

Abstract
Phenobarbital is the most commonly utilized drug for the treatment of neonatal seizures. However, mounting preclinical evidence suggests that even brief exposure to phenobarbital in the neonatal period can induce neuronal apoptosis, alterations in synaptic development, and long-lasting changes in behavioral functions. In the present report, we treated neonatal rat pups with phenobarbital and evaluated behavior in adulthood. Pups were treated initially with a loading dose (80mg/kg) on postnatal day (P)7 and with a lower dose (40 mg/kg) on P8 and P9. We examined sensorimotor gating (prepulse inhibition), passive avoidance, and conditioned place preference to cocaine when the animals reached adulthood. Consistent with our previous reports, we found that three days of neonatal exposure to phenobarbital significantly impaired prepulse inhibition as compared to vehicle-exposed control animals. Using a step-though passive avoidance paradigm, we found that animals exposed to phenobarbital as neonates and tested as adults showed significant deficits in passive avoidance retention as compared to matched controls, indicating impairment in associative memory and/or recall. Finally, we examined place preference conditioning in response to cocaine. Phenobarbital exposure did not alter the normal conditioned place preference associated with cocaine exposure. Our findings expand the profile of behavioral toxicity induced by phenobarbital.
Keywords: antiepileptic drug, seizure, toxicology, reward
1. Introduction
Phenobarbital (PB) is the most commonly utilized drug for the treatment of neonatal seizures [1–3] despite growing concerns about its efficacy [4,5] and safety in neonatal or infant populations. For example, prolonged early-life exposure to phenobarbital as a treatment for febrile seizures has been associated with reduced IQ [6,7]. Comparable studies examining shorter exposures have not been performed.
Preclinically, there is mounting evidence that even brief exposure to phenobarbital during early postnatal life can have long-lasting effects on brain development in rodents. For example, when given even once to postnatal day (P) 7 rats, phenobarbital induced a profound increase in neuronal apoptosis throughout a variety of cortical (e.g., frontal and parietal cortices) and subcortical structures (e.g., hippocampus, nucleus accumbens, amygdala, thalamus) [8–10]. This effect has been well documented by several groups, with the period of vulnerability lasting until ~P10–14 [8]. Early-life phenobarbital exposure is also associated with changes in the cortical proteome, including genes associated with synaptic function and regulation of oxidative stress [11].
Importantly, P7 exposure to PB also induces changes in nervous system function. For example, between P10 and P18 there is normally a robust increase in the number of functional excitatory and inhibitory synapses in striatum [12]. In contrast, when rats were exposed to PB on P7, striatal synaptic development was stunted [12,13]. Interestingly, when the timing of PB exposure was shifted to P10, normal maturation patterns were found [12].
We [12–16] and others [17–21] have also reported functional effects of early-life PB exposure on adult behavior. One of the most consistent findings is impaired spatial memory in PB-exposed animals tested as adults; exposure from P6–P10, P7–P14, or P2–P21 all disrupted adult spatial memory in the Morris Water Maze or radial arm maze [15,17,20,21]. Deficits in other memory tasks (i.e., delayed alternation [21], fear conditioning [13,15], reversal learning [12]) have also been reported after PB exposure in early life. Additional behavioral changes following acute or subacute neonatal exposure include impaired prepulse inhibition (PPI) [13–15], hypersensitivity to the locomotor-enhancing effects of amphetamine [14,16], decreased anxiety-like behavior, and reduced social exploration [15].
The present study had two objectives. The first objective was to better approximate a clinical schedule of PB exposure. Previous studies have either used acute [12,14] or prolonged [13,15,18,20,21] exposure to PB. While a single exposure is useful for examining the “worst case” scenario of drug toxicity (i.e., even a single dose is sufficient to alter behavior), it does not mirror clinical recommendations [3]. Conversely, longer exposures can exceed both the therapeutically relevant dose (due to drug accumulation) and the developmentally equivalent [22] time period during which treatment would occur. To reduce these confounds, here, we examined the effects of subacute administration of PB (P7, 8 and 9) on subsequent adult behavior. Pups received a loading dose on P7 and half doses on P8 and 9 to avoid drug accumulation.
