neutrophil recruitment via modulating T cell function and neutrophil-related bactericidal functions. These studies suggest that modulation of CXCL1 levels in tissues and blood could reduce bacterial burden in sepsis.
Introduction
Despite improvements in antibiotic therapies and critical care management, sepsis remains a leading cause of infectious death in intensive care units (1). The pathogenesis of sepsis is characterized by an early, overwhelming systemic pro-inflammatory response that leads to multiple organ damage; the later phases are characterized by anti-inflammatory responses and negative regulation of immune signaling pathways (2). However, the molecular and cellular mechanisms that regulate immune responses to polymicrobial sepsis are not completely understood. Prior studies have shown that deletion of nucleotide-binding oligomerization domain-2 (NOD2) receptors or the scavenger receptor class A leads to host protection (3, 4). In contrast, reduced survival and diminished production of cytokines and chemokineswas observed in Stat-2−/− mice during lipopolysaccharide (LPS)-mediated endotoxic shock, indicating a crucial role for STAT-2 in host defense (5).
Neutrophils are a pivotal arm of the innate immune response during polymicrobial sepsis, and play a critical role in bacterial elimination (6, 7). Impaired neutrophil transmigration is associated with increased mortality and higher bacterial burden in peritoneal exudates and blood, as demonstrated during sepsis induced by cecal ligation and puncture (CLP)(7, 8) whereas excessive influx of neutrophils can cause unwanted tissue damage and organ dysfunction (9). Our group has demonstrated an important role for CXCL1 [also known as keratinocyte cell-derived chemokine (KC)] in pulmonary defense during pneumonia caused by Klebsiella pneumoniae (10). CXCL1 was found to be critical for neutrophil-dependent bacterial elimination via induction of reactive oxygen species (11) and reactive nitrogen species in the lung (12). Neutrophil migration to multiple organs is impaired during severe sepsis, due to down-regulation of the CXCL1 receptor, CXCR2, resulting in failed pathogen clearance (13). These findings suggest a potential role for CXCR2, or its ligands, including CXCL1 (KC), CXCL2 (MIP-2), and CXCL5 (LIX), in controlling sepsis. To better understand the role of CXCL1 in neutrophil influx and function during fatal polymicrobial sepsis (PMS), we employed a murine model of PMS, induced by CLP in CXCL-/- mice. In addition, we addressed the role of bone marrow cell versus resident cell-derived CXCL1 in bacterial clearance following PMS through the use of bone marrow chimeras.
Materials and Methods
Animal studies
All animal experiments were approved by the Louisiana State University Animal Welfare Committee. CXCL1 gene-deficient (cxcl1−/−) and C57Bl/6 wild type (WT) male mice (8-12 weeks) were generated as previously described (10, 14). PMS was induced by the CLP method as previously described (15). In brief, male mice were anesthetized, the cecum was punctured with a 21-gauge needle, a small amount of fecal material was extruded through the puncture, and the cecum was repositioned into the peritoneal cavity. Animals with sham surgery underwent the same protocol without CLP. Survival of both cxcl1−/− and WT mice that underwent CLP or sham surgery was monitored every 12 h up to 10 days. In additional experiments, recombinant rIL-17A, or bovine serum albumen (BSA) control was injected intraperitoneally immediately following CLP, and survival was monitored for 10 days. Bacterial burdens were determined by enumerating bacterial numbers in colony forming units (CFUs) on tryptic soy agar plates as previously described (10). Cytokine and chemokine levels in peritoneal lavage fluid and serum were measured by a double-ligand enzyme-linked immunosorbent assay (ELISA) (10, 12).
Bone marrow chimeras
Generation of CXCL1 chimeras has been described in our prior reports (10, 12). We found that greater than 80% of blood leukocytes were derived from donor mice at the time of experiments.
MPO activity
An MPO assay was performed in the peritoneal fluid as described elsewhere (2,5,11,12). Briefly, 200 μl of peritoneal fluid was incubated with 800 μl hexadecyltrimethylammoniumbromide (HTAB) buffer and the mixture was transferred into microcentrifuge tube and centrifuged at 20,000 × g for 4 min. Seven microliters of supernatant was transferred into 96-well plate, and 200 μl of an O-dianisidine hydrochloride solution was added immediately. The MPO activity, measured as optical density at 450 nm, was expressed in units/ml of supernatant.
Western blotting
At the designated times, the lungs, livers, spleens, and kidneys were harvested and homogenized in 1 mL of phosphate buffered saline (PBS) containing 0.1% Triton X-100 supplemented with complete protease and phosphatase inhibitor cocktail as described (12).
Immunofluorescence and electron microscopy
Immunofluorescent of the cells from cxcl1−/− and WT mice was performed to detect Neutrophil Extracellular Trap (NET) formation as described previously (16). Scanning electron microscopy (16) (FEI Quanta 200, USA) was performed to examine NET formation in peritoneal neutrophils following the protocols described in a previous publication (16). E. coli (ATCC 25922) was used to stimulate neutrophils as described previously (17).
Flow cytometry
The detection of reactive oxygen species (11), OH-, and O2 - producing neutrophils in the peritoneal fluid from CLP WT and KO mice was performed as described earlier (18). For isolation and detection of IL-17 producing T-cells, lungs were first minced, digested with collagenase for 90 min, and made into a single-cell suspension, followed by staining. Spleens were homogenized and were made into single cell suspension by sieving. Cells were surface stained for markers of IL-17A–producing T cells (γδ, NK1.1, CD4, and CD8α). FlowJo software was used to analyze the data.
Th17 differentiation
The method for IL-17 differentiation has been previously reported (19). Recombinant CXCL1 (rCXCL1) (1 mg/mL) was added on days 1 and 3. Cells were washed and resuspended in PBS followed by blocking with Fc receptor blocking reagent at 3, 6 and 12 days post-stimulation. IL-17-producing cells were surface stained with anti-CD4 fluorescein isothiocyanate (FITC). Flowjo was used for data analysis. In another set of experiments, supernatant was collected for IL-17A or IL-17F assay by ELISA at 3 day post-stimulation with rCXCL1.