The second objective of this study was to examine a previously unexplored behavioral domain: psychostimulant reinforcement. We chose this measure because of the enhanced locomotor response PB-exposed rats display to psychostimulants [14,16], and because of the profound apoptosis that occurs in limbic structures that mediate reward [10]. As a basis for comparison we also examined PPI, which is impaired by both acute and chronic exposure, and thus serves as a positive control for the present study [13–15]. We also chose a step-through passive avoidance task as a measure of associative learning. Associative learning is impaired by chronic exposure [13,15], but has yet to be examined after acute or subacute exposure.
2. Materials and methods
2.1 Animals
Timed-pregnant Sprague-Dawley rats (Harlan Laboratories, Frederick, Maryland) were housed in the Georgetown University Division of Comparative Medicine. Animals were maintained in a temperature-controlled room (21°C) with a 12-h light cycle (0600-1800 lights on). Food and water were available ad libitum. A total of 30 pups (a mix of male and females) were used and date of parturition was designated P0 for all pups. Treatment was counterbalanced across litters and sex and all manipulations occurred during the light phase. One animal from the PB-treated group was excluded from data analysis because it was a statistical outlier using Dixon’s test (P<0.009). All experiments were approved by the Georgetown University Animal Care and Use Committee.
2.2 Drug Treatment
Phenobarbital sodium (5-ethyl-5-phenyl-1,3-diazinane-2,4,6-trione; Sigma) was dissolved in saline at a concentration of 8 mg/ml and administered intraperitoneally. Pups were treated on postnatal day (P)7, P8 and P9. We employed a loading dose of 80 mg/kg on P7, followed by 40 mg/kg on P8 and P9. Loading doses are commonly used clinically for neonates [23–25]. Doses were selected based on pharmacodynamic equivalence. 80 mg/kg was selected because this dose, but not lower doses, provides complete protection against seizures evoked by pentylenetetrazole in P7 rats [26]. Moreover, this dose, but not lower doses, prevented mortality associated with kainic acid treatment in P7 rats [27]. 40 mg/kg was selected because it is the lowest dose that provides complete protection against tonic seizures, and partial protection against clonic seizures evoked by pentylenetetrazol [26]. Control pups received equivalent volumes of vehicle.
2.3 Behavioral Testing
Animals were weaned into same sex cages of 2–3 rats at P21 and maintained until adulthood, when testing began (P60+). Prior to each behavioral test (described below), animals were allowed to acclimate to the testing room for at least 30 minutes. Tests were performed in the order described below.
2.3.1 Prepulse inhibition (PPI)
PPI testing was conducted as we have previously described [13–15,28]. Briefly, testing was conducted using the SR-LAB system (San Diego Instruments). Startle chambers were ventilated and illuminated, with continuous background noise (65 dB). Broadband noise pulses were generated by a high-frequency loudspeaker within the chamber. Each chamber contained a clear non-restrictive Plexiglas cylinder resting on a platform. A piezoelectric accelerometer attached to the platform detected motion produced by startle responses.
Animals were allowed to acclimatize within the Plexiglas chamber for 5 min. Following the acclimation period, a startle-inducing broadband noise pulse (“Pulse Alone”; 120 dB, 30 ms) was presented five times to habituate the animals to the testing procedure. These trials were excluded from data analysis. Rats were then presented with either the startle pulse alone (“Pulse Alone”) or 100 ms after a 30 ms prepulse (“Prepulse+Pulse”) that was 3, 6, or 12 dB above the background noise. Each session consisted of a total of 40 trials (10 Pulse Alone trials, 10 of each prepulse trial) presented in pseudorandom order. Trials were separated by an average of 15 s (range 5–25 s). Startle magnitude was calculated as the average of the startle responses to the pulse-alone trials. PPI was calculated according to the formula: .
2.3.2 One-trial step-through Passive Avoidance Task
As a measure of learning and memory, we employed a passive avoidance task. Conditioning and testing occurred in a standard rat shuttle cage (Coulbourn Instruments, H10-11R-PA). The cage (20″×10″×12″, width × depth × height) was divided evenly into a dark chamber and light chamber. The light chamber was illuminated by a ceiling mounted light bulb. A computer-controlled drop door separated the chambers.
On the first day of passive avoidance training, rats were placed into the light side of the apparatus (the door was closed to prevent entry to the dark side) for 180 s. On the second day, rats were placed into the light side of the apparatus. After 30 s, the door was lifted allowing access to the dark chamber. When the animal crossed into the dark chamber, the door was lowered (to prevent re-entry to the light chamber) and a mild footshock (0.4 mA, 2 s duration) was administered. Animals were removed from the dark side within 30 s of the conclusion of the conditioning trial.
On days 3 and 4, animals were tested for retention. They were placed in the light side of the chamber, and after 30 s the door to the dark side was lifted, allowing access. Latency to enter the dark chamber was recorded. If the rat did not enter the dark side within 300 s of the door opening, the trial was terminated and a latency of 300 s was assigned.
2.3.3 Conditioned Place Preference (CPP)
Rats were tested in a place preference conditioning paradigm as previously described [29]. The apparatus consisted of two triangular compartments that shared one wall, with a rectangular door connecting the two chambers. Distinctive tactile and visual cues were used to differentiate the two chambers: (1) pellet bedding with dots on the walls and (2) corn-cob bedding and vertical stripes on the walls. The location of the rat was monitored by a camera suspended over the chambers and tracked and recorded by AnyMaze (Stoelting).
In a pretreatment baseline preference test, rats were placed in the doorway at the center of the chamber facing the dotted chamber. The rats were allowed free access to both compartments for 10 min. Animals with an unconditioned bias (>75% preference) to either chamber were withdrawn from further experiments. 5 of 30 rats were removed on the basis of this criterion.
On the subsequent 4 days, the preferred (dotted, CS−) compartment was paired with administration of saline vehicle (1 ml/kg, ip) and the other compartment (stripes, CS+) with cocaine hydrochloride (10 mg/kg, ip; NIDA). This dose was selected to give a moderate place preference. Saline and cocaine treatments were given on alternate days (i.e., two doses of saline and two doses of cocaine). Animals were injected with cocaine or saline immediately prior to being placed into the respective chamber. Each conditioning session lasted 30 min, during which the rats were restricted to the respective conditioning chamber by keeping the door between compartments closed.
Following the four days of conditioning, animals were tested in a post-conditioning preference session. During this 10 min session, rats were allowed to freely explore both chambers. During a 10 min session, the animals were recorded by a camera and time spent in each compartment was tracked and recorded by AnyMaze. Data are presented as the % increase in time spent in the CS+ chamber (comparing the pre-test to the post-test).
2.3.4 Data Analysis
Data were analyzed using GraphPad Prism (GraphPad Software, La Jolla CA). Parametric data (i.e., prepulse inhibition) were analyzed using a two-way ANOVA with prepulse intensity as a repeated measure and drug treatment as a between subject variable. Startle amplitude was analyzed using an unpaired t test. Nonparametric data (i.e., passive avoidance retention scores and conditioned place preference scores) were analyzed using a Mann-Whitney-U test and Wilcoxon tests. P-values <0.05 were considered to be statistically significant. Results are presented graphically as mean + standard error of the mean (SEM).
3. Results
3.1 Body Weight
Control and PB groups had equivalent mean body weight before the onset of treatment (14.6 g). Rats treated with PB showed a mean 2% increase in body weight from P7 to P8, and a 9% increase from P7 to P9. In contrast, vehicle treated animals showed 15% increase from P7 to P8, and 34% increase from P7 to P9. Two-way analysis of variance revealed a significant main effect of age (postnatal day) [F2,64=102.7, P<0.0001], a significant main effect of treatment [F1,32=15.34, P=0.0004], and a significant treatment by age interaction [F2,64=40.16, P<0.0001]. Sidak-correct posthoc tests showed that vehicle-treated animals had significantly greater body weight than PB-treated animals on P8 and P9.