Analysis of NET-derived DNA
To analyze NET-derived DNA, peritoneal fluid was taken from the post-CLP or sham mice at 24h and subjected to agarose gel electrophoresis. To confirm whether extracellular DNA was present in the cell-free supernatant, peritoneal fluid was treated with DNase (50 mg/ mL) for 1 h and subjected to gel electrophoresis. The observation of DNA in the samples, but not in DNase-treated samples, was judged to be NET-derived DNA.
Determination of Reactive Oxygen Species (11), H2O2 and O2 production by neutrophils
ROS and H2O2 levels in peritoneal neutrophils of cxcl1−/− and WT mice after CLP were measured using the Fluorescent H2O2/Peroxidase Detection Kit.
Bacterial killing assay
A neutrophil-dependent killing assay was performed as reported earlier (12). Briefly, 1 × 106 neutrophils were suspended in RPMI 1640 with 10% v/v fetal bovine serum (FBS), and 1 × 106 opsonized bacteria were added (1 multiplicity of infection (MOI)). Samples were incubated at 37°C with continuous agitation. Samples were harvested at 60 min post-infection, treated with gentamycin (100 mg/mL) for 15 min to kill extracellular bacteria and plated to determine colony forming units (CFU)(12).
Data analysis
Data are expressed as mean +/-standard error (SE). Data were analyzed with the Student's t-test (between two groups) or with the two-way analysis of variance (ANOVA) (>2 groups). Survival curves were compared by the Wilcoxon rank sign test. Differences in values were defined as significant at a p value of less than 0.05. Experiments were repeated 2-3 times in each group in each time point.
Results
CXCL1 contributes to protection during polymicrobial sepsis
The role of CXCL1 in the pathogenesis of polymicrobial sepsis is unclear. To explore this, cxcl1−/− and WT mice were subjected to CLP, and the survival of animals was monitored up to 10 days post-CLP. As shown in Fig. 1A, cxcl1−/− mice displayed a reduction in survival rate compared to WT mice after CLP. To examine whether reduced survival of cxcl1−/− mice was due to an impaired ability to recruit immune cells to the peritoneum, total and differential cell counts as well as myeloperoxidase activity (MPO) activity were determined in the peritoneal fluid. In cxcl1−/− mice, total white blood cells (WBCs), neutrophil number, and MPO activity were attenuated at 24 h post-CLP as compared to WT mice (Fig. 1C). In the control (sham) group, no cellular influx was observed in the peritoneal cavity of either cxcl1−/− or WT mice (Fig. 1C). To determine whether the reduced recruitment of WBCs and neutrophils contributed to bacterial growth and subsequent dissemination to extraperitoneal organs, bacterial numbers in the peritoneal fluid, blood, and lung were enumerated 24 h post-CLP. As compared to WT mice, cxcl1−/− mice had a higher bacterial burden in all tissues (Fig. 1B). Moreover, neutrophil recruitment to the peritoneal cavity was reduced in cxcl1−/− mice following infection (Fig. 1C). As compared with WT mice transplanted with WT marrow cells, WT mice transplanted with KO marrow cells or KO mice transplanted with WT marrow cells showed no attenuation bacterial clearance in peritoneal fluid (PF), suggesting that both hematopoietic (radiosensitive) and non-hematopietic (radioresistant) cells are important for bacterial clearance (Fig. 1D).
Figure 1. CXCL1 deficient mice are more susceptible to polymicrobial sepsis (PMS) than WT mice and are defective for cellular infiltration and bacterial clearance.

A, Enhanced mortality of cxcl1−/− mice after CLP-induced PMS. cxcl1−/− and WT control mice were subjected to sham or PMS. The survival rates of mice were determined every 12 h until 10 days after PMS. The results are expressed as percent for 20 animals per group. Significance between groups was examined by Wilcoxon rank test.. B, Impaired bacterial clearance in cxcl1−/− mice. The CFUs were examined in peritoneal lavage fluid and blood or the homogenates obtained from lungs of cxcl1−/− as well as WT mice at 6 and 24 h post-PMS. The results are expressed as mean log of CFU/mL (n=5/group). C, cxcl1−/− mice have reduced total WBC and neutrophil accumulation at the peritoneum after PMS, as measured by direct cell counts (for total WBC and neutrophils) or MPO activity (for neutrophil influx). The results are expressed as mean/mL (n=5-8/group). D, Both hematopoietic and non-hematopoietic cells are essential for bacterial clearance. Bone marrow chimeras were generated as described in Methods and show bacterial CFUs at 24 h post-PMS or sham (n=5 mice/group). *, p<0.05; **, p<0.01; ***, p<0.001.
CXCL1 contributes to cytokine and chemokine production
Next, we determined the role of CXCL1 in the production of cytokines and chemokines in peritoneal fluid (PF) and serum, by assessing levels in WT and cxcl1−/− mice at 6 h and 24 h post-CLP. As shown in Fig. 2, cytokine and chemokine levels were reduced in PF and in serum samples of cxcl1−/− mice 24 h post-CLP. Intriguingly, the production of IL-17A and IL-17F in PF and serum of cxcl1−/− was reduced at 24 h post-CLP (Fig. 2).
Figure 2. Diminished production of cytokines and chemokines in cxcl1−/− mice after induction of PMS.

cxcl1−/− and their WT control mice were subjected to sham or PMS. The concentration (in pg/mL) of IL-6, MIP-2, IL-17A and IL-17F were quantified in the peritoneal fluid (A) and serum (B) for the time points of 6 and 24 h post-PMS. The results are expressed as mean ± SEM (n=5-8/group; *, p<0.05; **, p<0.01; ***, p<0.001).