When adult body weight was analyzed (i.e., weight immediately prior to CPP testing), the two groups displayed statistically equivalent weight (Student’s T Test, t=0.33, d.f.=23, P=0.75). The PB-exposed group had a mean (+/− SEM) body weight of 286.5+/−18.4g while the vehicle treated group weighed a mean of 295.6+/−21.04 g. Thus, although PB exposure slowed weight gain in neonatal pups, weights were equivalent to control animals by adulthood.
3.2 Prepulse Inhibition
PPI is a measure of sensorimotor gating, in which the normal startle response to a loud stimulus (i.e., pulse) is attenuated by a preceding weak stimulus (i.e., a prepulse). When tested as adults, vehicle exposed rats displayed a normal pattern of increasing PPI as a function of prepulse intensity. Animals treated with phenobarbital displayed significantly lower levels of PPI as compared to controls (Fig 1A). This effect was revealed by a two-way ANOVA with prepulse intensity as a within-subject factor and treatment as a between subject factor. This analysis revealed a main effect of prepulse intensity (F2,56=14.0, P<0.001), a main effect of treatment (F1,28=4.2, P=0.05), but no treatment-by-prepulse interaction (F2×56=0.17, P=0.85). Post-hoc analysis (Fisher’s LSD), revealed that PPI was significantly lower in PB-exposed, as compared to control pups at prepulses of 3 and 9 (Ps<0.05, 1-tailed), with a trend toward significance for PP12 (P<0.10). Startle amplitude (Fig 1B) did not differ as a function of neonatal treatment (t=1.59, df=28, P=0.12), suggesting that a change in startle amplitude cannot account for the deficits in PPI seen in PB treated pups.
Fig 1.
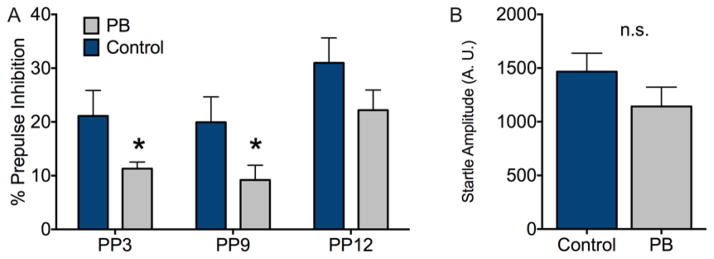
Adult rats exposed to phenobarbital as neonates display impaired PPI. Bars show mean + SEM. (A) Percent prepulse inhibition as a function of prepulse intensity. (B) Acoustic startle response to a 120dB noise pulse, expressed in arbitrary units. * = P<0.05
3.3 Passive Avoidance
We next evaluated passive avoidance learning (Fig 2). We found that the latency to enter the dark chamber on the conditioning day (i.e., before any footshock had been delivered) did not differ between treatment groups (Mann-Whitney U=94.5, P=0.35). After conditioning, animals were tested on two retention tests (separated by 24 h) and the performance on these tests was averaged. We found that adult rats exposed to PB as neonates displayed significantly lower passive avoidance performance than did control animals (Mann Whitney U=62, P<0.05, one-tailed), suggesting impaired learning and/or recall.
Fig 2.

Adult rats exposed to phenobarbital as neonates display impaired passive avoidance performance. Latency to enter the dark chamber was tested one and two days after initial conditioning and data were averaged for each subject. Y-axis shows the average latency to enter the dark (shock-paired chamber). Bars show mean + SEM. * = P<0.05
3.4 Conditioned Place Preference
Because we had previously found that P7 exposure to PB potentiated the locomotor-enhancing effects of psychostimulant exposure [14], we next examined place preference conditioning with cocaine to determine if PB exposure also changed the reinforcing properties of psychostimulants (Fig 3). We found that both PB-exposed and control animals displayed a significant (Ps<0.05, 1-sample t-test) increase in preference for the cocaine-paired chamber (CS+). Moreover, both groups exhibited equivalent (Student’s t-test t=0.07, df=22, P=0.94) percent increases in time spent in the CS+ chamber.
Fig 3.

Neonatal phenobarbital exposure does not alter place preference conditioning. Figure shows % increase in time spent in the CS+ chamber. (Time in CS+ on post-conditioning test minus time in CS+ on preconditioning test) / Time in CS+ during the preconditioning test]. Bars show mean + SEM.
4. Discussion
Here we found that subacute, clinically relevant neonatal exposure to phenobarbital resulted in behavioral alterations detected in adults.
Consistent with our previous studies, PB exposure was associated with deficits in prepulse inhibition. PPI is a measure of sensorimotor gating that is critically dependent on the limbic forebrain structures, including the hippocampus, amygdala, nucleus accumbens [30]. The fact that this phenotype is highly consistent across studies may be due to the distributed nature of the PPI network; PB exposure triggers apoptosis in many of the brain regions needed to support normal PPI.
Likewise, we found deficits in associative learning in adult rats exposed to PB as pups. We used the passive avoidance task as a measure of associative learning. While this task has not been previously examined after neonatal PB exposure, it shares features with fear conditioning, another associative learning task that is impaired by neonatal PB exposure [13,15]. Both tasks require the amygdala for normal acquisition [31–33], and the fact that PB induces neuronal apoptosis in the amygdala may be related to this deficit [10]. While we did not measure the pain threshold to foot shock in the present study, we observed that all rats displayed a response to footshock delivery during conditioning. This suggests that a difference in pain threshold is unlikely to account for the change in passive avoidance behavior we found. Because the passive avoidance task requires animals to inhibit a species-typical, prepotent response (i.e., to exit a brightly lit compartment in favor of a darkly lit compartment) deficits in frontal function can impair performance [34]. This raises the possibility that executive function / inhibitory control is impaired in PB-exposed rats. In support of this hypothesis, the prefrontal cortex is also vulnerable to antiepileptic drug-induced neuronal apoptosis [10]. It would be particularly interesting to examine frontal function in PB-exposed animals in future studies.
Surprisingly, we did not detect a change in the reinforcing properties of psychostimulant exposure. We had hypothesized that the hypersensitivity to the locomotor effects of psychostimulants seen after P7 exposure to PB [14,16] would be accompanied by increased sensitivity to the reinforcing features of these drugs. Our study was designed to induce a mild place preference allowing us to detect both increases and decreases in conditioning. We have previously utilized a similar study design with gestational exposure to fluoxetine, which increases adult sensitivity to cocaine CPP [29]. The lack of effect of PB exposure on CPP in the present study is also interesting in light of the deficits in associative learning seen with passive avoidance. The dissociation between these two tasks indicates that animals do not have a global deficit in associative learning, but rather a deficit that is specific to the type of conditioning (i.e., aversive but not appetitive).
Together, the data in the present study add to a growing literature suggesting that PB exposure in early life can have long-lasting effects on nervous system function. These data, which show deficits in sensorimotor gating and associative learning, are consistent with prior reports. These data also provide the first functional examination of reward circuitry in animals exposed to PB, and indicate that the rewarding properties of psychostimulants are unchanged by early life PB exposure. P7–P9 in the rat corresponds to the brain growth spurt, which occurs during late gestational and the early postnatal period in humans [22]. Thus, our data may be relevant to both gestational and postnatal exposure. Gestational exposure is of interest because phenobarbital is still widely used in developing countries for the control of epilepsy in adults [35–37].
With respect to postnatal exposure, phenobarbital is used clinically to treat neonatal seizures or as an intervention following hypoxia-ischemia. However, our present studies were conducted in otherwise normal rat pups, i.e., animals without seizures. Thus, it remains an open question as to what profile of developmental alterations would occur when both seizures and phenobarbital are present. Some evidence suggests that repeated electroshock seizures in neonatal rats does not alter the pro-apoptotic action of phenobarbital [38]. Moreover, phenobarbital does not protect against the long-term cognitive sequelae in juvenile rats [39], while sodium valproate does. However, other data suggest that phenobarbital may have neuroprotective effects against seizure-induced injury in adult animals [40] and hypoxic-ischemic injury in neonatal rats [41]. The continued examination of dose, age, and factors (i.e., history of seizures or hypoxia) that may modify susceptibility to PB-induced behavioral toxicity is an area in need of further research. In parallel, we suggest that identification of anticonvulsant drugs or adjunct treatments that minimize or avoid neurotoxicity should remain a high priority.