CXCL1 activates NF-κB, MAPK, and NADPH oxidase, and induces ICAM-1 expression
Activation of both Nuclear Factor κ B (NF-κB) and Mitogen-activated Protein Kinase (MAPK) is critical for controlling bacterial infections (20, 21). NF-κB activation was reduced in cxcl1−/− mice after CLP at both 6 and 24 h, as assessed by reduced phosphorylation of NF-κB/p65 (Ser 536), , and the degradation of IκBα in the liver, lung, and spleen (Fig. 3). Similarly, cxcl1−/− mice showed reduced activation of the p38, Extracellular Signal-regulated Kinase (ERK), and c-Jun N-terminal Kinase (JNK) MAPKs in the liver, lung and spleen at 24 h post-CLP (Fig. 3). At 6 h post-CLP, cxcl1−/− mice exhibited reduced activation of MAPK. However, activation of p38 in the lung and spleen; JNK in the liver, lung, and spleen were the same as WT at 6 h post-CLP (Fig. 3).
Figure 3. Activation of NF-κB, MAPK, and NADPH oxidase, and expression of adhesion molecule ICAM-1 were impaired in cxcl1−/− mice following PMS.

A, CXCL1 is essential for the activation of NF-κB during PMS induction. Phosphorylation of NF-κB/p65(ser 536), and IκBα in the homogenates from liver, lung and spleen kidney from cxcl1−/− as compared to WT mice. B, Attenuated activation of MAPKs in the organs of cxcl1−/− mice. Phosphorylated p38-MAPK, ERK, and JNK in the homogenates from different organs from cxcl1−/− and WT mice were probed with their respective antibodies. C, Expression of adhesion molecule ICAM-1 but not VCAM-1 is reduced in cxcl1−/− mice. The expression level of ICAM-1 and VCAM-1 were determined in the organs of cxcl1−/− and WT control mice following PMS for time points 6 and 24 h. D, The activation of NADPH oxidase is impaired in cxcl1−/− mice. The expression levels of Nox2, p22phox, p67phox, and p47phox were determined in the homogenates of organs of cxcl1−/− and WT control mice by immunobloting at 6 and 24 h post-PMS. GAPDH or total p38 expression levels were assessed in all samples as internal loading control and the blots are representative of three independent experiments with similar results (A, B, C, and D). Densitometric analysis of 3 separate blots from 3 independent experiments is shown in parenthesis as mean ± SE. (n=4-6/group; *, p<0.05; **, p<0.01; ***, p<0.001).
We next examined whether defective bacterial clearance in the organs of cxcl1−/− mice was due to reduced activation of NADPH (nicotinamide adenine dinucleotide phosphate) oxidase, an enzyme complex consisting of membrane subunits (gp91phox and p22phox) and cytoplasmic subunits (p47phox, p67phox, and p40phox, and rac) that are assembled upon cellular activation to produce microbicidal ROS (22). We observed that CXCL1 deficiency impairs the expression of p22phox, p67phox, and p47phox, as well as Nox2, in different organs post-CLP (Fig. 3).
Cell adhesion molecules are important for neutrophil migration, thus we investigated the expression levels of Intercellular Adhesion Molecule-1 (ICAM-1) and Vascular Cell Adhesion Protein-1 (VCAM-1) after CLP. We found that the expression of ICAM-1, but not VCAM-1, was diminished in the liver, lung, and spleen from cxcl1−/− mice, in comparison to their WT counterparts at 6 h, as well as 24 h post-CLP (Fig. 3).
CXCL1 mediates IL-17A and IL-17F production via activation of CD4, CD8, NK, and γδ cells
Since both IL-17A and IL-17F were reduced in PF and serum of cxcl1−/− mice following CLP, the specific T cell subsets that produce IL-17A or IL-17F in the peritoneum following CLP were determined. Our results show that CD4, CD8, natural killer (NK) or NK T cells, and γδ cells all produce IL-17A and IL-17F in the lungs, and spleen of WT mice 24 h after sepsis (Fig. 4A-B). There were fewer IL-17A and IL-17F-producing cells in cxcl1−/− mice following sepsis compared to WT mice (Figs. 4A-B). In order to confirm the positive regulation of IL-17A and IL-17F by CXCL1, we treated cxcl1−/− mice with recombinant CXCL1 (rCXCL1). Administration of rCXCL1/KC immediately following CLP rescued IL-17A but not IL-17F producing, T cell subsets in the lungs, kidney and spleen of cxcl1−/− mice (Fig. 4). Since the enhanced number of T cell subsets in cxcl1−/− mice following CXCL1 administration may be due to T cell recruitment to the lungs and/or IL-17 differentiation by rCXCL1. To explore these possibilities, we performed Th17differentiation assays using naïve CD4+ T cells obtained from WT and KO mice. Intriguingly, we specifically found that rCXCL1 enhances Th17 (IL-17A-producing) (Fig. 5) but not Th1 or Th2 differentiation (data not shown). Moreover, we collected supernatants at 3 days post-stimulation with rCXCL1 and found that differentiated cells produce substantial IL-17A and IL-17F (Fig. 5D), validating the data related to Th17 differentiation using flow cytometry.
Figure 4. CD4, CD8, NK, and γδ cells are the major source of CXCL1-mediated IL-17A and IL-17F production and rCXCL1 rescues T cell subsets in cxcl1−/− mice in response to PMS.
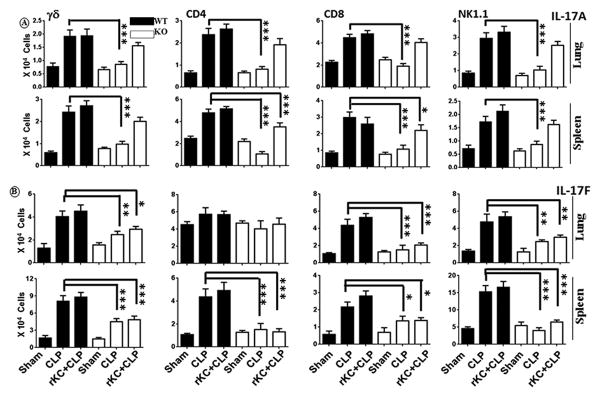
Intracellular IL-17A (A) and IL-17F (B) in gated CD4+, CD8a, γδ, and NK1.1 cells from lung and spleen of WT, cxcl1−/− mice, and cxcl1−/− mice after rCXCL1/KC administration at 24 h post-PMS was analyzed by flow cytometry. The results are expressed as mean ± SE from three independent experiments (n=5-8/group; *, p<0.05; **, p<0.01; ***, p<0.001).