Highlights.
Neonatal rats were treated with phenobarbital and their behavior assessed as adults
Neonatal phenobarbital exposure caused deficits in prepulse inhibition in adulthood
Neonatal phenobarbital exposure impaired passive avoidance learning in adulthood
Neonatal phenobarbital exposure did not alter cocaine place preference conditioning
Footnotes
Disclosures: None of the authors has any conflict of interest to disclose. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bartha AI, Shen J, Katz KH, Mischel RE, Yap KR, Ivacko JA, et al. Neonatal seizures: multicenter variability in current treatment practices. Pediatr Neurol. 2007;37:85–90. doi: 10.1016/j.pediatrneurol.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 2.Glass HC, Kan J, Bonifacio SL, Ferriero DM. Neonatal seizures: treatment practices among term and preterm infants. Pediatr Neurol. 2012;46:111–5. doi: 10.1016/j.pediatrneurol.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guidelines on Neonatal Seizures. World Health Organization; 2012. [PubMed] [Google Scholar]
- 4.Sankar R, Painter MJ. Neonatal seizures: after all these years we still love what doesn’t work. Neurology. 2005;64:776–7. doi: 10.1212/01.WNL.0000157320.78071.6D. [DOI] [PubMed] [Google Scholar]
- 5.Scher MS, Alvin J, Gaus L, Minnigh B, Painter MJ. Uncoupling of EEG-clinical neonatal seizures after antiepileptic drug use. Pediatr Neurol. 2003;28:277–80. doi: 10.1016/s0887-8994(02)00621-5. [DOI] [PubMed] [Google Scholar]
- 6.Farwell JR, Lee YJ, Hirtz DG, Sulzbacher SI, Ellenberg JH, Nelson KB. Phenobarbital for febrile seizures--effects on intelligence and on seizure recurrence. N Engl J Med. 1990;322:364–9. doi: 10.1056/NEJM199002083220604. [DOI] [PubMed] [Google Scholar]
- 7.Sulzbacher S, Farwell JR, Temkin N, Lu AS, Hirtz DG. Late cognitive effects of early treatment with phenobarbital. Clin Pediatr (Phila) 1999;38:387–94. doi: 10.1177/000992289903800702. [DOI] [PubMed] [Google Scholar]
- 8.Bittigau P, Sifringer M, Genz K, Reith E, Pospischil D, Govindarajalu S, et al. Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proc Natl Acad Sci U A. 2002;99:15089–94. doi: 10.1073/pnas.222550499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bittigau P, Sifringer M, Ikonomidou C. Antiepileptic drugs and apoptosis in the developing brain. Ann N Y Acad Sci. 2003;993:103–114. doi: 10.1111/j.1749-6632.2003.tb07517.x. discussion 123–124. [DOI] [PubMed] [Google Scholar]
- 10.Forcelli PA, Kim J, Kondratyev A, Gale K. Pattern of antiepileptic drug-induced cell death in limbic regions of the neonatal rat brain. Epilepsia. 2011;52:e207–211. doi: 10.1111/j.1528-1167.2011.03297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaindl AM, Koppelstaetter A, Nebrich G, Stuwe J, Sifringer M, Zabel C, et al. Brief alteration of NMDA or GABAA receptor-mediated neurotransmission has long term effects on the developing cerebral cortex. Mol Cell Proteomics. 2008;7:2293–310. doi: 10.1074/mcp.M800030-MCP200. [DOI] [PubMed] [Google Scholar]