Figure 5. Recombinant CXCL1 increases T cell differentiation and the production of IL-17A.

A-C, Naive CD4+ T cells from WT and KO mice were isolated and stimulated with PMA in the presence or absence of recombinant CXCL1 as described in Materials and Methods, and a representative dot blot is shown. A, Quantitation of IL-17A producing CD4+ T cells (n=5/group). B, Production of IL-17A, IL-17A/F (heterodimer) or IL-17F at the protein level by differentiated CD4+ T cells from WT and KO mice after PMA stimulation in the absence or presence of recombinant CXCL1 at 3 days post-stimulation. (n=5-6/group; *, p<0.05; **, p<0.01; ***, p<0.001).
Recombinant IL-17A administration rescues host defense in cxcl1−/− mice
Previous studies have shown that early intraperitoneal injection of chemokines after CLP results in rapid neutrophil recruitment and subsequently lower peritoneal bacterial burdens (7). To determine whether exogenous CXCL1 or IL-17A administration rescues neutrophil-mediated host defense in cxcl1−/− mice, cxcl1−/− mice were subjected to CLP, and immediately followed byintraperitoneal administration with either saline, CXCL1 or rIL-17A, and assessed for survival through day 10. Intriguingly, CXCL1 or IL-17A-treated cxcl1−/− mice showed improved survival rates compared to saline-treated controls (Fig. 6A-B). Furthermore, cxcl1−/− mice treated with rCXCL1 or rIL-17A showed decreased bacterial burden in blood and peritoneum and increased neutrophil counts and cytokine/chemokines expression in peritoneal fluid of cxcl1−/− mice (Data not shown; Figs. 6C-I). These results suggest that IL-17A controls neutrophil-dependent host defense during sepsis via the production of neutrophil chemoattractants such as CXCL2/MIP-2 and CXCL5/LIX.
Figure 6. Enhanced mortality, higher bacterial burden, reduced leukocyte recruitment, and attenuated cytokine/chemokine production in peritoneal fluid (PF) are rescued in cxcl1−/− mice following PMS after rCXCL1 or rIL-17A administration.

A-B, cxcl1−/− and WT control mice were subjected to sham surgery or PMS and PBS, rCXCL1 or rIL-17A was immediately injected intraperitoneally. Survival was assessed every 12 h up to 10 days after PMS. The results are expressed as percentage for 20 animals per group. Significance between groups was examined by Wilcoxon rank test. C, Bacterial clearance in cxcl1−/− mice after CLP is rescued by exogenous rIL-17A administration. CFUs were determined in the peritoneal fluid and blood of cxcl1−/− mice mice at 6 and 24 h post-PMS. D, Total WBC and neutrophil accumulation at peritoneum in cxcl1−/− mice following PMS are rescued after administration of rIL-17A. E, Production of cytokine and chemokines production in PF of are rescued after administration of rIL-17A. The results are expressed as mean ± SE (n=5-8/group; *, p<0.05; **, p<0.01; ***, p<0.001).
ROS, H2O2, and O2 production by neutrophils is attenuated in cxcl1−/− mice
Efficient function of neutrophils is critical to bacterial elimination from tissues and NADPH activity in neutrophils is shown to be crucial in this process. To examine NADPH oxidase activity in cxcl1−/− mice after CLP, we measured ROS production in peritoneal fluid and in neutrophils, because these cells are a primary source for ROS production (21). These results are consistent with data indicating a low level of ROS and H2O2 production by peritoneal neutrophils and in the peritoneal fluid of cxcl1−/− mice (Fig. 7A). We also found reduced production of ROS, H2O2, and O2 by peritoneal neutrophils from cxcl1−/− micevia FACS analysis (Fig. 7B).
Figure 7. ROS production by neutrophils of cxcl1−/− mice is attenuated during PMS.

A, the production of total ROS and H2O2 in peritoneal fluid and by peritoneal neutrophils of cxcl1−/− and WT control mice following PMS induction were quantified based upon relative fluorescence intensity using commercial kits. The results are expressed as mean ± SE (n=5-8/group). B, ROS+, H2O2+ and OH./O2-+ neutrophils were quantitated in the peritoneal fluid from cxcl1−/− and WT control mice after PMS by FACS using anti-Gr1/Ly6G Ab for staining neutrophils. The results are expressed as mean ± SE (n=5-8/group; *, p<0.05; **, p<0.01; ***, p<0.001).
cxcl1−/− neutrophils exhibit decreased NET formation and NET-mediated bacterial killing
The antibacterial function of neutrophils is not only mediated by intracellular killing, but also by the formation of NETs, termed NETosis and subsequent extracellular killing (23). NETs are composed of DNA studded with many granular proteins that have antimicrobial activity, and their formation is mediated by the induction of ROS in neutrophils (23). Decondensed chromatin with NET morphology was shown to have high levels of histone citrullination (Cit-H3 or H3-Cit), indicating that this event is a marker of NETosis (18). In this regard, we isolated peritoneal neutrophils from cxcl1−/− and WT mice following CLP and examined them for NET formation and NET-mediated bacterial killing. For evidence of NET formation, we performed agarose gel electrophoresis of peritoneal fluid in the absence and presence of DNase. The results showed increased DNA content in the fluid from WT mice compared to cxcl1−/− mice (Fig. 8A). The presence of extracellular DNA in the fluid of both groups of mice was further confirmed by the disappearance of DNA bands after DNase treatment in the gel (Fig. 8A), suggesting different levels of NET formation in both groups during sepsis. Kinetic analysis of NET formation showed a reduction in NET formation rate by cxcl1−/− neutrophils through the initial 8 h (Fig. 8B). To further demonstrate unequal NET formation in the neutrophils from cxcl1−/− and WT mice, we visualized the DNA in neutrophils from both groups after staining with Sytox green nucleic acid stain and anti-citrullinated histone H3 Ab up to 8 hours post-CLP. Utilizing fluorescence microscopy, long string-like extracellular DNA with citrullinated H3 was most evident in WT neutrophils (indicated by arrowheads) (Fig. 8C). The percentage of neutrophils positive for extracellular DNA and citrullinated H3 was reduced among cxcl1−/− mice compared to WT mice (Fig. 8D). Scanning Electron Micrograph analyses showed typical NET structures (cables, threads, and globular domains), as defined by Brinkmann et al.(18), associated with peritoneal neutrophils derived from WT mice after CLP; those structures were largely undetected with peritoneal neutrophils derived from cxcl1−/− mice (Fig. 8E). Furthermore, the expression of citrullinated H3 and peptidylarginine deiminase-4 (PAD-4), associated with NET formation, was reduced in neutrophils from cxcl1−/− mice compared to WT mice after CLP (Fig. 8F).
Figure 8. cxcl1−/− neutrophils exhibit decreased NET formation and NET-mediated bacterial killing in PMS.

A, Agarose gel electrophoresis of peritoneal fluid of cxcl1−/− mice exhibited reduced amounts of extracellular DNA compared to their WT counterpart after PMS. B, Kinetic analysis of NET formation by peritoneal neutrophils harvested from cxcl1−/− and WT mice. Relative fluorescent intensity was determined to evaluate NET formation each hour until 8 h of in vitro culture of neutrophils (n=6-9/group). C, Peritoneal neutrophils harvested from cxcl1−/− or WT mice after PMS, allowed to undergo NET formation and fixed at 8 h. Neutrophils were stained with Sytox green DNA stain and citrullinated histone H3 Ab to visualize citrullinated DNA after the cells were fixed with 4% (v/v) paraformaldehyde. Scale bars, 20 μm. Long strands of DNA (arrowhead) are evidence of NET formation. Images presented are representative images of three independent experiments (n=5-7/group). D, A total of 20 images were selected from one experiment and quantified for the presence of NET-positive neutrophils from cxcl1−/− and WT mice. The results are expressed as mean ± SE (n=5-8/group). E, Evaluation of NET formation by SEM. Peritoneal neutrophils harvested from cxcl1−/− and WT mice after induction of PMS were analyzed by SEM. Presence of long thread-like structures (arrowhead) is evidence of NET formation. Scale bars, 20 μm. Images presented are representative of two independent experiments (n=5-8/group). F, Western blot of PAD-4 and cittrullinated-H3 in the peritoneal neutrophils from cxcl1−/− and WT mice at 24 h post-PMS. This blot is representative of 3 independent experiments with similar results. G-H, Bone marrow neutrophils from cxcl1−/− mice exhibited diminished extracellular bacterial killing activity. Bacterial killing capacity of E. coli-infected bone marrow neutrophils from WT and CXCL1-deficient mice was determined by assessing extracellular (G) and intracellular CFUs (H) at 60 min after infection with E. coli (MOI 1). Relative phagocytosis of E. coli-infected WT and CXCL1-deficient neutrophils at 60 min post-treatment (MOI 1). A total of 4–5 mice/group were used. I, The percentage of NET-forming bone marrow neutrophils following LPS and E. coli stimulations were quantified after staining with Sytox green DNA stain and anti-α H3-Cit antibody as described above. The results are expressed as mean ± SE from two independent experiments (n=5-8/group: *, p<0.05; **, p<0.01; ***, p<0.001).
To determine whether CXCL1-mediated NET formation is essential for bacterial killing, E. coli was incubated with cxcl1−/− or WT neutrophils, and bacterial killing was analyzed. We found a decrease in NET-mediated bacterial killing and relative phagocytosis in cxcl1−/− neutrophils compared with WT neutrophils (Figs. 8G-H). Because DNase can be used to prevent NET formation, neutrophils were pretreated with DNase in one set of experiments. WT neutrophils without DNase treatment showed effective NET-mediated bacterial killing compared to the DNase-treated control, where as no difference in bacterial killing was observed in the DNase-treated cxcl1−/− neutrophils (Fig. 8G). In addition, the percentage of neutrophils positive for extracellular DNA and citrullinated H3 was lower among cxcl1−/− mice following LPS stimulation (10 ng or 100 ng) or E. coli (MOI 1) infection (Fig. 8I).
Discussion
Brisk neutrophil migration to the site of infection and local ROS production are multistep processes and pivotal events in host defense during bacterial infection (24). The severity of sepsis induced by CLP correlates with decreased neutrophil recruitment and consequent diminished bactericidal activity in the peritoneum (7, 8, 25-27). Recruitment and activation of neutrophils has been demonstrated to be critical for controlling CLP-induced polymicrobial sepsis (PMS) by suppressing bacterial growth in the peritoneum and extraperitoneal organs (7).
In this report, we investigated the role of CXCL1 in neutrophil recruitment and function during sepsis induced by CLP. Mice deficient in CXCL1 exhibited a reduction in total leukocytes and neutrophils in the peritoneum after CLP, and a reduction in bacterial clearance from the peritoneum and extra-peritoneal organs, leading to reduced survival. These findings are consistent with previous studies, which showed a critical protective role for CXCL1 in pulmonary host defense against Klebsiella pneumoniae infection (10). Inhibition of CXCL1 attenuates lung neutrophil accumulation following intratracheal LPS administration (28). Moreover, CXCL1-transgenic mice that constitutively express lung CXCL1 show enhanced survival after K. pneumoniae challenge, as well as enhanced neutrophil recruitment and bacterial clearance in the lungs (28). In addition, CXCL1/CXCL1 mRNA was found to be strongly and rapidly induced in the liver and lungs, which was associated with heightened neutrophil infiltration into these organs (29).