- 12.Forcelli PA, Janssen MJ, Vicini S, Gale K. Neonatal exposure to antiepileptic drugs disrupts striatal synaptic development. Ann Neurol. 2012 doi: 10.1002/ana.23600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Forcelli PA, Janssen MJ, Stamps LA, Sweeney C, Vicini S, Gale K. Therapeutic strategies to avoid long-term adverse outcomes of neonatal antiepileptic drug exposure. Epilepsia. 2010;51 (Suppl 3):18–23. doi: 10.1111/j.1528-1167.2010.02603.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhardwaj S, Forcelli P, Palchik G, Gale K, Srivastava LK, Kondratyev A. Neonatal exposure to phenobarbital potentiates schizophrenia-like behavioral outcomes in the rat. Neuropharmacology. 2012 doi: 10.1016/j.neuropharm.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forcelli PA, Kozlowski R, Snyder C, Kondratyev A, Gale K. Effects of neonatal antiepileptic drug exposure on cognitive, emotional, and motor function in adult rats. J Pharmacol Exp Ther. 2012;340:558–66. doi: 10.1124/jpet.111.188862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Forcelli PA. Doctoral Dissertation. Georgetown University; 2011. Sequelae of Neonatal Antiepileptic Drug Exposure. [Google Scholar]
- 17.Stefovska VG, Uckermann O, Czuczwar M, Smitka M, Czuczwar P, Kis J, et al. Sedative and anticonvulsant drugs suppress postnatal neurogenesis. Ann Neurol. 2008;64:434–45. doi: 10.1002/ana.21463. [DOI] [PubMed] [Google Scholar]
- 18.Pick C, Yanai J. Long-term reduction in spontaneous alternations after early exposure to phenobarbital. Int J Dev Neurosci. 1984;2:223–8. doi: 10.1016/0736-5748(84)90016-9. [DOI] [PubMed] [Google Scholar]
- 19.Pick C, Yanai J. Long term reduction in eight arm maze performance after early exposure to phenobarbital. Int J Dev Neurosci. 1985;3:223–7. doi: 10.1016/0736-5748(85)90027-9. [DOI] [PubMed] [Google Scholar]
- 20.Rogel-Fuchs Y, Newman ME, Trombka D, Zahalka EA, Yanai J. Hippocampal cholinergic alterations and related behavioral deficits after early exposure to phenobarbital. Brain Res Bull. 1992;29:1–6. doi: 10.1016/0361-9230(92)90002-f. [DOI] [PubMed] [Google Scholar]
- 21.Pereira de Vasconcelos A, Colin C, Desor D, Divry M, Nehlig A. Influence of early neonatal phenobarbital exposure on cerebral energy metabolism and behavior. Exp Neurol. 1990;108:176–87. doi: 10.1016/0014-4886(90)90025-n. [DOI] [PubMed] [Google Scholar]
- 22.Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Hum Dev. 1979;3:79–83. doi: 10.1016/0378-3782(79)90022-7. [DOI] [PubMed] [Google Scholar]
- 23.Ouvrier RA, Goldsmith R. Phenobarbitone dosage in neonatal convulsions. Arch Dis Child. 1982;57:653–7. doi: 10.1136/adc.57.9.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Filippi L, la Marca G, Cavallaro G, Fiorini P, Favelli F, Malvagia S, et al. Phenobarbital for neonatal seizures in hypoxic ischemic encephalopathy: a pharmacokinetic study during whole body hypothermia. Epilepsia. 2011;52:794–801. doi: 10.1111/j.1528-1167.2011.02978.x. [DOI] [PubMed] [Google Scholar]
- 25.Turhan AH, Atici A, Okuyaz C, Uysal S. Single Enteral Loading Dose of Phenobarbital for Achieving Its Therapeutic Serum Levels in Neonates. Croat Med J. 2010;51:215–8. doi: 10.3325/cmj.2010.51.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kubova H, Mares P. Anticonvulsant effects of phenobarbital and primidone during ontogenesis in rats. Epilepsy Res. 1991;10:148–55. doi: 10.1016/0920-1211(91)90007-3. [DOI] [PubMed] [Google Scholar]
- 27.Velísek L, Kubová H, Velísková J, Mares P, Ortová M. Action of antiepileptic drugs against kainic acid-induced seizures and automatisms during ontogenesis in rats. Epilepsia. 1992;33:987–93. doi: 10.1111/j.1528-1157.1992.tb01748.x. [DOI] [PubMed] [Google Scholar]