The relative contribution of myeloid cells versus stromal cells in neutrophil accumulation following PMS is unclear. Myeloid cells in tissues produce a battery of neutrophil chemotactic substances such as CXCL1 (10, 11) and MIP-2 (12, 13) and resident cells, including epithelial and endothelial cells, produce other neutrophil chemoattractants, such as LIX (14). The findings in this investigation are consistent with our previous studies, demonstrating that hematopoietic and nonhematopoietic cell-derived CXCL1 is essential for neutrophil-dependent bacterial clearance (10, 12)
We found that the local and systemic inflammatory response to PMS was dependent on CXCL1, and correlated with higher cellular influx, bacterial clearance, and reduced mortality. In addition, the expression of key inflammatory cytokines and chemokines was dependent upon CXCL1 during PMS. Interestingly, we observed that the expression of IL-1β, IL-6, LIX, and IL-17A was mediated by CXCL1 even during the early phase of sepsis (6 h). These findings are in agreement with previous studies that showed the expression of CXCL2 and CXCL5, but not TNF-α, to be dependent on CXCL1 expression in K. pneumoniae-infected lungs (10).
Activation of transcription factors is a central feature of inflammation and host response (20), and NF-κB is a well-studied transcription factor (21). We found reduced activity of NF-κB in the organs from cxcl1−/− mice post-CLP. Although NF-κB activity is important for survival after PMS (4, 30), it is questionable whether this phenomenon contributes to efficient neutrophil recruitment and bacterial clearance. NF-κB activation has been demonstrated to be essential for neutrophil recruitment due to its contribution to the expression of adhesion molecules, such as ICAM-1 (31). Our results reveal a strong correlation between CXCL1 expression after PMS, NF-κB activation, and ICAM-1 expression. This suggests that CXCL1-mediated NF-κB activation plays a crucial role in neutrophil recruitment at early and late phases of polymicrobial sepsis.
MAPKs are important enzymes that enhance the expression of pro-inflammatory cytokines, chemokines, adhesion molecules, and antibacterial effectors by activating transcription factors, such as AP-1, c-Jun, and STAT-1 (32). Our results show reduced activation of p38 (at 24 h post-CLP), JNK, and ERK MAP kinases in the organs from cxcl1−/− mice. These results are consistent with previous findings that showed a crucial role for CXCL1 in MAPK activation during K. pneumonaie pulmonary infection (10)
IL-17-producing CD4+ T cells control bacterial infection by secretion of neutrophil chamoattractants and granulopoietic factors such as CXC chemokine CXCL2/MIP-2 and G-CSF, which cause neutrophil recruitment (33). We found not only γδ T cells, but also CD4, CD8, and NK1.1 cells as major IL-17-producers in WT mice 24 h post-PMS. This suggests that CXCL1 regulates both IL-17A and IL-17F expression by major T cell subsets during sepsis. This possibility was confirmed when we performed intracellular staining for IL-17A in T cell subsets. Because we observed increased numbers of IL-17A-producing CD4 cells and increased IL-17A and IL-17F secretion after treatment with recombinant CXCL1, our results suggest that CXCL1 is an essential chemokine for differentiation of naïve CD4 T-cells to Th17. To validate this hypothesis, we differentiated Th0 cells to Th1, Th2 and Th17 in vitro in the absence and presence of CXCL1. Intriguingly, our findings demonstrated that CXCL1 augments Th17 differentiation but not Th1 or Th2 differentiation (data not shown). The limitation of this study however noted. It is not possible to use either Gram-positive or Gram-negative bacterium in these cultures because these bacteria induced substantial cell death (data not shown).
To examine whether rIL-17A can restore the protective effect on cxcl1−/− mice, we administered each of these chemokines immediately after PMS, resulting in enhanced survival of cxcl1−/− mice. Additionally, enhanced neutrophil influx and bacterial clearance at the peritoneum were observed following rIL-17 administration. Our observation of host protection by treatment with IL-17 in cxcl1−/− mice agrees in part with a previous investigation that demonstrated reduced survival rates, lower neutrophil influx, and bactericidal capacity in IL-17R gene deficient mice following PMS (though cytokine and chemokine expression was enhanced in these mice) (8).
We observed reduced expression and activation of NADPH oxidase components Nox2, p22phox, p67phox, and p47phox in cxcl1−/− mice following PMS, which may due to the reduced accumulation of neutrophils at the infectious focus in cxcl1−/− mice. These observations are in agreement with a previous investigation that demonstrated reduced expression of NADPH oxidase components following neutrophil depletion in WT mice after K. pneumoniae challenge (12). CXCL1-mediated activation and expression of NADPH oxidase is consistent with ROS production by neutrophils, as we observed decreased ROS production in peritoneal fluid and by peritoneal neutrophils in cxcl1−/− mice compared to WT counterparts. Additionally, we observed a substantial reduction of ROS+, H2O2+, and O2+ producing neutrophils from cxcl1−/− mice following PMS. These findings are consistent with a previous study that demonstrated an essential role of CXCL1 in Klebsiella-induced expression of ROS by neutrophils (12).
NETosis is a unique host defense mechanism employed by neutrophils to trap and kill extracellular pathogens (34). In the present study, we found a strong correlation between reduced NADPH oxidase-mediated ROS production and reduced NET formation by neutrophils from cxcl1−/− mice following PMS. Additionally, NET formation and NET-positive peritoneal neutrophils were reduced in cxcl1−/− mice compared to WT. To confirm this result, we observed reduced formation of typical NET structures associated with cxcl1−/− neutrophils by SEM. In addition, we observed reduced E. coli killing by bone marrow neutrophils treated with DNase, confirming the link between NET formation and bacterial killing. The results linking neutrophil ROS production with NETosis and subsequent bacterial killing are consistent with several prior reports (18, 35, 36). Our finding that neutrophils harvested from cxcl1−/− mice are deficient for bacterial killing confirms the crucial role of CXCL1 in the containment of infection by migrating neutrophils.