- 28.Forcelli PA, West EA, Murnen AT, Malkova L. Ventral pallidum mediates amygdala-evoked deficits in prepulse inhibition. Behav Neurosci. 2012 doi: 10.1037/a0026898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Forcelli PA, Heinrichs SC. Teratogenic effects of maternal antidepressant exposure on neural substrates of drug-seeking behavior in offspring. Addict Biol. 2008;13:52–62. doi: 10.1111/j.1369-1600.2007.00078.x. [DOI] [PubMed] [Google Scholar]
- 30.Swerdlow NR, Geyer MA, Braff DL. Neural circuit regulation of prepulse inhibition of startle in the rat: current knowledge and future challenges. Psychopharmacology (Berl) 2001;156:194–215. doi: 10.1007/s002130100799. [DOI] [PubMed] [Google Scholar]
- 31.Roozendaal B, Koolhaas JM, Bohus B. The central amygdala is involved in conditioning but not in retention of active and passive shock avoidance in male rats. Behav Neural Biol. 1993;59:143–9. doi: 10.1016/0163-1047(93)90873-g. [DOI] [PubMed] [Google Scholar]
- 32.Wilensky AE, Schafe GE, LeDoux JE. The amygdala modulates memory consolidation of fear-motivated inhibitory avoidance learning but not classical fear conditioning. J Neurosci Off J Soc Neurosci. 2000;20:7059–66. doi: 10.1523/JNEUROSCI.20-18-07059.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilensky AE, Schafe GE, LeDoux JE. Functional inactivation of the amygdala before but not after auditory fear conditioning prevents memory formation. J Neurosci Off J Soc Neurosci. 1999;19:RC48. doi: 10.1523/JNEUROSCI.19-24-j0006.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jinks AL, McGregor IS. Modulation of anxiety-related behaviours following lesions of the prelimbic or infralimbic cortex in the rat. Brain Res. 1997;772:181–90. doi: 10.1016/s0006-8993(97)00810-x. [DOI] [PubMed] [Google Scholar]
- 35.Perucca E. Treatment of epilepsy in developing countries. BMJ. 2007;334:1175–6. doi: 10.1136/bmj.39065.460208.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ilangaratne NB, Mannakkara NN, Bell GS, Sander JW. Phenobarbital: missing in action. Bull World Health Organ. 2012;90:871–871A. doi: 10.2471/BLT.12.113183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kwan P, Brodie MJ. Phenobarbital for the treatment of epilepsy in the 21st century: a critical review. Epilepsia. 2004;45:1141–9. doi: 10.1111/j.0013-9580.2004.12704.x. [DOI] [PubMed] [Google Scholar]
- 38.Kim J. Doctoral Dissertation. Georgetown Universitty; 2007. Effects of repeated brief seizures and antiepileptic drugs in the developing rat brain. [Google Scholar]
- 39.Bolanos AR, Sarkisian M, Yang Y, Hori A, Helmers SL, Mikati M, et al. Comparison of valproate and phenobarbital treatment after status epilepticus in rats. Neurology. 1998;51:41–8. doi: 10.1212/wnl.51.1.41. [DOI] [PubMed] [Google Scholar]
- 40.Sutula T, Cavazos J, Golarai G. Alteration of long-lasting structural and functional effects of kainic acid in the hippocampus by brief treatment with phenobarbital. J Neurosci Off J Soc Neurosci. 1992;12:4173–87. doi: 10.1523/JNEUROSCI.12-11-04173.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barks JD, Liu Y-Q, Shangguan Y, Silverstein FS. Phenobarbital augments hypothermic neuroprotection. Pediatr Res. 2010;67:532–7. doi: 10.1203/PDR.0b013e3181d4ff4d. [DOI] [PMC free article] [PubMed] [Google Scholar]