The data in this study illustrate a number of advancements in our understanding of the biological role of CXCL1 in host defense during PMS. CXCL1 appears to be essential for host survival, activation of NF-κB, MAPK, and NADPH oxidase, expression of cytokines and chemokines essential for host defense, ROS production, as well as NET formation by peritoneal neutrophils and NET-mediated bacterial killing (Fig. 9). More importantly, CXCL1 regulates IL-17A production by enhancing Th17 differentiation in order to control neutrophil influx via the production of CXCL2/MIP-2 and CXCL5/LIX (Fig. 9).
Figure 9. Hypothetical scheme for CXCL1-mediated signaling cascades leading to bacterial clearance in the organs in response to PMS.

Bacteria in the peritoneum during polymicrobial sepsis interact with pattern recognition receptors (1) resulting in CXCL1 production by hematopoietic and resident cells (2). CXCL1 controls neutrophil recruitment (one arm) and neutrophil function (other arm). In one arm, CXCL1 activates NF-κB, MAPK (3) and upregulation of cell adhesion molecules (ICAM-1) along with the production of cytokines/chemokines (4) in order to induce neutrophil recruitment to the tissues from the bloodstream (5). In the second arm, CXCL1 regulates the production of IL-17 (IL-17A) (6) resulting in the production of CXCL2/MIP-2 and IL-6 (7) in order to induce neutrophil recruitment (8). In the third arm, CXCL1 modulates neutrophil function (9) via activating NADPH oxidase (9) leading to the production of ROS and eventual NETosis (9) and augmenting phagocytosis (9). Both of these arms are important for bacterial clearance in organs (10).
Acknowledgments
We thank Sergio Lira at Mount Sinai Medical Center for providing the cxcl1-/- mice. The authors thank the Lung biology Members, including Pankaj Baral and Ritwij Kulkarni for helpful discussions and critical reading of the manuscript. We thank Marilyn Dietrich for FACS, Julia Sokolova for Scanning Electron Microscopy and Pete Mottram for immunofluorescence microscopy.
Funding Source: Supported by a Scientist Award from the Flight Attendant Medical Research Institute (CIA-113043); and a grant from the NIH (2R01 HL-091958) to SJ
References
- 1.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Critical care medicine. 2001;29:1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Surbatovic M, Jevdjic J, Veljovic M, Popovic N, Djordjevic D, Radakovic S. Immune Response in Severe Infection: Could Life-Saving Drugs Be Potentially Harmful? TheScientificWorldJournal. 2013;2013:961852. doi: 10.1155/2013/961852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oh SJ, Kim JH, Chung DH. NOD2-mediated suppression of CD55 on neutrophils enhances C5a generation during polymicrobial sepsis. PLoS pathogens. 2013;9:e1003351. doi: 10.1371/journal.ppat.1003351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ozment TR, Ha T, Breuel KF, Ford TR, Ferguson DA, Kalbfleisch J, Schweitzer JB, Kelley JL, Li C, Williams DL. Scavenger receptor class a plays a central role in mediating mortality and the development of the pro-inflammatory phenotype in polymicrobial sepsis. PLoS pathogens. 2012;8:e1002967. doi: 10.1371/journal.ppat.1002967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alazawi W, Heath H, Waters JA, Woodfin A, O'Brien AJ, Scarzello AJ, Ma B, Lopez-Otalora Y, Jacobs M, Petts G, Goldin RD, Nourshargh S, Gamero AM, Foster GR. Stat2 loss leads to cytokine-independent, cell-mediated lethality in LPS-induced sepsis. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:8656–8661. doi: 10.1073/pnas.1221652110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fearon DT, Locksley RM. The instructive role of innate immunity in the acquired immune response. Science. 1996;272:50–53. doi: 10.1126/science.272.5258.50. [DOI] [PubMed] [Google Scholar]
- 7.Craciun FL, Schuller ER, Remick DG. Early enhanced local neutrophil recruitment in peritonitis-induced sepsis improves bacterial clearance and survival. Journal of immunology. 2010;185:6930–6938. doi: 10.4049/jimmunol.1002300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Freitas A, Alves-Filho JC, Victoni T, Secher T, Lemos HP, Sonego F, Cunha FQ, Ryffel B. IL-17 receptor signaling is required to control polymicrobial sepsis. Journal of immunology. 2009;182:7846–7854. doi: 10.4049/jimmunol.0803039. [DOI] [PubMed] [Google Scholar]
- 9.Weiss SJ. Tissue destruction by neutrophils. The New England journal of medicine. 1989;320:365–376. doi: 10.1056/NEJM198902093200606. [DOI] [PubMed] [Google Scholar]
- 10.Cai S, Batra S, Lira SA, Kolls JK, Jeyaseelan S. CXCL1 regulates pulmonary host defense to Klebsiella Infection via CXCL2, CXCL5, NF-kappaB, and MAPKs. Journal of immunology. 2010;185:6214–6225. doi: 10.4049/jimmunol.0903843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stearns-Kurosawa DJ, Osuchowski MF, Valentine C, Kurosawa S, Remick DG. The pathogenesis of sepsis. Annual review of pathology. 2011;6:19–48. doi: 10.1146/annurev-pathol-011110-130327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Batra S, Cai S, Balamayooran G, Jeyaseelan S. Intrapulmonary administration of leukotriene B(4) augments neutrophil accumulation and responses in the lung to Klebsiella infection in CXCL1 knockout mice. Journal of immunology. 2012;188:3458–3468. doi: 10.4049/jimmunol.1101985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ishii M, Asano K, Namkoong H, Tasaka S, Mizoguchi K, Asami T, Kamata H, Kimizuka Y, Fujiwara H, Funatsu Y, Kagawa S, Miyata J, Ishii K, Nakamura M, Hirai H, Nagata K, Kunkel SL, Hasegawa N, Betsuyaku T. CRTH2 is a critical regulator of neutrophil migration and resistance to polymicrobial sepsis. Journal of immunology. 2012;188:5655–5664. doi: 10.4049/jimmunol.1102330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shea-Donohue T, Thomas K, Cody MJ, Aiping Z, Detolla LJ, Kopydlowski KM, Fukata M, Lira SA, Vogel SN. Mice deficient in the CXCR2 ligand, CXCL1 (KC/GRO-alpha), exhibit increased susceptibility to dextran sodium sulfate (DSS)-induced colitis. Innate immunity. 2008;14:117–124. doi: 10.1177/1753425908088724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nature protocols. 2009;4:31–36. doi: 10.1038/nprot.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Douda DN, Jackson R, Grasemann H, Palaniyar N. Innate immune collectin surfactant protein D simultaneously binds both neutrophil extracellular traps and carbohydrate ligands and promotes bacterial trapping. Journal of immunology. 2011;187:1856–1865. doi: 10.4049/jimmunol.1004201. [DOI] [PubMed] [Google Scholar]
- 17.Balamayooran T, Batra S, Balamayooran G, Cai S, Kobayashi KS, Flavell RA, Jeyaseelan S. Receptor-interacting protein 2 controls pulmonary host defense to Escherichia coli infection via the regulation of interleukin-17A. Infection and immunity. 2011;79:4588–4599. doi: 10.1128/IAI.05641-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A. Novel cell death program leads to neutrophil extracellular traps. The Journal of cell biology. 2007;176:231–241. doi: 10.1083/jcb.200606027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boniface K, Bak-Jensen KS, Li Y, Blumenschein WM, McGeachy MJ, McClanahan TK, McKenzie BS, Kastelein RA, Cua DJ, de Waal Malefyt R. Prostaglandin E2 regulates Th17 cell differentiation and function through cyclic AMP and EP2/EP4 receptor signaling. The Journal of experimental medicine. 2009;206:535–548. doi: 10.1084/jem.20082293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annual review of immunology. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 21.Winterbourn CC, Kettle AJ. Redox reactions and microbial killing in the neutrophil phagosome. Antioxidants & redox signaling. 2013;18:642–660. doi: 10.1089/ars.2012.4827. [DOI] [PubMed] [Google Scholar]
- 22.Segal BH, Grimm MJ, Khan AN, Han W, Blackwell TS. Regulation of innate immunity by NADPH oxidase. Free radical biology & medicine. 2012;53:72–80. doi: 10.1016/j.freeradbiomed.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Papayannopoulos V, Zychlinsky A. NETs: a new strategy for using old weapons. Trends in immunology. 2009;30:513–521. doi: 10.1016/j.it.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 24.Choi EY, Santoso S, Chavakis T. Mechanisms of neutrophil transendothelial migration. Frontiers in bioscience. 2009;14:1596–1605. doi: 10.2741/3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mercer-Jones MA, Heinzelmann M, Peyton JC, Wickel D, Cook M, Cheadle WG. Inhibition of neutrophil migration at the site of infection increases remote organ neutrophil sequestration and injury. Shock. 1997;8:193–199. doi: 10.1097/00024382-199709000-00007. [DOI] [PubMed] [Google Scholar]
- 26.Moreno SE, Alves-Filho JC, Rios-Santos F, Silva JS, Ferreira SH, Cunha FQ, Teixeira MM. Signaling via platelet-activating factor receptors accounts for the impairment of neutrophil migration in polymicrobial sepsis. Journal of immunology. 2006;177:1264–1271. doi: 10.4049/jimmunol.177.2.1264. [DOI] [PubMed] [Google Scholar]
- 27.Giangola MD, Yang WL, Rajayer SR, Nicastro J, Coppa GF, Wang P. Growth Arrest-Specific Protein 6 (Gas6) Attenuates Neutrophil Migration and Acute Lung Injury in Sepsis. Shock. 2013 doi: 10.1097/SHK.0b013e3182a588c1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsai WC, Strieter RM, Wilkowski JM, Bucknell KA, Burdick MD, Lira SA, Standiford TJ. Lung-specific transgenic expression of KC enhances resistance to Klebsiella pneumoniae in mice. Journal of immunology. 1998;161:2435–2440. [PubMed] [Google Scholar]
- 29.Salkowski CA, Detore G, Franks A, Falk MC, Vogel SN. Pulmonary and hepatic gene expression following cecal ligation and puncture: monophosphoryl lipid A prophylaxis attenuates sepsis-induced cytokine and chemokine expression and neutrophil infiltration. Infection and immunity. 1998;66:3569–3578. doi: 10.1128/iai.66.8.3569-3578.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu X, Zhang P, Bao Y, Han Y, Wang Y, Zhang Q, Zhan Z, Meng J, Li Y, Li N, Zhang WJ, Cao X. Zinc finger protein ZBTB20 promotes Toll-like receptor-triggered innate immune responses by repressing IkappaBalpha gene transcription. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:11097–11102. doi: 10.1073/pnas.1301257110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Su Y, Lei X, Wu L, Liu L. The role of endothelial cell adhesion molecules P-selectin, E-selectin and intercellular adhesion molecule-1 in leucocyte recruitment induced by exogenous methylglyoxal. Immunology. 2012;137:65–79. doi: 10.1111/j.1365-2567.2012.03608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dong C, Davis RJ, Flavell RA. MAP kinases in the immune response. Annual review of immunology. 2002;20:55–72. doi: 10.1146/annurev.immunol.20.091301.131133. [DOI] [PubMed] [Google Scholar]
- 33.Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, Shellito JE, Bagby GJ, Nelson S, Charrier K, Peschon JJ, Kolls JK. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. The Journal of experimental medicine. 2001;194:519–527. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaplan MJ, Radic M. Neutrophil extracellular traps: double-edged swords of innate immunity. Journal of immunology. 2012;189:2689–2695. doi: 10.4049/jimmunol.1201719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lim MB, Kuiper JW, Katchky A, Goldberg H, Glogauer M. Rac2 is required for the formation of neutrophil extracellular traps. Journal of leukocyte biology. 2011;90:771–776. doi: 10.1189/jlb.1010549. [DOI] [PubMed] [Google Scholar]
- 36.Brinkmann V, Zychlinsky A. Beneficial suicide: why neutrophils die to make NETs. Nature reviews Microbiology. 2007;5:577–582. doi: 10.1038/nrmicro1710. [DOI] [PubMed] [Google Scholar]